

Abstract
The mechanisms regulating proximal tubule ammonia metabolism are incompletely understood. The present study addressed the role of the proximal tubule basolateral electrogenic Na+-coupled bicarbonate cotransporter (NBCe1; Slc4a4) in renal ammonia metabolism. We used mice with heterozygous and homozygous NBCe1 gene deletion and compared these mice with their wild-type littermates. Because homozygous NBCe1 gene deletion causes 100% mortality before day 25, we studied mice at day 8 (±1 day). Both heterozygous and homozygous gene deletion caused a gene dose-related decrease in serum bicarbonate. The ability to lower urinary pH was intact, and even accentuated, with NBCe1 deletion. However, in contrast to the well-known effect of metabolic acidosis to increase urinary ammonia excretion, NBCe1 deletion caused a gene dose-related decrease in ammonia excretion. There was no identifiable change in proximal tubule structure by light microscopy. Examination of proteins involved in renal ammonia metabolism showed decreased expression of phosphate-dependent glutaminase and phosphoenolpyruvate carboxykinase, key enzymes in proximal tubule ammonia generation, and increased expression of glutamine synthetase, which recycles intrarenal ammonia and regenerates glutamine. Expression of key proteins involved in ammonia transport outside of the proximal tubule (rhesus B glycoprotein and rhesus C glycoprotein) was not significantly changed by NBCe1 deletion. We conclude from these findings that NBCe1 expression is necessary for normal proximal tubule ammonia metabolism.
Keywords: acid-base, ammonia, electrogenic sodium-coupled bicarbonate cotransporter 1, proximal tubule
ammonia1 metabolism plays a critical role in the maintenance of acid-base homeostasis. Under basal conditions, renal ammonia excretion is the predominant component of net acid excretion (66, 68, 69). Furthermore, in response to acid-base disturbances such as metabolic acidosis (32), electrolyte disorders such as hypokalemia (5), and drug-induced increases in renal acid excretion such as chronic lithium administration (64), changes in ammonia excretion are the predominant component of the increase in net acid excretion (63, 67, 69).
The pathways underlying regulation of renal ammonia metabolism remain incompletely understood. However, findings from humans with a specific acid-base disturbance, familial forms of proximal renal tubular acidosis (pRTA), may provide important insights into a novel mechanism regulating renal ammonia production. This condition is characterized by clinical evidence of abnormal proximal tubule bicarbonate transport leading to the development of spontaneous metabolic acidosis, without abnormalities in glucose, phosphate, or amino acid transport. The appropriate renal response to metabolic acidosis is well known to involve increased ammonia excretion. However, carefully performed studies examining patients with familial pRTA showed they do not have the increase in ammonia excretion rates expected in view of their underlying metabolic acidosis (9, 39). We believe that these findings indicate that individuals with familial pRTA have abnormal renal ammonia metabolism.
Familial pRTA could theoretically be a disorder of any of several proteins involved in proximal tubule bicarbonate transport. However, extensive genetic testing of these individuals has, to date, identified abnormalities in only a single gene, that for the basolateral electrogenic Na+-coupled bicarbonate transporter (NBCe1) (13, 14, 22–24, 58, 60). NBCe1 is a member of the SLC4 family of integral membrane proteins, i.e., Slc4a4. It is located in the proximal tubule basolateral plasma membrane, where it mediates electrogenic transport of Na+ and bicarbonate (1, 29, 48). Transport studies have suggested that NBCe1 is the primary mechanism of proximal tubule basolateral bicarbonate transport required for normal proximal tubule bicarbonate reabsorption (1, 49, 50).
Because defects in NBCe1 cause pRTA in people and people with pRTA have inappropriately low urinary ammonia excretion, we speculated that NBCe1 expression was necessary for normal renal ammonia metabolism. To examine this question, we used mice with deletion of the NBCe1 gene, which develop spontaneous metabolic acidosis (16). Because NBCe1 deletion in these mice causes 100% perinatal mortality (16), we examined mice at day 8 ± 1. We determined the effects of NBCe1 deletion on systemic acid-base homeostasis and urinary ammonia excretion and on the expression of proteins involved in renal ammonia metabolism. Our results indicate that NBCe1 expression is necessary for normal renal ammonia metabolism and that its deletion results in changes in the expression of multiple proteins involved in proximal tubule ammonia metabolism.
METHODS
Animals.
Mice with heterozygous NBCe1 deletion were obtained from original colonies generated by Dr. Gary Shull (University of Cincinnati) (16). Heterozygous mice were bred in the University of Florida Cancer and Genetics Transgenic Animal Facility by trained personnel. All mice were genotyped using DNA obtained from tail-clip specimens using standard techniques, as previously described (5, 16, 33, 35, 36). Mice were studied at day 8 ± 1. Mice were euthanized after the induction of anesthesia using inhalant isoflurane and opening of the chest cavity. All animal experiments were approved by the University of Florida College of Medicine and the North Florida/South Georgia Veterans Health System Institutional Animal Care and Use Committees.
Plasma analyses.
Blood was obtained by cannulation of the heart, drawn in a heparinized syringe, and immediately analyzed for Na+, K+, and bicarbonate concentrations using a Siemens microanalytic blood gas analyzer (RAPIDLab 348 analyzer, Siemens).
Urinary experiments.
Urine was collected by bladder puncture after euthanasia or by immediate aspiration after spontaneous voiding during the anesthesia process and was frozen for subsequent analysis. Urine ammonia was measured using a modification of a commercially available kit (A7553, Pointe Scientific, Canton, MI) as previously described (6, 32). Urine creatinine was measured by the modified version of the Jaffe reaction, in which creatinine was treated with an alkaline picrate solution to yield a colored complex. Samples were loaded on a 96-well microplate and read on the microplate reader (SpectraMax 190, Molecular Devices) and analyzed using the microplate reader software (SoftMax Pro 6.3, Molecular Devices). Urine osmolality was measured using a Wescor 5500 Vapor Pressure Osmometer.
We used a novel approach to assess urine pH in 2-μl samples that we developed. Urine pH was measured using the color reaction of pH strips (ColorpHast, EMD Chemicals). Briefly, 2 μl of urine were spotted on pH strips, and the color reaction was quantified using RGB analysis of digital micrographs of the pH strips. Calibration was performed using known pH standards over the expected urine pH range of 4–7. Multivariable linear regression of the RGB color characteristics was then used to determine urine pH.
Antibodies.
Affinity-purified antibodies to rhesus B glycoprotein (Rhbg) and rhesus C glycoprotein (Rhcg) generated in our laboratory have been previously characterized (6, 25, 32, 41, 61, 65). Dr. Norman Curthoys (Colorado State University) graciously provided antibodies to phosphate-dependent glutaminase (PDG). Antibodies to phosphoenolpyruvate carboxykinase (PEPCK) were obtained from Cayman Chemical (Ann Arbor, MI), antibodies to glutamine synthetase were obtained from Chemicon (Temecula, CA), and antibodies to Na+/H+ exchanger 3 (NHE3) were obtained from StressMarq Biosciences (Victoria, BC, Canada). Antibodies to β-actin (13E5) were obtained from Cell Signaling Technology (Danvers, MA).
Protein preparation.
Animals were anesthetized with inhalant isoflurane. The kidneys were rapidly removed, snap frozen in liquid nitrogen, and stored frozen at −70°C until use. Tissues were homogenized in T-PER Tissue Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL) using microtube pestles (USA Scientific, Ocala, FL), and protein was extracted according to the manufacturer's recommended procedures. An aliquot was used for total protein quantification using a BCA assay, and the remainder was stored frozen at −70°C until use.
