

Abstract
Development of therapeutic molecules with clinical efficacy as modulators of defective CFTR includes efforts to identify potentiators that can overcome or repair the gating defect in mutant CFTR channels. This has taken a great leap forward with the identification of the potentiator VX-770, now available to patients as “Kalydeco.” Other small molecules with different chemical structure also are capable of potentiating the activity of either wild-type or mutant CFTR, suggesting that there are features of the protein that may be targeted to achieve stimulation of channel activity by structurally diverse compounds. However, neither the mechanisms by which these compounds potentiate mutant CFTR nor the site(s) where these compounds bind have been identified. This knowledge gap partly reflects the lack of appropriate experimental models to provide clues toward the identification of binding sites. Here, we have compared the channel behavior and response to novel and known potentiators of human CFTR (hCFTR) and murine (mCFTR) expressed in Xenopus oocytes. Both hCFTR and mCFTR were blocked by GlyH-101 from the extracellular side, but mCFTR activity was increased with GlyH-101 applied directly to the cytoplasmic side. Similarly, glibenclamide only exhibited a blocking effect on hCFTR but both blocked and potentiated mCFTR in excised membrane patches and in intact oocytes. The clinically used CFTR potentiator VX-770 transiently increased hCFTR by ∼13% but potentiated mCFTR significantly more strongly. Our results suggest that mCFTR pharmacological sensitivities differ from hCFTR, which will provide a useful tool for identifying the binding sites and mechanism for these potentiators.
Keywords: human CFTR, murine CFTR, potentiator, blocker, single-channel recording
cftr, the cystic fibrosis transmembrane conductance regulator, functions as a chloride ion channel. Mutations of CFTR directly cause the inherited disease cystic fibrosis (CF); CFTR also plays an important role in polycystic kidney disease and secretory diarrhea (8, 28). CFTR contains five functional domains: two membrane-spanning domains (MSDs) with six transmembrane helices (TM) in each MSD, two nucleotide-binding domains (NBD1, NBD2), and a regulatory domain (R), which is unique to CFTR and carries multiple protein kinase A (PKA) consensus sites (11, 23, 25).
Expressible CFTR clones from seven species have been produced, including Xenopus, pig, mouse, ferret, killifish, shark, and human CFTRs (27). Multiple genetically engineered murine CF models have been developed since the CFTR gene was identified (12, 36). Several groups have demonstrated that wild-type murine CFTR (mCFTR) has biophysical characteristics clearly different from wild-type human CFTR (hCFTR), but both require phosphorylation of their R domain by protein kinase A or C (PKA or PKC) and ATP binding in the NBDs for activation (14, 15). hCFTR and mCFTR share a high degree of sequence identity (∼78%): identity at the amino acid level is 81% in NBD1 and 84% in NBD2. The murine DelF508 (ΔF508)-CFTR mutant was at least partially processed and successfully localized to the apical membrane in differentiated airway epithelium, unlike ΔF508-hCFTR (22). hCFTR opens mainly to the full (f) open state with subconductance state 1 (s1) and subconductance state 2 (s2) as very rare events, whereas mCFTR exhibits mainly the s1 state with very brief transitions to the f state when recorded in transfected CHO-K1, Cos-7, and HEK293 cells (14, 15, 22). The unique s1 state of mCFTR was proposed to arise primarily from the NBDs (24). However, it is not known whether mCFTR exhibits additional features that are different from hCFTR, such as reaction to CFTR blockers including the most well-known and widely studied hCFTR pore blockers glibenclamide, GlyH-101, and diphenylamine-2-carboxylate (DPC). Both GlyH-101 and DPC block hCFTR in a voltage- and concentration-dependent manner with time independence, suggesting that both blockers have a single binding site or binding pocket in the CFTR pore that is accessible either from the extracellular side (GlyH-101) or cytoplasmic side (DPC) (4, 10, 32, 41). Glibenclamide blocks hCFTR in a voltage-, concentration-, and time-dependent manner (1, 5, 39). Meanwhile, the sole clinically used potentiator, VX-770, has been developed and studied in hCFTR, but the binding sites and functional mechanism and effects of VX-770 on mCFTR remain unclear (29, 31).
To gain a better understanding of the possible functional and pharmacological differences between hCFTR and mCFTR, we endeavored to study the channel behavior and the effects of glibenclamide, GlyH-101, DPC, and VX-770 with the hypothesis that the effects of these chemicals differ on mCFTR vs. hCFTR. Our results identify multiple differences between hCFTR and mCFTR behavior and suggest that the functional mechanisms of VX-770 and GlyH-101 may be understood via cross-species comparison of hCFTR and mCFTR.
MATERIALS AND METHODS
Preparation of oocytes and cRNA.
Human CFTR cRNAs for electrophysiology experiments were prepared from constructs encoding WT-hCFTR in the pgemhe vector (hCFTR/pGEMHE kindly provided by Dr. D. Gadsby, Rockefeller University). Murine CFTR (mCFTR/pcDNA 3.1) was kindly provided by Dr. A. P. Naren (University of Cincinnati) and subcloned into the pGEMHE vector (mCFTR/pGEMHE). The mutants of mCFTR and hCFTR used in this study were prepared by using site-directed mutagenesis with the Quikchange protocol (Stratagene, La Jolla, CA), and all mutant constructs were verified by sequencing across the entire open reading frame before use. Xenopus laevis oocytes were injected in a range of 0.4–10 ng of CFTR cRNAs and were incubated at 17°C in modified Liebovitz's L-15 media with the addition of HEPES (pH 7.5), penicillin, and streptomycin. Recordings were made 24–96 h after the injection of cRNAs. Methods of animal handling and oocyte collection were in accordance with the NIH guidelines and the protocol was approved by the Institutional Animal Care and Use Committee of Emory University (4, 37).