Immunoblot procedures.
Immunoblot analysis was performed as we have previously described (6, 26, 34). Briefly, 20 μg of renal protein were electrophoresed on a 10% PAGE ReadyGel (Bio-Rad, Hercules, CA). Gels were then transferred electrophoretically to nitrocellulose membranes, blocked with 5 g/dl nonfat dry milk in Blotto buffer (50 mM Tris, 150 mM NaCl, 5 mM Na2EDTA, and 0.05% Tween 20, pH 7.6), and incubated at 4°C overnight with primary antibody diluted in nonfat dry milk. Loading and transfer equivalence were assessed with Ponceau S staining. After being washed, membranes were exposed to secondary antibody (goat anti-rabbit IgG, Millipore, Billerica, MA) conjugated to horseradish peroxidase at a dilution of 1:5,000. Sites of antibody-antigen reaction were visualized using enhanced chemiluminescence (SuperSignal West Pico Substrate, Pierce) and a Kodak Image Station 440CF digital imaging system. In selected experiments, blots were stripped, and expression of the housekeeping protein β-actin was determined. Band density was quantified using Kodak 1D (version 5.0) software (Kodak Scientific Imaging, New Haven, CT). Band density was normalized such that mean density in wild-type tissues was 100. The absence of saturation was confirmed by examining pixel intensity distribution in all immunoblots.
Tissue preparation for immunolocalization and histology.
Mice were anesthetized with inhalant isoflurane. The kidneys were removed, bisected, and preserved by immersion fixation in periodate-lysine-2% paraformaldehyde for 48 h at 4°C. Kidney samples were embedded in polyester wax made using polyethylene glycol 400 distearate (Polysciences, Warrington, PA) with 10% 1-hexadecanol, and 3-μm-thick sections were cut and mounted on gelatin-coated glass slides.
Histology.
For histological examination, sections were dewaxed and stained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI) and eosin (Richard-Allan Scientific) using standard methods, dehydrated, and then mounted with Permount (Fisher).
Immunohistochemistry.
Immunolocalization was accomplished using standard immunoperoxidase procedures in our laboratory (5, 35, 36, 62). Briefly, sections were dewaxed in ethanol, rehydrated, and then rinsed in PBS. For some experiments, antigen retrieval was accomplished by immersing slides in Trilogy (Cell Marque, Rocklin, CA) and heating to 88–96°C for 1 h. Endogenous peroxidase activity was blocked by incubating sections in 3% H2O2 in distilled water for 45 min. Sections were blocked for 15 min with serum-free Protein Block (Dako Cytomation) and then incubated at 4°C overnight with primary antibody. Sections were washed in PBS and incubated for 30 min with polymer-linked peroxidase-conjugated goat anti-rabbit IgG (MACH2, Biocare Medical, Concord, CA), washed again with PBS, and then exposed to diaminobenzidine for 5 min. Sections were washed in distilled water, dehydrated with xylene, mounted, and observed by light microscopy. Comparisons of labeling were made only between sections from the same immunohistochemistry experiment.
Sections were examined on a Nikon E600 microscope equipped with differential interference contrast optics and photographed using a DXM1200F digital camera and ACT-1 software (Nikon) or a Leica DM2000 microscope and photographed using a Leica DFC425 digital camera and Leica DFC Twain Software and LAS application suite (Leica Microsystems, Buffalo Grove, IL). Color adjustment was performed using Adobe Photoshop CS2 and CS5 software (Adobe Systems, San Jose, CA).
Statistics.
Results are presented as means ± SE; n values are numbers of animals studied. Statistical analyses were performed using ANOVA with a post hoc t-test. P values of <0.05 were taken as statistically significant.
RESULTS
Physiological data.
The effects of NBCe1 deletion on plasma electrolytes was determined to confirm the development of metabolic acidosis associated with pRTA. In wild-type 8-day-old pups, plasma bicarbonate averaged 26.4 ± 1.0 mmol/l; this was decreased significantly with heterozygous NBCe1 deletion to 19.8 ± 1.9 mmol/l and was decreased significantly further by homozygous NBCe1 deletion to 10.3 ± 0.6 mmol/l (n = 7, 7, and 6, respectively; P < 0.001 for all comparisons). Figure 1 shows these findings. In contrast to the effects on plasma bicarbonate, plasma Na+ and K+ concentrations were not significantly altered by genotype on day 8. Thus, NBCe1 deletion results in spontaneous metabolic acidosis in mice on day 8.
Fig. 1.
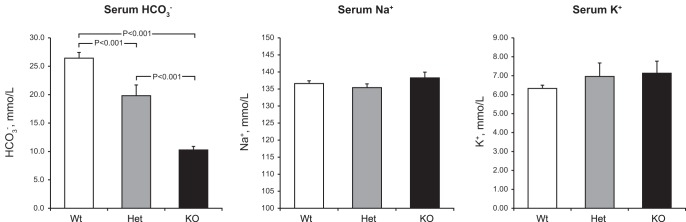
Effect of electrogenic Na+-coupled bicarbonate (HCO3−) cotransporter (NBCe1) deletion on serum electrolytes. Left: effects on serum HCO3−. NBCe1 deletion induced a gene dose-dependent decrease in serum HCO3−. n = 7, 7, and 6 for wild-type (Wt), heterozygous (Het), and homozygous NBCe1 deletion [knockout (KO)], respectively. Middle and right: effects on serum Na+ (middle) and K+ (right). NBCe1 deletion did not significantly alter either serum Na+ or K+. n = 5, 5, and 4 for wild-type, heterozygous, and homozygous NBCe1 deletion, respectively, for Na+ and n = 7, 7, and 5 for wild-type, heterozygous, and homozygous NBCe1 deletion, respectively, for K+.
Under normal circumstances, the appropriate renal response to metabolic acidosis is to increase urinary ammonia excretion. In contrast, despite the presence of spontaneous metabolic acidosis, mice with heterozygous NBCe1 deletion exhibited a significant decrease in ammonia excretion and mice with homozygous deletion had a further decrease in urinary ammonia excretion (Fig. 2). Thus, NBCe1 deletion causes an abnormal pattern of urinary ammonia excretion.
Fig. 2.
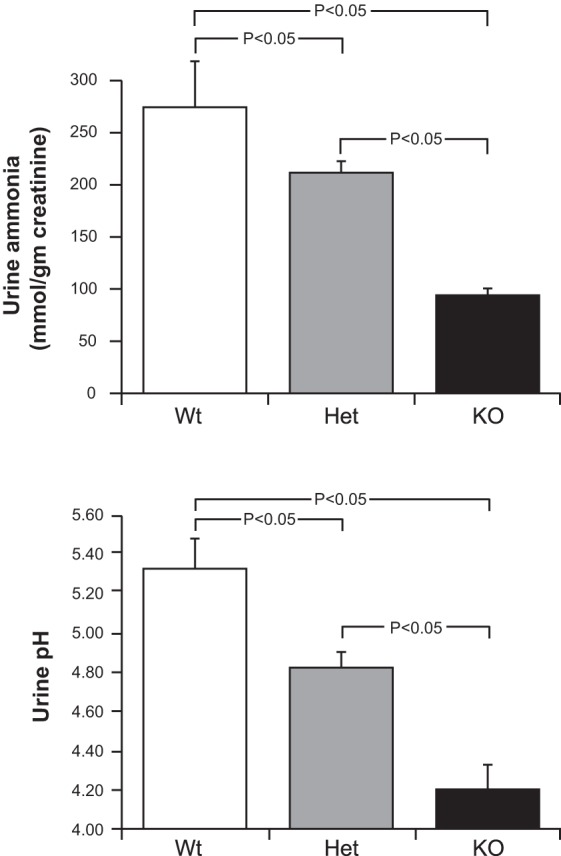
Effect of NBCe1 deletion on urinary parameters. Top: effects on urine ammonia. Despite the associated metabolic acidosis with NBCe1 gene deletion, ammonia excretion decreased with heterozygous gene deletion and was suppressed significantly further with homozygous deletion. n = 26, 44, and 16 for wild-type, heterozygous, and homozygous NBCe1 deletion, respectively. Bottom: effects on urine pH. Urine pH was significantly more acidic (lower) in mice with heterozygous NBCe1 deletion than in wild-type mice and was decreased significantly further by homozygous NBCe1 deletion. n = 18, 29, and 11 for wild-type, heterozygous, and homozygous NBCe1 deletion, respectively.
Urinary ammonia excretion is stimulated by an acidic2 urine pH and is inhibited by urine alkalinization. pRTA can be associated with either acidic or alkaline urine depending on the serum bicarbonate concentration (10, 21, 56). Figure 2 shows that NBCe1 deletion in these mouse pups was associated with increasingly acidic urine pH and that there was a gene-dose relationship. These findings indicate that in pups with NBCe1 deletion, by postnatal day 8, bicarbonaturia was not present. More important, though, is that the decreased ammonia excretion observed with NBCe1 deletion cannot be attributed to an inability to acidify the urine.
A substantial proportion (60–80%) of urinary ammonia is secreted by the collecting duct (20, 63). To screen further for generalized defects in collecting duct function, we examined urine osmolality. NBCe1 deletion did not alter osmolality significantly (wild-type pups: 596 ± 74 mosm/kg H2O, heterozygous pups: 693 ± 51 mosm/kg H2O, and knockout pups: 726 ± 34 mosm/kg H2O, n = 6, 12, and 5, respectively, P = not signficant). The finding that heterozygous and knockout pups, like wild-type pups, can concentrate urine to at least approximately twice that of plasma osmolality, in combination with intact urine acidification, suggests that a generalized defect in collecting duct function is not present.
Effect of NBCe1 deletion on proximal tubule structure.
These profound effects of NBCe1 deletion on ammonia metabolism could indicate either that NBCe1 has a critical role in renal ammonia metabolism or that its deletion induces significant nonspecific changes in the proximal tubule that inhibit normal function. To test the latter possibility, we examined kidney structure by histological examination. By light microscopy, proximal tubule segments in NBCe1 knockout kidneys appeared normal (Fig. 3). Overall renal anatomy appeared relatively unchanged, with the exception of mild dilation of some collecting duct segments in the renal cortex. Thus, the effect of NBCe1 gene deletion to cause abnormal renal ammonia metabolism does not appear to be the result of a generalized disturbance in renal anatomy.
Fig. 3.
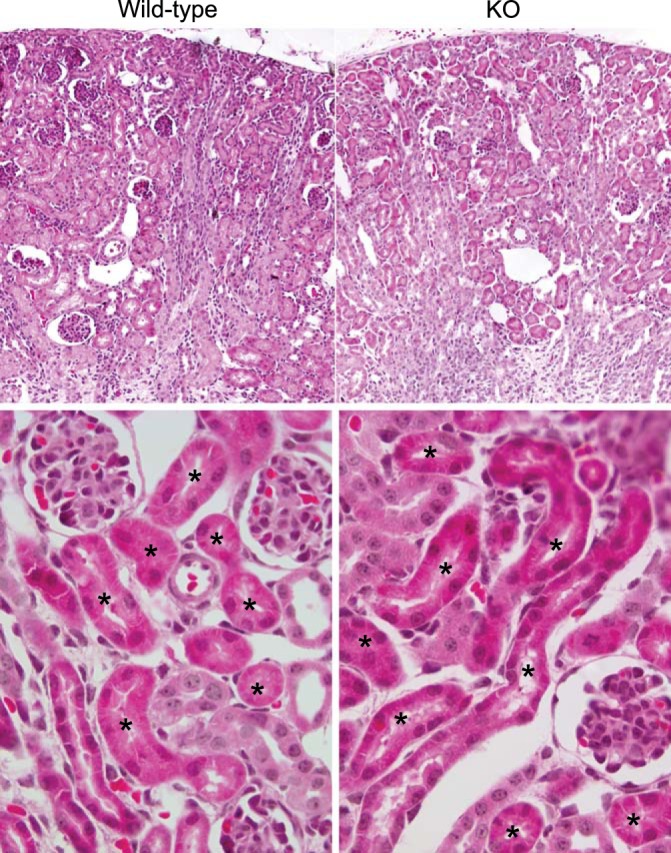
Effect of NBCe1 deletion on renal morphology. Low-power micrographs (top) and high-power micrographs (bottom) of hematoxylin and eosin-stained kidney sections from wild-type and homozygous NBCe1 deletion (KO) mice. There was no detectable difference in proximal tubule morphology as a consequence of NBCe1 deletion. *Proximal tubule segments.
Expression of proteins involved in proximal tubule ammonia generation.
To begin to determine the mechanism through which NBCe1 deletion decreases ammonia excretion, we examined the expression of key proteins involved in ammonia metabolism. Two important proteins involved in ammoniagenesis are PDG and PEPCK (12, 69). The normal response to metabolic acidosis is increased expression of both (6, 12, 32, 34, 36). However, NBCe1 deletion, despite the presence of metabolic acidosis, decreased both PDG expression (Fig. 4) and PEPCK expression (Fig. 5); immunohistochemistry confirmed the latter was due to decreased proximal tubule PEPCK expression (Fig. 5).
Fig. 4.
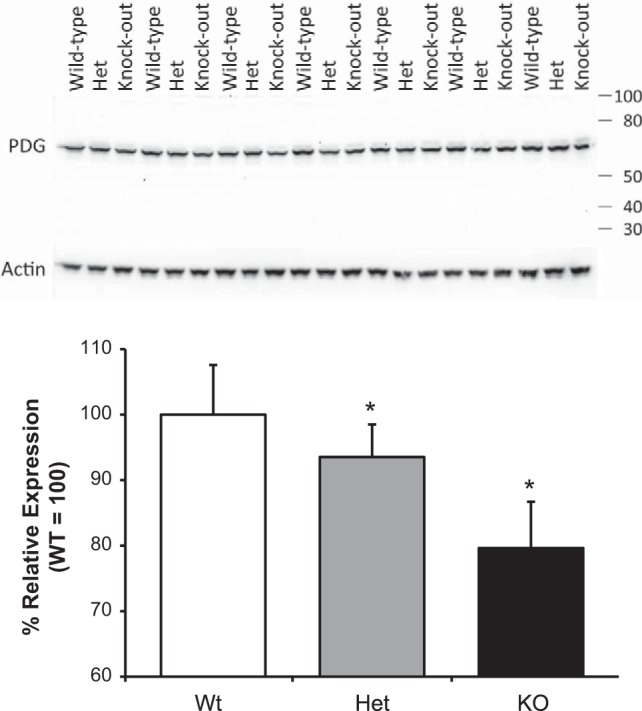
Effect of NBCe1 deletion on phosphate-dependent glutaminase (PDG) expression. Top: effects of NBCe1 deletion on PDG expression. Middle: expression of the “housekeeping” protein β-actin in the same blot showing that changes in PDG expression were not due to changes in protein loading or transfer. Bottom: quantitative analysis of PDG expression relative to β-actin expression. NBCe1 deletion resulted in significant inhibition of PDG expression. n = 7 for each genotype. *P < 0.05.