Electrophysiology.
For single-channel recording, CFTR channels were studied in excised, inside-out patches at room temperature (22–23°C) (18, 20, 32). Oocytes were prepared for study by shrinking in hypertonic solution followed by manual removal of the vitelline membrane. Pipettes were pulled from borosilicate glass (Sutter Instrument, Novato, CA) and had resistances averaging ∼10 MΩ when filled with chloride-containing pipette solution (in mM): 150 N-methyl-d-glucamine (NMDG)-Cl, 5 MgCl2, 10 TES (pH 7.5). Typical seal resistances were 200 GΩ or greater. Channels were activated by excision into cytoplasmic solution containing (in mM) 150 NMDG-Cl, 1.1 MgCl2, 2 Tris-EGTA, 10 TES, 1 MgATP (adenosine 5′-triphosphate magnesium), and 127.6 U/ml PKA (pH 7.5). CFTR currents were measured with an Axopatch 200B amplifier (Molecular Devices; Sunnyvale, CA), and were recorded at 10 kHz to DAT tape. For subsequent analysis, records were played back and filtered with a four-pole Bessel filter (Warner Instruments; Hamden, CT) at a corner frequency of 1 kHz and acquired by using a Digidata 1322A interface and computer at 500 Hz with pClamp 8.2 (Molecular Devices). For display, single-channel records were filtered digitally to 100 Hz. pClamp version 10.2 was used to analyze single-channel currents and to make all-points amplitude histograms with a bin width of 0.01 pA, which were fit with Gaussian distributions. The open-duration analysis was performed on records from patches containing one to three active CFTR channels and only isolated single openings were measured for the analysis. Open-duration histograms were made and fit with a single exponential function with Igor version 4.10 (WaveMetrics; Lake Oswego, OR). Apparent open probability was measured with Clampfit 10.2.
For inside-out macropatch recording, patch pipette resistances were 1–2 MΩ and the same symmetrical 150 mM Cl− solutions were used as for single-channel recording. Macropatch recordings were performed with an Axopatch 200B amplifier operated by pClamp 8.2 software, filtered at 100 Hz with a four-pole Bessel filter, and acquired at 2 kHz. We used two protocols in the project: 1) a voltage-ramp protocol was applied every 5 s, holding at membrane potential (VM) = 0 mV then stepping up to +100 mV for 50 ms followed by a ramp down to −100 mV over 300 ms then a step back to 0 mV; and 2) a voltage-step protocol was used to observe time-dependent glibenclamide-induced block, which included holding at VM = 0 mV, step to VM = +80 mV, to VM = −120 mV, to VM = +100 mV, then back to 0 mV (1, 4).
Standard two-electrode voltage-clamp (TEVC) techniques were used to study the blocking effects of GlyH-101 on both hCFTR and mCFTR and their mutants with application of reagents to the extracellular side. Each oocyte was injected with CFTR cRNA along with cRNA encoding the β2-adrenergic receptor (18, 19). Pipette resistances measured 0.5–1.4 MΩ when filled with 3 M KCl and measured in standard ND96 bath solution that contained (in mM) 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES (pH 7.5). CFTR channels were activated by exposure to 10 μM isoproterenol (ISO) alone and alternatively assayed in the presence or absence of different concentrations of GlyH-101 in the bath solution. To measure block of CFTR by glibenclamide and DPC, a step protocol was used with holding potential −30 mV, and membrane potential stepped to potentials ranging from −140 mV to +80 mV in 20-mV increments, holding at each for a period of 80 ms. Currents were acquired with an Axoclamp 900A amplifier and Clampex 10.2 software, and current data were digitized at 2 kHz.
Fractional change of current (Fc) at steady-state in response to exposure to chemicals was calculated according to the equation
where Ic and Ib represent the steady-state control currents and currents in the presence of chemicals.
Source of reagents.
Unless otherwise noted, all reagents were obtained from Sigma Chemical (St. Louis, MO). L-15 medium was from GIBCO-BRL (Gaithersburg, MD), GlyH-101 was from Calbiochem (Billerica, MA), and PKA was from Promega (Madison, WI); 1 μl PKA is equal to 127.6 units (U). GlyH-101 was prepared as a stock solution at 50 mM in DMSO. VX-770 was from Selleckchem (Houston, TX) and was prepared as a stock solution at 10 mM in DMSO. Glibenclamide and DPC were initially prepared as 0.5 M stocks in DMSO as previously reported (1, 2, 26). All chemicals were diluted to a final concentration in recording solution immediately before use.
Statistical analysis.
Unless otherwise noted, values given are means ± SE. Statistical analyses were performed by the t-test for unpaired or paired measurements in SigmaPlot 12.3 (San Jose, CA). For multiple comparisons, we performed one-way ANOVA plus Dunnett's multiple comparison post hoc test using Prism (La Jolla, CA). P < 0.05 was considered significantly different. *P < 0.05; **P < 0.01; #P < 0.001.