Fig. 5.
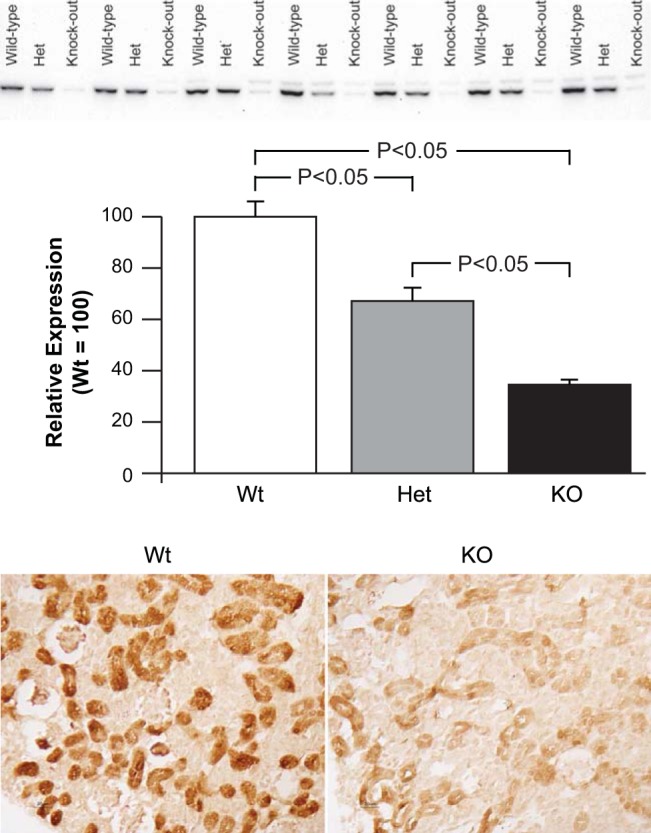
Effect of NBCe1 deletion on phosphoenolpyruvate carboxykinase (PEPCK) expression. Top: effects of NBCe1 deletion on PEPCK expression by immunoblot analysis. Heterozygous NBCe1 deletion significantly decreased PEPCK expression despite the associated metabolic acidosis. Homozygous NBCe1 deletion resulted in more marked loss of PEPCK expression. n = 7 for each genotype. Bottom: effects of NBCe1 deletion on PEPCK expression by immunohistochemistry. Compared with PEPCK immunolabel in wild-type mice, there was very weak PEPCK immunolabel in the proximal tubule of homozygous NBCe1 deletion (KO) mice.
Effect of NBCe1 deletion on proximal tubule ammonia recycling.
In addition to generating ammonia, the proximal tubule also has an ammonia recycling process. The enzyme glutamine synthetase recycles ammonia by catalyzing the reaction of NH4+ with glutamate to regenerate glutamine (11, 62). In normal mice, both metabolic acidosis and hypokalemia decrease glutamine synthetase expression; this contributes to the increase in net ammoniagenesis (11, 62, 72). However, in mice with NBCe1 deletion, glutamine synthetase expression is increased significantly despite the associated metabolic acidosis (Fig. 6). Moreover, immunohistochemistry showed that this increase occurred in proximal tubule cells (Fig. 6). Thus, NBCe1 expression is necessary for the normal regulation of glutamine synthetase expression. Moreover, the increased glutamine synthetase expression with NBCe1 deletion shows that the decreased PDG and PEPCK expression does not occur because of a generalized decrease in proximal tubule protein expression.
Fig. 6.
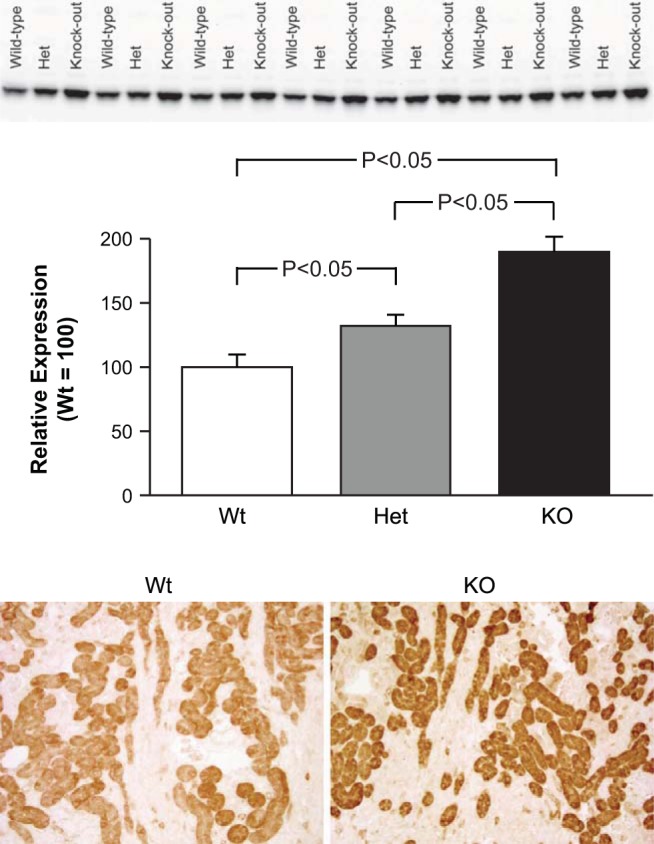
Effect of NBCe1 deletion of glutamine synthetase expression. Top: effects of NBCe1 on glutamine synthetase expression by immunoblot analysis. Heterozygous NBCe1 deletion significantly increased glutamine synthetase expression, and homozygous deletion (KO) increased expression significantly further. This was in contrast to the expected inhibition of glutamine synthetase expression in response to metabolic acidosis. n = 7 for each genotype. Bottom: representative immunohistochemistry of glutamine synthetase expression in the cortex. Homozygous deletion (KO) increased glutamine synthetase immunolabel in proximal tubule segments.
Effect of NBCe1 deletion on NHE3 expression.
NHE3 is thought to be the primary protein involved in proximal tubule ammonia secretion (7, 67). Numerous studies have shown that metabolic acidosis increases NHE3 expression (2, 31, 71), likely contributing to the increased luminal ammonia secretion observed with metabolic acidosis (43–45). In contrast, in mice with NBCe1 deletion, either heterozygous or homozygous, despite the presence of metabolic acidosis, there was a tendency for decreased NHE3 expression, although this did not reach statistical significance (Fig. 7). Examination of expression of the housekeeping protein β-actin showed no significant differences, indicating that the variability in NHE3 expression cannot be explained by genotype-specific differences in protein loading or transfer for the immunoblot analysis (data not shown). We speculate that the observed interanimal variations in NHE3 expression may reflect interanimal variability in the developmental pattern of NHE3 expression (3).
Fig. 7.
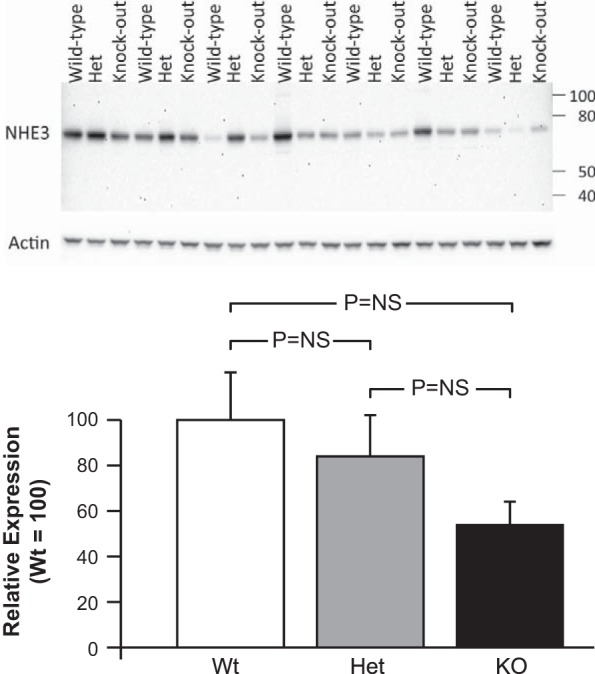
Na+/H+ exchanger 3 (NHE3) expression and the effect of NBCe1 deletion. Top: immunoblot analysis of NHE3 expression in kidneys from mice with intact (Wt), heterozygous NBCe1 deletion (Het), and homozygous NBCe1 deletion (KO). The apparent molecular mass for mouse NHE3 of ∼75 kDa was similar to that which we and others have previously reported (33, 36, 40). Middle: expression of the housekeeping protein β-actin in the same blot showing that differences in NHE3 expression were not due to differences in protein loading or transfer. Bottom: quantitative analysis. NBCe1 deletion caused a trend for a gene dose-related decrease of NHE3 expression, but the differences were not statistically significant (NS).
Expression of proteins involved in collecting duct ammonia transport.
Collecting duct ammonia secretion involves ammonia transport mediated largely by Rhbg and Rhcg. Immunohistochemistry showed that expression and localization of each of these was not detectably altered by NBCe1 deletion and that the numbers and distribution of intercalated cells were normal (Fig. 8). Thus, NBCe1 deletion appears to cause abnormalities in ammonia metabolism and transport specific to the proximal tubule.
Fig. 8.
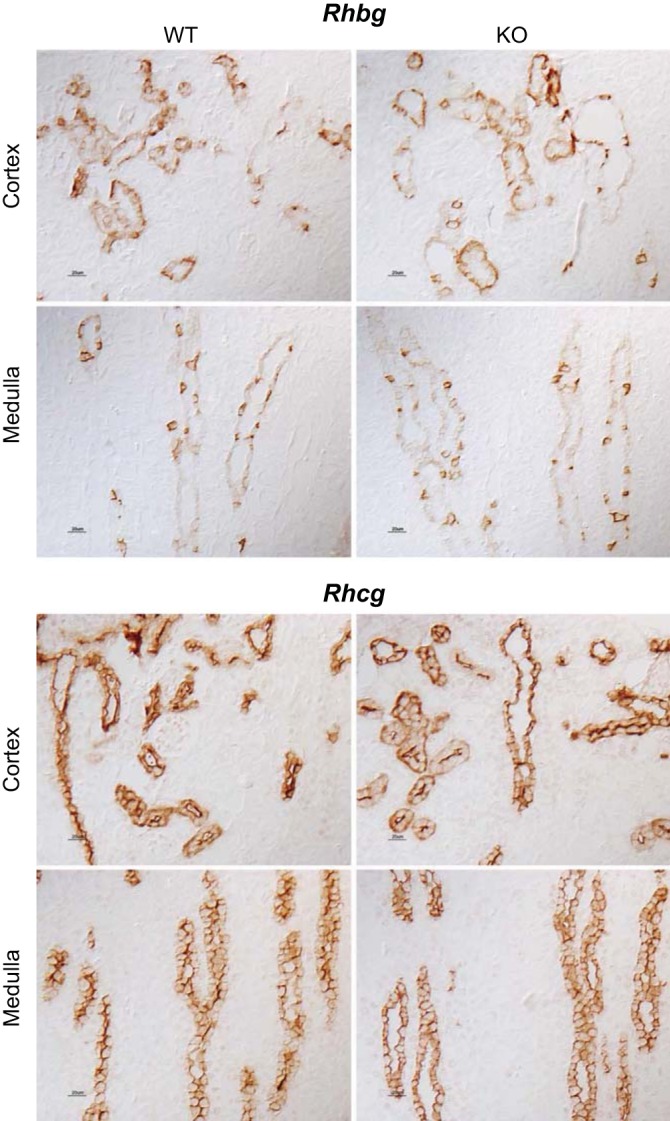
Effect of NBCe1 deletion on proteins involved in collecting duct ammonia secretion. Top: rhesus B glycoprotein (Rhbg) localization in the cortex and medulla of mice with intact NBCe1 expression and with NBCe1 deletion. There was no detectable difference in Rhbg immunolabel in either the cortex or medulla as a result of NBCe1 deletion. Bottom: rhesus C glycoprotein (Rhcg) localization in the cortex and medulla of mice with intact NBCe1 expression and with NBCe1 deletion. There was no detectable difference in Rhcg immunolabel in the cortex or medulla as result of NBCe1 deletion.
DISCUSSION
The present study examined the effect of deletion of NBCe1 (Slc4a4) on renal ammonia metabolism. The experiments presented in this report demonstrate that NBCe1 deletion suppresses renal ammonia excretion despite the simultaneous spontaneous metabolic acidosis and intact, even accentuated, ability to acidify the urine. The suppressed ammonia excretion is associated with abnormal expression of multiple proteins involved in proximal tubule ammonia metabolism but no significant effect on expression of collecting duct ammonia transport proteins. These findings indicate that NBCe1 expression is necessary for normal proximal tubule ammonia metabolism.
Genetic abnormalities in NBCe1 are the most common causes of familial forms of pRTA in humans (23, 29, 30). The spontaneous metabolic acidosis that occurs has been attributed to abnormalities in proximal tubule bicarbonate reabsorption. When serum bicarbonate is normal, this abnormality in proximal tubule bicarbonate transport results in bicarbonate delivery to more distal epithelial segments that is beyond their capability to reabsorb, leading to bicarbonaturia and net urine alkali excretion. Eventually, urinary alkali excretion decreases the arterial bicarbonate concentration to the extent that the filtered load of bicarbonate is below the renal reabsorptive threshold. At this point, distal epithelial segments are able to reabsorb the remaining luminal bicarbonate, further urinary bicarbonate losses cease, and the urine can become acidic (21, 47, 57).
The present study identified an additional cause of the metabolic acidosis in a mouse model of pRTA due to NBCe1 defects, namely, inadequate ammonia metabolism and excretion. Ammonia metabolism and excretion are necessary to generate sufficient new bicarbonate to buffer endogenous acid production. In the present study, despite the presence of metabolic acidosis, net ammonia excretion was suppressed by NBCe1 deletion. This indicates a failure of both the normal basal ammonia metabolism necessary to buffer acid loads from endogenous acid production and a failure of the increased ammonia excretion expected in response to metabolic acidosis.