RESULTS
mCFTR single channels behave somewhat differently from hCFTR.
All of the published studies of mCFTR have been done in mammalian cell lines, and whether mCFTR exhibits species-specific behavior in oocytes remains unknown. We recorded single hCFTR and mCFTR channel currents in 150 mM Cl− symmetrical solution at VM = −100 mV. Representative current traces are shown in Fig. 1. mCFTR single-channel behavior exhibited several divergent aspects from hCFTR. Unlike hCFTR, which opens mainly to the f state with rare s1 and s2 states (Fig. 1A), mCFTR opens to multiple open states, s1, s2, and f (Fig. 1, B and C). The amplitude of s1 was ∼25% and s2 was ∼65% of f, which is different from the ratios of s1 and s2 to f in WT-, R334C-, R352A-, and R347A-hCFTR (s1 is ∼40% and s2 is ∼70% of f) (21, 27, 28). The single-channel amplitude of the s1 state in mCFTR expressed in oocytes was not as low as previously reported (14, 15, 24). The single-channel amplitude of f in mCFTR (0.66 ± 0.01 pA, n = 15) was significantly smaller than that of hCFTR (0.75 ± 0.02 pA, n = 6). As we previously reported, R334C-, R347A-, and R352A-hCFTR generally open from the closed state (c), to s1, then opened to s2 and f states (6, 37, 40). In contrast, mCFTR seems to randomly open to the s1, s2, or f state (Fig. 1B) in no clear order. Mean burst duration of mCFTR was significantly more brief than that of hCFTR (Fig. 1D). mCFTR exhibited inward rectification of the current-voltage (I–V) relationship whereas hCFTR exhibited a linear I–V relationship under the same experimental conditions (Fig. 1, E and F).
Fig. 1.

Human CFTR (hCFTR) and murine (mCFTR) differ in single-channel behavior. Representative single-channel current traces are shown for hCFTR (A) and mCFTR (B and C), along with their all-points amplitude histograms, from inside-out membrane patches excised from Xenopus oocytes, with symmetrical 150 mM Cl− solution in the presence of 1 mM MgATP and 127.6 U/ml PKA. All traces were recorded at VM = −100 mV. c, Closed state; s1, subconductance state 1; s2, subconductance state 2; f, full open state. Solid lines in histograms are fit results to a Gaussian function. D: mean burst durations of hCFTR (n = 5 patches) and mCFTR (n = 8 patches). #P < 0.001 compared with hCFTR. For mCFTR, we considered a transition from c to any open level (s1, s2, or f) as an opening. Representative current-voltage (I–V) curves of hCFTR (E) and mCFTR (F) recorded in symmetrical 150 mM Cl− solutions with the inside-out macropatch technique by using the ramp protocol as described in materials and methods; n = 8 for both hCFTR and mCFTR.
VX-770 potentiated mCFTR significantly better than hCFTR.
To date, VX-770 is the only Food and Drug Administration approved prescription potentiator of CFTR, available to patients for the treatment of CF under the trade name Kalydeco. It has been reported that VX-770 potentiates G551D-hCFTR by increasing open probability and enhancing ATP-independent activity (7, 29, 31). We hypothesized that VX-770 would potentiate mCFTR similar to hCFTR. Because VX-770 modifies hCFTR intracellularly, we utilized the inside-out macropatch technique with a voltage-ramp protocol. Representative current recordings of hCFTR potentiated by VX-770 under different concentrations of MgATP and PKA are shown in Fig. 2. During 5-min exposure, 10 μM VX-770 increased wild-type hCFTR current by ∼13% (13.3 ± 0.06%, n = 7), measured at VM = −100 mV in the presence of 1 mM MgATP and 127.6 U/ml PKA (standard conditions) (Fig. 2A). We observed effects of 10 μM VX-770 on hCFTR under both low-PKA and low-ATP conditions that would lead to reduced activity of hCFTR. Under standard conditions, 25.5 U PKA/ml with 1 mM ATP activated CFTR current to ∼65% of the maximum current (34). The EC50 for ATP-dependent activation of hCFTR in presence of standard PKA concentrations is ∼20 μM (data not shown), similar to previous reports (9, 35). hCFTR current reached plateau in ∼10–15 min under both low-PKA (25.5 U/ml PKA + 1 mM MgATP) or low-ATP (50 μM MgATP + 127.6 U/ml PKA) conditions. Representative current traces are shown in Fig. 2, B and C. During 5-min exposure, 10 μM VX-770 increased hCFTR current to a similar level as standard conditions after being activated with ∼20-min exposure of ATP and PKA. Summary data are shown in Fig. 2D. The data suggest that VX-770 mildly potentiated hCFTR regardless of CFTR phosphorylation level and open probability.
Fig. 2.

Cytoplasmic VX-770 weakly potentiated hCFTR. Representative macropatch currents of hCFTR recorded in inside-out mode with symmetrical 150 mM Cl− solution under the following experimental conditions with or without 10 μM VX-770: 1 mM MgATP + 127.6 U/ml PKA (A), 1 mM MgATP + 25.5 U/ml PKA (B), and 50 μM MgATP + 127.6 U/ml PKA (C). A voltage-ramp protocol described in the materials and methods section was applied every 5 s. D: summary data for fractional increase of hCFTR current by VX-770 under each set of conditions; n = 4–7 each.