The observation that NBCe1 deletion in mice causes abnormal ammonia excretion parallels observations in humans with familial forms of pRTA. In humans, the normal renal response to metabolic acidosis involves increased renal ammonia excretion (18, 38, 46). However, in people with familial pRTA, despite their spontaneous metabolic acidosis, ammonia excretion is not significantly different than that observed in normal individuals with a normal acid-base balance (9, 39). Moreover, experimental metabolic acidosis induced by an exogenous acid load for 3 days increases ammonia excretion by 45 mmol/day in normal individuals but only by ∼20 mmol/day in individuals with familial pRTA (9). Thus, not only do humans with familial pRTA have abnormal basal ammonia metabolism, but their response to an exogenous acid load is suppressed.
In humans with familial pRTA, the most common genetic defect is in NBCe1. Defects have been identified that alter multiple components of NBCe1 function, including trafficking to the plasma membrane, trafficking to the apical rather than basolateral plasma membrane, changes in substrate affinity, and the production of nonfunctional protein (13, 14, 22, 24, 58, 60, 73, 74). At present, no studies have directly correlated specific abnormalities in the NBCe1 gene with specific defects in human ammonia metabolism. However, the present study of mice with genetic deletion of NBCe1, interpreted in conjunction with evidence from humans with familial pRTA, strongly suggests that NBCe1 is necessary for normal ammonia metabolism and excretion, both in mice and in humans.
Renal ammonia metabolism involves renal ammoniagenesis coupled to epithelial cell ammonia transport. NBCe1 expression is limited to the proximal tubule (54), and the present study shows that its deletion results in abnormal expression of multiple proteins involved in multiple components of proximal tubule ammonia metabolism. Two key proteins involved in ammoniagenesis are PDG and PEPCK, and NBCe1 deletion impairs the normal expression of both. Importantly, not only do mice with NBCe1 deletion not exhibit increased PDG and PEPCK expression, as occurs normally with metabolic acidosis (12, 59), but steady-state protein expression is actually decreased. The suppression of PEPCK expression, a cytosolic protein, is much greater than the suppression of PDG, an intramitochondrial protein. However, suppressed PEPCK expression is likely to cause feedback inhibition of upstream ammonia metabolism. This feedback inhibition would likely increase the concentration of upstream metabolites, including glutamate, and elevated levels of glutamate cause substantial inhibition of PDG activity (55, 70). Thus, NBCe1 deletion likely suppresses ammonia excretion through substantial inhibition of both PDG- and PEPCK-mediated ammonia generation.
Proximal tubule ammonia metabolism also involves a mechanism that “reverses” normal ammoniagenesis. The protein glutamine synthetase catalyzes the reaction of NH4+ with glutamate, resulting in the regeneration of glutamine and release of H+ (62). Changes in glutamine synthetase expression occur normally in conditions of altered ammonia metabolism, and metabolic acidosis typically induces decreased glutamine synthetase expression and activity (11, 32, 36, 72). By decreasing ammonia utilization for regeneration of glutamine, decreased glutamine synthetase expression in metabolic acidosis contributes to the increase in net ammonia generation. In contrast, NBCe1 deletion, despite the spontaneous metabolic acidosis, induces increased glutamine synthetase expression. Not only does this reflect abnormal regulation of glutamine synthetase expression, but also the increased glutamine synthetase expression likely contributes to decreased net ammoniagenesis and net ammonia excretion observed in these mice.
NBCe1 deletion results in a significantly lower urine pH. This significantly lower urine pH likely in part reflects the effects of metabolic acidosis to stimulate urine acidification. We suggest that the increased degree of urine acidification is also likely due in part to inadequate proximal tubule ammoniagenesis leading to inadequate ammonia available for collecting duct ammonia secretion. In particular, collecting duct ammonia secretion, the source of 60–80% of urinary ammonia, involves parallel H+ and NH3 secretion, and the secreted H+ is buffered by the secreted NH3 (15, 17, 27, 28). Thus, inadequate availability of ammonia for secretion as a result of the impairment of proximal tubule ammonia metabolism likely contributes to the excessively acidic urine pH observed in response to NBCe1 deletion.
pRTA is frequently associated with hypokalemia, but hypokalemia was not present in NBCe1 heterozygous or homozygous deletion mice. The early age of the animals studied in this report likely explains the lack of hypokalemia. The major site of K+ secretion is the collecting duct, and collecting duct K+ secretion is not present at birth and does not become evident until 4 wk of age (53). This lack of net K+ secretion appears to result from an absence of apical secretory K+ channels (4, 75) and from an increased expression of K+-reabsorbing H+-K+-ATPases (19). Thus, incomplete mechanisms of collecting duct K+ secretion may explain the lack of hypokalemia in mice with NBCe1 deletion at day 8.
NBCe1 deletion is likely to have multiple effects on the renal proximal tubule. Because NBCe1 is a major mechanism of basolateral bicarbonate exit (8, 13, 14, 16, 22–24, 37, 51, 52, 74) and because changes in its activity alter intracellular pH (42), its deletion may increase intracellular pH. These changes in intracellular pH could have direct or indirect effects to alter either the expression or activity of proteins involved in proximal tubule ammonia metabolism. Similarly, because NBCe1 is also an electrogenic transporter (8, 13, 14, 16, 22–24, 37, 51, 52, 74) and changes in its activity alter membrane potential (42), we postulate that its deletion may also alter intracellular electronegativity. This could have direct effects on cellular processes and, because intracellular electronegativity can alter intracellular Ca2+ regulation, may have direct and indirect effects mediated through intracellular Ca2+. Finally, we cannot exclude effects on the other second messenger systems involved in proximal tubule ion transport.
In summary, NBCe1 deletion results in an impairment of multiple components of renal ammonia metabolism that are localized to the proximal tubule. These include suppression of urine ammonia excretion despite an intact, even exaggerated, ability to acidify the urine in combination with abnormal expression of multiple proximal tubule proteins (PDG, PEPCK, and glutamine synthetase) involved in ammonia metabolism. In contrast, there is no apparent abnormality in expression of proteins involved in ammonia transport in the collecting duct. Thus, not only is NBCe1 necessary for proximal tubule reabsorption of filtered bicarbonate, its expression is also necessary for normal renal ammonia metabolism.
GRANTS
This work was supported by National Institutes of Health Grants R01-DK-045788 (to I. D. Weiner) and EY-017732 (to M. F. Romero) and by Department of Veterans Affairs Grant 1I01BX000818 (to I. D. Weiner).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.E.H., G.O., and H.-W.L. performed experiments; M.E.H., G.O., H.-W.L., M.F.R., J.W.V., and I.D.W. analyzed data; M.E.H., G.O., H.-W.L., J.W.V., and I.D.W. interpreted results of experiments; M.E.H., G.O., H.-W.L., J.W.V., and I.D.W. edited and revised manuscript; M.E.H., G.O., H.-W.L., M.F.R., J.W.V., and I.D.W. approved final version of manuscript; H.-W.L., J.W.V., and I.D.W. conception and design of research; H.-W.L. and I.D.W. prepared figures; I.D.W. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Sharon W. Matthews and Tanisha Thomas of the University of Florida College Of Medicine Electron Microscopy Core Laboratory for assistance with the light microscopic experiments.
Footnotes
Ammonia exists in two molecular forms, NH3 and NH4+. When referring specifically to the molecular form NH3, we state specifically “NH3.” When referring specifically to NH4+, we either state specifically “NH4+.” In this report, the term “ammonia” refers to the combination of both molecular forms.
M. E. Handlogten and G. Osis contributed equally to this work.
When using the terms “acidify” and “acidic” in reference to urine, we are referring only to urine pH. In other words, a “more acidic” urine indicates a urine with a lower urine pH. These terms do not indicate differences in either net acid excretion or titratable acid excretion.
REFERENCES
- 1.Aalkjaer C, Frische S, Leipziger J, Nielsen S, Praetorius J. Sodium coupled bicarbonate transporters in the kidney, an update. Acta Physiol Scand 181: 505–512, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Ambuhl PM, Amemiya M, Danczkay M, Lotscher M, Kaissling B, Moe OW, Preisig PA, Alpern RJ. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 271: F917–F925, 1996. [DOI] [PubMed] [Google Scholar]
- 3.Baum M, Biemesderfer D, Gentry D, Aronson PS. Ontogeny of rabbit renal cortical NHE3 and NHE1: effect of glucocorticoids. Am J Physiol Renal Fluid Electrolyte Physiol 268: F815–F820, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Benchimol C, Zavilowitz B, Satlin LM. Developmental expression of ROMK mRNA in rabbit cortical collecting duct. Pediatr Res 47: 46–52, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Bishop JM, Lee HW, Handlogten ME, Han KH, Verlander JW, Weiner ID. Intercalated cell-specific Rh B glycoprotein deletion diminishes renal ammonia excretion response to hypokalemia. Am J Physiol Renal Physiol 304: F422–F431, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop JM, Verlander JW, Lee HW, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the rhesus glycoprotein, Rh B glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol 299: F1065–F1077, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bobulescu IA, Moe OW. Luminal Na+/H+ exchange in the proximal tubule. Pflügers Arch 458: 5–21, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boron WF, Boulpaep EL. Intracellular pH regulation in the renal proximal tubule of the salamander. Basolateral HCO3− transport. J Gen Physiol 81: 53–94, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brenes LG, Sanchez MI. Impaired urinary ammonium excretion in patients with isolated proximal renal tubular acidosis. J Am Soc Nephrol 4: 1073–1078, 1993. [DOI] [PubMed] [Google Scholar]
- 10.Brenes LG, Brenes JN, Hernandez MM. Familial proximal renal tubular acidosis: a distinct clinical entity. Am J Med 63: 244–252, 1977. [DOI] [PubMed] [Google Scholar]
- 11.Conjard A, Komaty O, Delage H, Boghossian M, Martin M, Ferrier B, Baverel G. Inhibition of glutamine synthetase in the mouse kidney: a novel mechanism of adaptation to metabolic acidosis. J Biol Chem 278: 38159–38166, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol 9: 1627–1638, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demirci FY, Chang MH, Mah TS, Romero MF, Gorin MB. Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol Vis 12: 324–330, 2006. [PubMed] [Google Scholar]
- 14.Dinour D, Chang MH, Ji Satoh Smith BL, Angle N, Knecht A, Serban I, Holtzman EJ, Romero MF. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem 279: 52238–52246, 2004. [DOI] [PubMed] [Google Scholar]
- 15.DuBose TD, Good DW, Hamm LL, Wall SM. Ammonium transport in the kidney: new physiological concepts and their clinical implications. J Am Soc Nephrol 1: 1193–1203, 1991. [DOI] [PubMed] [Google Scholar]
- 16.Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 cotransporter. J Biol Chem 282: 9042–9052, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Good DW, Knepper MA. Ammonia transport in the mammalian kidney. Am J Physiol Renal Fluid Electrolyte Physiol 248: F459–F471, 1985. [DOI] [PubMed] [Google Scholar]
- 18.Goodman AD, Lemann J Jr, Lennon EJ, Relman AS. Production, excretion and net balance of fixed acid in patients with renal acidosis. J Clin Invest 44: 495–506, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurkan S, Estilo GK, Wei Y, Satlin LM. Potassium transport in the maturing kidney. Pediatr Nephrol 22: 915–925, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol Renal Fluid Electrolyte Physiol 253: F595–F605, 1987. [DOI] [PubMed] [Google Scholar]
- 21.Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant 27: 4273–4287, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igarashi T, Inatomi J, Sekine T, Cha SH, Kanai Y, Kunimi M, Tsukamoto K, Satoh H, Shimadzu M, Tozawa F, Mori T, Shiobara M, Seki G, Endou H. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet 23: 264–266, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol 13: 2171–2177, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Inatomi J, Horita S, Braverman N, Sekine T, Yamada H, Suzuki Y, Kawahara K, Moriyama N, Kudo A, Kawakami H, Shimadzu M, Endou H, Fujita T, Seki G, Igarashi T. Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflügers Arch 448: 438–444, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Kim HY, Verlander JW, Bishop JM, Cain BD, Han KH, Igarashi P, Lee HW, Handlogten ME, Weiner ID. Basolateral expression of the ammonia transporter family member, Rh C glycoprotein, in the mouse kidney. Am J Physiol Renal Physiol 296: F545–F555, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293: F1238–F1247, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Knepper MA. NH4+ transport in the kidney. Kidney Int 40: S95–S102, 1991. [PubMed] [Google Scholar]
- 28.Knepper MA, Packer R, Good DW. Ammonium transport in the kidney. Physiol Rev 69: 179–249, 1989. [DOI] [PubMed] [Google Scholar]
- 29.Kurtz I, Zhu Q. Structure, function, and regulation of the SLC4 NBCe1 transporter and its role in causing proximal renal tubular acidosis. Curr Opin Nephrol Hypertens 22: 572–583, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurtz I, Zhu Q. Proximal renal tubular acidosis mediated by mutations in NBCe1-A: unraveling the transporter's structure-functional properties. Front Physiol 4: 350, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laghmani K, Preisig PA, Alpern RJ. The role of endothelin in proximal tubule proton secretion and the adaptation to a chronic metabolic acidosis. J Nephrol 15, Suppl 5: S75–S87, 2002. [PubMed] [Google Scholar]
- 32.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C Glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HW, Osis G, Handlogten ME, Guo H, Verlander JW, Weiner ID. Effect of dietary protein restriction on renal ammonia metabolism. Am J Physiol Renal Physiol 308: F1463–F1473, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee HW, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID. Effect of intercalated cell-specific Rh C glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 299: F369–F379, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HW, Verlander JW, Bishop JM, Handlogten ME, Han KH, Weiner ID. Renal ammonia excretion in response to hypokalemia: effects of collecting duct-specific Rh C glycoprotein deletion. Am J Physiol Renal Physiol 304: F410–F421, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HW, Verlander JW, Handlogten ME, Han KH, Weiner ID. Effect of collecting duct-specific deletion of both Rh B glycoprotein (Rhbg) and Rh C glycoprotein (Rhcg) on renal response to metabolic acidosis. Am J Physiol Renal Physiol 306: F389–F400, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SK, Boron WF, Parker MD. Substrate specificity of the electrogenic sodium/bicarbonate cotransporter NBCe1-A (SLC4A4, variant A) from humans and rabbits. Am J Physiol Renal Physiol 304: F883–F899, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemann J Jr, Lennon EJ, Goodman AD, Litzow JR, Relman AS. The net balance of acid in subjects given large loads of