We next tested the effects of VX-770 on mCFTR under standard conditions (1 mM MgATP +127.6 U/ml PKA). Surprisingly, we found that mCFTR failed to reach plateau even after 50 min of exposure to standard conditions (Fig. 3A). From 20 to 25 min, mCFTR current increased further 0.26 ± 0.07-fold (n = 5). mCFTR channels were activated more rapidly under high-PKA conditions (1 mM MgATP + 638 U/ml PKA) (Fig. 3B); current reached plateau in ∼15 min.
Fig. 3.
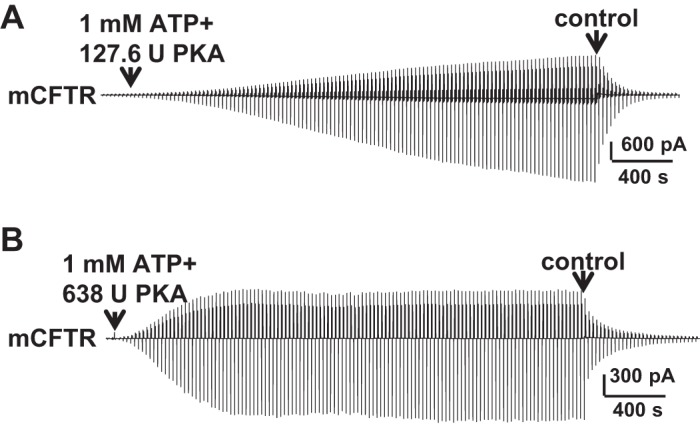
Full activation of mCFTR required high PKA concentration. Representative macropatch currents of mCFTR recorded in the inside-out mode with symmetrical 150 mM Cl− solution under standard conditions (1 mM MgATP + 127.6 U/ml PKA) (A) and high-PKA conditions (1 mM MgATP + 638 U/ml PKA) (B), via a ramp protocol applied every 5 s as in Fig. 2.
We then asked whether 0.5 μM VX-770 potentiated mCFTR activated under standard conditions or high-PKA conditions; representative current traces are shown in Fig. 4, A and B. After mCFTR was activated under high-PKA conditions, channels were exposed to 0.5 μM VX-770 in the presence of ATP + PKA for 5 min. mCFTR current was increased 0.84 ± 0.09-fold under high-PKA conditions (n = 4), even at this low VX-770 concentration, suggesting that VX-770 potentiated mCFTR when channels were fully activated.
Fig. 4.

Effects of VX-770 on mCFTR. A: representative macropatch currents of mCFTR recorded under high-PKA conditions (1 mM MgATP + 638 U/ml PKA) in the absence and presence of 0.5 μM VX-770. mCFTR channels were activated by 20 min in high-PKA conditions followed by addition of 0.5 μM VX-770 for 5 min. B: mCFTR was activated under standard conditions (1 mM MgATP + 127.6 U/ml PKA) for 20 min, then exposed to 0.5 μM VX-770 along with ATP + PKA for 5 min. C: mCFTR currents were potentiated by VX-770 to a similar degree under high- and low-PKA conditions, suggesting that the effect was not sensitive to channel activity level. D: the concentration dependence of the increase in mCFTR current induced by VX-770 is shown; n = 4–6 patches at each concentration. The solid line indicates the fit with a 1-site, ligand-saturation-binding equation giving an apparent Kd = 7.32 nM at VM = −100 mV. E: representative single-channel current traces of mCFTR from 1 patch recorded under the same conditions as Fig. 1. −VX-770, 1 mM MgATP + 127.6 U/ml PKA; +VX-770, 200 nM VX-770 + 1 mM MgATP + 127.6 U/ml PKA. F: apparent open probability (NPo) of mCFTR channels in the absence (−VX-770) and presence (+VX-770) of 200 nM VX-770. **P < 0.01 compared with control conditions (−VX-770).
mCFTR channels also were activated under standard conditions for 20 min and then exposed to 1 mM ATP + 127.6 U/ml PKA with different concentration of VX-770 for 5 min. Fractional increases in mCFTR current induced by VX-770 were calculated after correction for the average current increase over 5 min in the absence of VX-770 (see above); 0.5 μM VX-770 potentiated mCFTR with similar efficacy under high-PKA conditions and standard PKA conditions (Fig. 4C) (P = 0.46). VX-770 potentiated mCFTR with Kd = 7.32 nM at VM = −100 mV in standard conditions (Fig. 4D), demonstrating increased sensitivity of mCFTR to VX-770-induced potentiation relative to hCFTR.
To further determine the mechanism of action of VX-770 on mCFTR, we performed single-channel recording of mCFTR in the absence (−VX-770) and in the presence of 200 nM VX-770 (+VX-770) (Fig. 4E). VX-770 did not affect the unique single burst structure of mCFTR but significantly enhanced open probability of mCFTR (Fig. 4F). These data suggest that VX-770 potentiates CFTR channels by affecting gating in a species-specific manner. Consequently, the cross-species comparison between hCFTR and mCFTR presents a strong tool that may facilitate the identification of VX-770 binding sites and functional mechanisms.
Effects of GlyH-101 on mCFTR differ from hCFTR.
GlyH-101 is a well-known open pore blocker of hCFTR that gains its binding pocket for this function from the extracellular space (26). We have previously reported with the TEVC technique that the apparent Kd for GlyH-101 at hCFTR is 0.98 μM at VM = +80 mV and 4.9 μM at VM = −60 mV (4). We hypothesized that GlyH-101 block of mCFTR would be similar to that of hCFTR.
We first investigated the effect of 2.5 μM GlyH-101 on hCFTR with mutations at selected amino acids that are also conserved in mCFTR, selected as follows: 1) R334 sits in the outer mouth of the CFTR pore, attracts Cl− into the pore, and directly affects ion conduction, which might affect GlyH-101 binding in the pore; 2) T338 is located in the narrow part of the hCFTR pore and has been suggested as a possible binding site for GlyH-101 (21); 3) R352 forms a salt bridge with D993 and D924 and plays a key role in maintaining the open pore architecture of hCFTR (3, 37); consequently, mutation R352A disrupts the R352-D993-D924 salt bridge and may affect the GlyH-101 binding site by altering the hCFTR open pore architecture. Representative current traces and summary data are shown in Fig. 5. GlyH-101 at 2.5 μM blocked WT-hCFTR 28% at VM = −60 mV in the continuing presence of 10 μM ISO (used to activate CFTR via the β2-adrenergic receptor; see materials and methods), but the blocking effect was completely lost with both the R334A and R334C mutations (Fig. 5). In contrast, 2.5 μM GlyH-101 exhibited strengthened block of both T338A- and R352A-hCFTR (Fig. 5, C and D). These data suggest that 1) R334 might contribute to GlyH-101 binding and function in CFTR's outer mouth; 2) in disagreement with the report from Norimatsu and coworkers, T338 might not be a direct binding site for GlyH-101 (21); and 3) loss of the R352-D993-D924 salt bridge and therefore maintenance of pore architecture did not weaken the GlyH-101 blocking effect, but rather strengthened its function, suggesting that the altered pore architecture did not preclude GlyH-101 from reaching its binding site in hCFTR. In fact, GlyH-101 is a large molecule, so it is highly possible that it is not able to get into the narrow part of the CFTR pore (between F337 and S341 in TM6), which has a diameter <4 Å (1).
Fig. 5.

Effects of GlyH-101 on wild-type (WT)- (A), R334C- (B), and T338A-hCFTR (C). Representative whole oocyte [2-electrode voltage-clamp (TEVC)] current traces were recorded at VM = −60 mV in ND96 solution, in oocytes expressing CFTR with the β2-adrenergic receptor. After activation with 10 μM isoproterenol (ISO), oocytes were exposed to 2.5 μM GlyH-101 for ∼50 s in the continuing presence of ISO. Dashed lines at bottom of the trace indicate the maximum plateau current in each trace. Summary data for all variants tested under the same conditions are shown in D. Some error bars are too small to view. #P < 0.001 compared with wild-type hCFTR; n = 4–6.
We next examined the concentration dependence of GlyH-101 effects on mCFTR wild type and mutants. Surprisingly, we found that under the same experimental conditions as used for hCFTR, GlyH-101 not only blocked mCFTR but also potentiated it. A representative current trace is shown in Fig. 6A, and the dashed line at bottom of the trace indicates the maximum current of mCFTR activated by ISO alone before addition of GlyH-101. As shown, GlyH-101 initially blocked mCFTR current, similar to its effect on hCFTR. However, upon washout and the rapid recovery from pore block, current rebound past the control level (dashed line) was consistently seen in mCFTR. In these experiments, cells were exposed to extracellular GlyH-101 for only ∼50 s; the magnitude of rebound was larger when incubation time was lengthened, suggesting that potentiation reflected a fraction of GlyH-101 permeating the membrane.
Fig. 6.

GlyH-101 affects mCFTR differently from hCFTR. Representative current trace recorded by TEVC from 1 oocyte expressing mCFTR at VM = −60 mV. The oocyte was exposed to 25 μM GlyH-101 in the continuing presence of 10 μM ISO for ∼50 s. Dashed line at bottom of the trace shows the maximum plateau current before GlyH-101. With use of this protocol, it is apparent that GlyH-101 both inhibits and potentiates mCFTR. The concentration-dependent inhibition and potentiation of mCFTR by GlyH-101 at VM = −60 mV are shown in B and C, respectively; solid lines indicate the fit with a ligand-binding, 1-site saturation equation, giving apparent Kd = 32.4 μM for inhibition and apparent Kd = 103.6 μM for potentiation by extracellular GlyH-101, with ∼50-s exposure. D: representative current trace of R334A-mCFTR recorded at VM = −60 mV. Extracellular 25 μM GlyH-101 failed to block but only potentiated R334A-mCFTR. Summary data for fractional inhibition (E) and fractional potentiation (F) of WT-, V100L-, R147H-, I201V-, R334A-, and T338A-mCFTR with 25 μM GlyH-101. *P < 0.05 and #P < 0.001 compared with wild-type mCFTR; n = 4–6 each.