acid or alkali. J Clin Invest 44: 507–517, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int 58: 1267–1277, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Li HC, Du Z, Barone S, Rubera I, McDonough AA, Tauc M, Zahedi K, Wang T, Soleimani M. Proximal tubule specific knockout of the Na+/H+ exchanger NHE3: effects on bicarbonate absorption and ammonium excretion. J Mol Med 91: 951–963, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mak DO, Dang B, Weiner ID, Foskett JK, Westhoff CM. Characterization of transport by the kidney Rh glycoproteins, RhBG and RhCG. Am J Physiol Renal Physiol 290: F297–F305, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muto S, Miyata Y, Imai M, Asano Y. Troglitazone stimulates basolateral rheogenic Na+/HCO3− cotransport activity in rabbit proximal straight tubules. Exp Nephrol 9: 191–197, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Nagami GT. Enhanced ammonia secretion by proximal tubules from mice receiving NH4Cl: role of angiotensin II. Am J Physiol Renal Physiol 282: F472–F477, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Nagami GT. Ammonia production and secretion by S3 proximal tubule segments from acidotic mice: role of ANG II. Am J Physiol Renal Physiol 287: F707–F712, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Nagami GT, Sonu CM, Kurokawa K. Ammonia production by isolated mouse proximal tubules perfused in vitro: effect of metabolic acidosis. J Clin Invest 78: 124–129, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pitts RF. The role of ammonia production and excretion in regulation of acid-base balance. N Engl J Med 284: 32–38, 1971. [DOI] [PubMed] [Google Scholar]
- 47.Rocher LL, Tannen RL. The clinical spectrum of renal tubular acidosis. Annu Rev Med 37: 319–331, 1986. [DOI] [PubMed] [Google Scholar]
- 48.Romero MF. Molecular pathophysiology of SLC4 bicarbonate transporters. Curr Opin Nephrol Hypertens 14: 495–501, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO3− transporters. Pflügers Arch 447: 495–509, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Romero MF, Chen AP, Parker MD, Boron WF. The SLC4 family of bicarbonate transporters. Mol Aspects Med 34: 159–182, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romero MF, Fong P, Berger UV, Hediger MA, Boron WF. Cloning and functional expression of rNBC, an electrogenic Na+-HCO3− cotransporter from rat kidney. Am J Physiol Renal Physiol 274: F425–F432, 1998. [DOI] [PubMed] [Google Scholar]
- 52.Romero MF, Hediger MA, Boulpaep EL, Boron WF. Expression cloning and characterization of a renal electrogenic Na+/HCO3− cotransporter. Nature 387: 409–413, 1997. [DOI] [PubMed] [Google Scholar]
- 53.Satlin LM. Postnatal maturation of potassium transport in rabbit cortical collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 266: F57–F65, 1994. [DOI] [PubMed] [Google Scholar]
- 54.Schmitt BM, Biemesderfer D, Romero MF, Boulpaep EL, Boron WF. Immunolocalization of the electrogenic Na+-HCO3− cotransporter in mammalian and amphibian kidney. Am J Physiol Renal Physiol 276: F27–F38, 1999. [DOI] [PubMed] [Google Scholar]
- 55.Shapiro RA, Morehouse RF, Curthoys NP. Inhibition by glutamate of phosphate-dependent glutaminase of rat kidney. Biochem J 207: 561–566, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soriano JR, Boichis H, Stark H, Edelmann CM. Proximal renal tubular acidosis. A defect in bicarbonate reabsorption with normal urinary acidification. Pediatr Res 1: 81–98, 1967. [DOI] [PubMed] [Google Scholar]
- 57.Soriano JR. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 13: 2160–2170, 2002. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki M, Van Paesschen W, Stalmans I, Horita S, Yamada H, Bergmans BA, Legius E, Riant F, De Jonghe P, Li Y, Sekine T, Igarashi T, Fujimoto I, Mikoshiba K, Shimadzu M, Shiohara M, Braverman N, Al-Gazali L, Fujita T, Seki G. Defective membrane expression of the Na+-HCO3− cotransporter NBCe1 is associated with familial migraine. Proc Natl Acad Sci USA 107: 15963–15968, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor L, Curthoys NP. Glutamine metabolism: role in acid-base balance. Biochem Mol Biol Educ 32: 291–304, 2004. [DOI] [PubMed] [Google Scholar]
- 60.Toye AM, Parker MD, Daly CM, Lu J, Virkki LV, Pelletier MF, Boron WF. The human NBCe1-A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. Am J Physiol Cell Physiol 291: C788–C801, 2006. [DOI] [PubMed] [Google Scholar]
- 61.Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID. Localization of the ammonium transporter proteins, Rh B glycoprotein and Rh C glycoprotein, in the mouse kidney. Am J Physiol Renal Physiol 284: F323–F337, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Verlander JW, Chu D, Lee HW, Handlogten ME, Weiner ID. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal Physiol 305: F701–F713, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weiner ID, Hamm LL. Molecular mechanisms of renal ammonia transport. Annu Rev Physiol 69: 317–340, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weiner ID, Leader JP, Bedford JJ, Verlander JW, Ellis G, Vos F, de Jong S, Walker RJ. Effects of chronic lithium administration on renal acid excretion in humans and rats. Physiol Rep. 2014. December 11;2(12). pii: e12242. doi: 10.14814/phy2.12242. Print 2014 Dec 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weiner ID, Miller RT, Verlander JW. Localization of the ammonium transporters, Rh B glycoprotein and Rh C glycoprotein, in the mouse liver. Gastroenterology 124: 1432–1440, 2003. [DOI] [PubMed] [Google Scholar]
- 66.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol 10: 1444–1458, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weiner ID, Verlander JW. Role of NH3 and NH4+ transporters in renal acid-base transport. Am J Physiol Renal Physiol 300: F11–F23, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weiner ID, Verlander JW. Renal acidification mechanisms. In: Brenner and Rector's The Kidney, edited by Taal M, Chertow GM, Marsden PA, Skorecki K, Yu AS, Brenner BM. Philadelphia, PA: Elsevier Saunders, 2012, p. 293–325. [Google Scholar]
- 69.Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Comp Physiol 3: 201–220, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welbourne TC, Mu X. Extracellular glutamate flux regulates intracellular glutaminase activity in LLC-PK1-F+ cells. Am J Physiol Cell Physiol 268: C1418–C1424, 1995. [DOI] [PubMed] [Google Scholar]
- 71.Wu MS, Biemesderfer D, Giebisch G, Aronson PS. Role of NHE3 in mediating renal brush border Na+-H+ exchange. Adaptation to metabolic acidosis. J Biol Chem 271: 32749–32752, 1996. [DOI] [PubMed] [Google Scholar]
- 72.Xue Y, Liao SF, Son KW, Greenwood SL, McBride BW, Boling JA, Matthews JC. Metabolic acidosis in sheep alters expression of renal and skeletal muscle amino acid enzymes and transporters. J Anim Sci 88: 707–717, 2010. [DOI] [PubMed] [Google Scholar]
- 73.Zhu Q, Kao L, Azimov R, Newman D, Liu W, Pushkin A, Abuladze N, Kurtz I. Topological location and structural importance of the NBCe1-A residues mutated in proximal renal tubular acidosis. J Biol Chem 285: 13416–13426, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu Q, Shao XM, Kao L, Azimov R, Weinstein AM, Newman D, Liu W, Kurtz I. Missense mutation T485S alters NBCe1-A electrogenicity causing proximal renal tubular acidosis. Am J Physiol Cell Physiol 305: C392–C405, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zolotnitskaya A, Satlin LM. Developmental expression of ROMK in rat kidney. Am J Physiol Renal Physiol 276: F825–F836, 1999. [DOI] [PubMed] [Google Scholar]