We analyzed the concentration-dependent inhibition and potentiation of mCFTR by GlyH-101; summary data are shown in Fig. 6, B and C. At VM = −60 mV, the apparent Kd for inhibition of mCFTR was 32.4 μM and the apparent Kd for potentiation of mCFTR (with ∼50-s exposure) was 103.6 μM extracellular GlyH-101. Hence the blocking function of GlyH-101 on mCFTR is relatively weak compared with hCFTR; however, the inhibition may be underestimated owing to concurrent potentiation. We then tested block by 25 μM GlyH-101 of five mCFTR mutants selected as follows: 1) R334 and T338 are conserved in both mCFTR and hCFTR and both amino acids are important in GlyH-101 function in hCFTR (see above); 2) V100 and I201 sit in the extracellular side of CFTR and are not conserved between hCFTR and mCFTR; therefore, we mutated V100 in mCFTR to the L of hCFTR and mutated I201 in mCFTR to the V of hCFTR; 3) R147 is predicted to be in the cytoplasmic aspect of the pore and so we mutated it to H as in hCFTR. A representative current trace of R334A-mCFTR is shown in Fig. 6D. GlyH-101 completely lost its blocking effect and only exhibited the potentiation function at R334A-mCFTR. Summary data for inhibition are shown in Fig. 6E. GlyH-101 displayed significantly strengthened block of V100L-mCFTR and distinctly weakened block of I201V-mCFTR. These data suggest that 1) R334 may contribute to GlyH-101 binding and function in mCFTR as in hCFTR, 2) I201 might be involved in GlyH-101-mediated inhibition, 3) V100 and T338 are probably not directly involved in GlyH-101 blocking function, and 4) R147 clearly is not involved in GlyH-101-mediated extracellular blockade of the pore.
In addition to inhibition of mCFTR, GlyH-101 potentiated WT-mCFTR and the same five mutant mCFTR variants to an almost identical level, as shown in Fig. 6F. Hence, those mutations that impacted pore block by GlyH-101 did not generally impact potentiation by this compound. GlyH-101 was originally identified as a membrane impermeant chemical, but it actually permeates into cells to ∼60% in 2 h (16). It is unlikely that GlyH-101 both blocks and potentiates mCFTR by binding to the same sites in the pore and therefore we hypothesized that GlyH-101 potentiated mCFTR from the cytoplasmic side in a manner different from its blocking effect from the extracellular side.
To investigate the response to cytoplasmic GlyH-101 in hCFTR and mCFTR, we used inside-out macropatch recordings in symmetrical 150 mM Cl− solution. As shown in Fig. 7A, hCFTR was not potentiated by 0.5 μM GlyH-101 when it was applied onto the cytoplasmic side under standard conditions (n = 3). Similar results were also seen in hCFTR under low-PKA and low-ATP conditions (data not shown). In contrast, 5-min exposure to 20 nM GlyH-101 under high-PKA conditions (1 mM MgATP and 638 U/ml PKA) strongly and rapidly potentiated fully activated mCFTR (Fig. 7B); mCFTR current increased 0.48 ± 0.13-fold (n = 5) in 5 min under high-PKA conditions. We then tested the concentration-dependent potentiation of mCFTR by GlyH-101 under standard conditions. Representative current traces are shown in Fig. 7C and summary data are shown in Fig. 7D. mCFTR channels were activated under standard conditions for 20 min and then exposed to ATP + PKA with different concentrations of GlyH-101 for 5 min. Fractional increases in mCFTR current induced by GlyH-101 were calculated after correction for the average current increase over 5 min in the absence of GlyH-101; the data were fit with a one-site, ligand saturation-binding equation, resulting in an apparent Kd = 0.6 nM. At 20 nM, GlyH-101 potentiated mCFTR channels to a similar degree in high-PKA and standard PKA conditions (P = 0.29). These data strongly support our hypothesis that GlyH-101 potentiates mCFTR from the cytoplasmic side, in contrast to the channel inhibition effect that was observed with drug applied extracellularly. During ∼50 s exposure to GlyH-101 in TEVC experiments, very little GlyH-101 likely permeated into the oocytes and potentiated mCFTR while simultaneously extracellular drug blocked the mCFTR pore. Consequently, the blocking efficiency of mCFTR by GlyH-101 was underestimated because of the potentiation function and therefore the apparent Kd for inhibition of mCFTR was dramatically higher than that for hCFTR.
Fig. 7.

GlyH-101 potentiated mCFTR from the cytoplasmic side but not hCFTR. Representative macropatch currents of hCFTR recorded in the inside-out mode with symmetrical 150 mM Cl− solution. A voltage-ramp protocol was applied every 5 s as in Fig. 2. Control, intracellular solution only; ATP + PKA, 1 mM MgATP and 127.6 U/ml PKA; ATP + PKA + GlyH-101, 0.5 μM GlyH-101 + 1 mM MgATP + 127.6 U/ml PKA. B: representative macropatch currents from mCFTR recorded in inside-out mode under high-PKA conditions. After the inside-out macropatch was formed, mCFTR was fully activated with cytoplasmic 1 mM MgATP + 638 U/ml PKA for 20 min, followed by addition of 20 nM GlyH-101 for 5 min. C: representative macropatch currents from mCFTR recorded in inside-out mode with 1 mM MgATP + 127.6 U/ml PKA for 20 min followed by addition of 5 nM GlyH-101 for 5 min. D: concentration-dependent increase in mCFTR current induced by GlyH-101 at VM = −100 mV under standard PKA conditions is shown; solid line is the fit with a 1-site, ligand-saturation-binding equation, giving apparent Kd = 0.6 nM at VM = −100 mV. E: representative single-channel currents of mCFTR from the same patch before and after exposure to 5 nM GlyH-101, recorded under the same conditions as Fig. 1. −GlyH, 1 mM MgATP + 127.6 U/ml PKA; + GlyH, 5 nM GlyH-101 + 1 mM MgATP + 127.6 U/ml PKA. F: apparent open probability of mCFTR in the absence (−GlyH) and presence (+GlyH) of 5 nM GlyH-101. *P < 0.05 compared with control condition (−GlyH).
We next investigated possible mechanisms by which GlyH-101 potentiates mCFTR by utilizing single-channel recording at VM = −100 mV in 150 mM Cl− solution. Representative traces recorded from one patch in the absence (−GlyH) and presence (+GlyH) of GlyH-101 are shown in Fig. 7E. GlyH-101 did not change the unique single-channel burst behavior of mCFTR but significantly increased its open probability (Fig. 7F). These results suggest that GlyH-101 affects mCFTR gating to potentiate channel opening.
VX-770 slowed hCFTR closing but accelerated mCFTR closing.
Although VX-770 had very little effect on hCFTR steady-state currents, we found that exposure to 10 μM VX-770 dramatically slowed hCFTR deactivation after subsequent removal of ATP, PKA, and VX-770. A representative current trace is shown in Fig. 8A. The deactivation time course of hCFTR after VX-770 incubation was fitted with a single exponential function with time constant 833.33 s as shown in Fig. 8B. The kinetics of CFTR deactivation after exposure to VX-770 were significantly slower than without VX-770 pretreatment (Fig. 8C). This phenomenon was not observed in mCFTR. As shown in Fig. 8, D–F, 0.5 μM VX-770 preincubation accelerated mCFTR deactivation (time constant significantly smaller than ATP and PKA alone, without exposure to VX-770). This suggests that VX-770 might stabilize the dimerization of NBDs in hCFTR and destabilize the dimerization of NBDs in mCFTR. GlyH-101 pretreatment exhibited no effects on deactivation of either hCFTR or mCFTR after removal of ATP and PKA (data not shown).
Fig. 8.

Rate of deactivation of current upon removal of PKA and ATP after exposure to VX-770 was increased for mCFTR but decreased for hCFTR. Representative current trace for hCFTR after exposure to 10 μM VX-770 (A) or mCFTR after exposure to 0.5 μM VX-770 (D), recorded under standard conditions. B and E show single exponential fits to the time courses of current deactivation at VM −100 mV for hCFTR (B) and mCFTR (E). Summary data for mean deactivation time constants in the absence and presence of VX-770 are shown in C and F. #P < 0.001 compared with no-VX-770-treatment conditions (−VX-770) (n = 4 each).
VX-770 did not exhibit ATP-independent potentiation at hCFTR or mCFTR.
VX-770 was suggested first by the Bear group and then by the Hwang group to potentiate hCFTR in an ATP-independent manner (7,13). To determine whether VX-770 can stimulate CFTR in the absence of ATP, hCFTR was first activated by ATP + PKA and then fully deactivated by removal of ATP and PKA; hCFTR then was successfully reactivated by exposure to 1 mM ATP alone, suggesting that most channels remained phosphorylated after several minutes without PKA (Fig. 9A). Similar experiments were done with 10 μM VX-770 alone. After hCFTR channels were allowed to close for a similar time period, exposure to VX-770 alone failed to reactivate hCFTR (Fig. 9B). The data indicate that, unlike previous reports, VX-770 did not stimulate hCFTR in an ATP-independent manner. Similar experiments were performed with mCFTR (Fig. 9, C and D). mCFTR was activated with ATP + PKA for 20 min then fully deactivated upon removal of ATP and PKA. mCFTR was successfully reactivated by exposure to ATP alone (Fig. 9C), whereas exposure to VX-770 alone failed to reactivate mCFTR (Fig. 9D), indicating lack of ATP-independent stimulation by VX-770. The difference between our data and that of Jih and Hwang (13) may be due to protocol difference where they observed and reported ATP-independent potentiation by VX-770 immediately after removal of cytosolic ATP; given the channel closing rate described above, it is unlikely that the patch was truly free of ATP and even less likely that the NBDs had fully dedimerized and released the unhydrolyzed ATP from the degenerate ATP-binding site in their studies. GlyH-101 also did not potentiate mCFTR in an ATP-independent manner (data not shown).
Fig. 9.
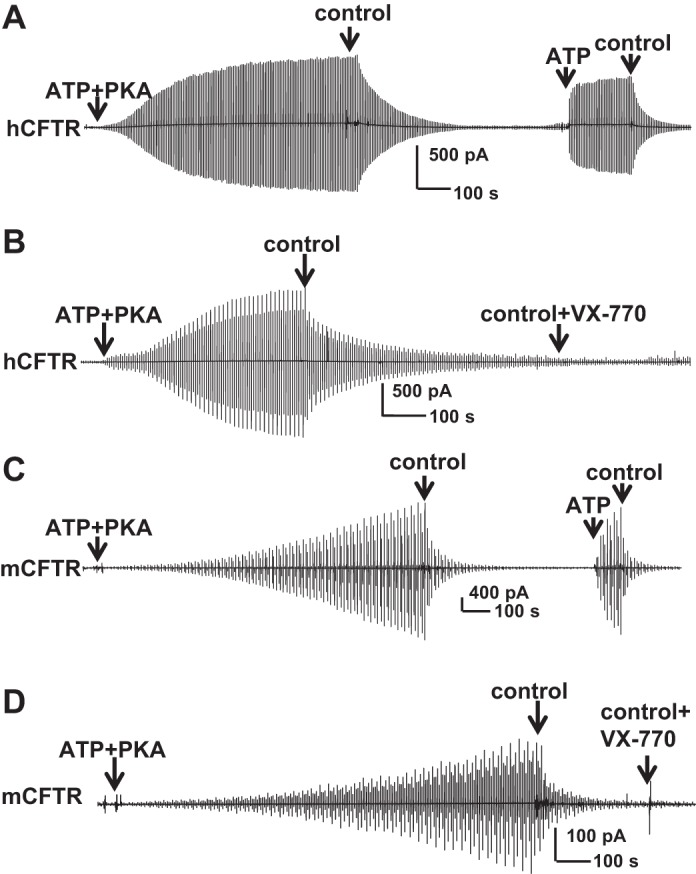
VX-770 exhibited no ATP-independent potentiation of hCFTR or mCFTR. A and B: representative inside-out macropatch hCFTR currents recorded with a ramp protocol as in Fig. 2. hCFTR channels were fully activated with 1 mM MgATP + 127.6 U/ml PKA and then allowed to fully deactivate after removal of ATP and PKA. Currents were reactivated with exposure to 1 mM MgATP alone (A), but not 10 μM VX-770 alone (B). C and D: representative inside-out macropatch mCFTR currents as in A and B. mCFTR channels were activated with 1 mM MgATP + 127.6 U/ml PKA for 20 min then allowed to fully deactivate after removal of ATP and PKA. mCFTR currents were reactivated with exposure to 1 mM MgATP alone (C) but not 0.5 μM VX-770 alone (D).
Glibenclamide blocked as well as potentiated mCFTR.
Glibenclamide is a well-characterized small molecule open pore blocker of hCFTR that enters the pore from the cytoplasmic side (39). We hypothesized that glibenclamide would block mCFTR similarly to hCFTR. Representative current traces from hCFTR in the absence and presence of 10 μM glibenclamide are shown in Fig. 10A. The Kd for glibenclamide-induced time-dependent block of hCFTR at VM = −120 mV was previously reported to be 26.5 μM (5). Representative mCFTR current traces in the absence and presence of 10 μM glibenclamide are shown in Fig. 10B. Glibenclamide exhibited concentration- and time-dependent block of mCFTR with apparent Kd = 7.01 μM at VM = −120 mV (Fig. 10C). The results suggest that glibenclamide more strongly blocks mCFTR compared with hCFTR. The fractional block of mCFTR reached plateau at 50 μM glibenclamide; surprisingly, the fractional block of mCFTR by 200 μM glibenclamide was significantly lower than that at 50 μM, suggesting that glibenclamide, like GlyH-101, not only blocked but also potentiated mCFTR (P < 0.05, 200 μM compared with 50 μM glibenclamide). At high concentrations of glibenclamide, potentiation and inhibition of mCFTR may have occurred concurrently.
Fig. 10.

Glibenclamide (Glib) blocked as well as potentiated mCFTR. Representative macropatch current of hCFTR from 1 patch in the absence of glibenclamide (black) and in the presence of 50 μM glibenclamide (red) in 150 mM Cl− symmetrical solution. −Glib, 1 mM MgATP alone; + Glib, 50 μM glibenclamide + 1 mM MgATP. The voltage-step protocol is described in materials and methods and consisted of these phases: pulses to VM = +80 mV, −120 mV, and +100 mV. CFTR channels were previously phosphorylated with PKA and 1 mM MgATP. Dashed lines represent zero current level. Representative macroscopic current of mCFTR from the same patch in the absence and presence of glibenclamide (10 μM in B, and 20 nM in C), with the same recording conditions as A. D: concentration dependence of steady-state block by glibenclamide at VM = −120 mV. The apparent Kd = 7.01 μM for block was obtained from a fit with a 1-site, ligand-saturation-binding equation (red line, n = 4–6 patches for each concentration). E: concentration-dependent potentiation of mCFTR by glibenclamide at VM = +100 mV. Apparent Kd = 4.71 nM for potentiation was obtained from a fit by using the same equation (red line, n = 4–6 patches for each concentration).
To separate the potentiation effects of glibenclamide from inhibition, we recorded mCFTR at lower glibenclamide concentration and at depolarizing potentials where inhibition is not effective. Representative current traces and summary data are shown in Fig. 10, D and E, respectively, indicating Kd = 4.71 nM for the rather weak glibenclamide-mediated potentiation of mCFTR at VM= +100 mV. At the Kd for potentiation there was no inhibitory effect of glibenclamide on mCFTR. This behavior also was observed in whole oocytes studied with the TEVC technique. As shown in Fig. 11, hCFTR only exhibited the blocking effect by glibenclamide applied extracellularly (Fig. 11A), whereas mCFTR was blocked at negative potentials but potentiated weakly at positive potentials by glibenclamide (Fig. 11, B and C).
Fig. 11.
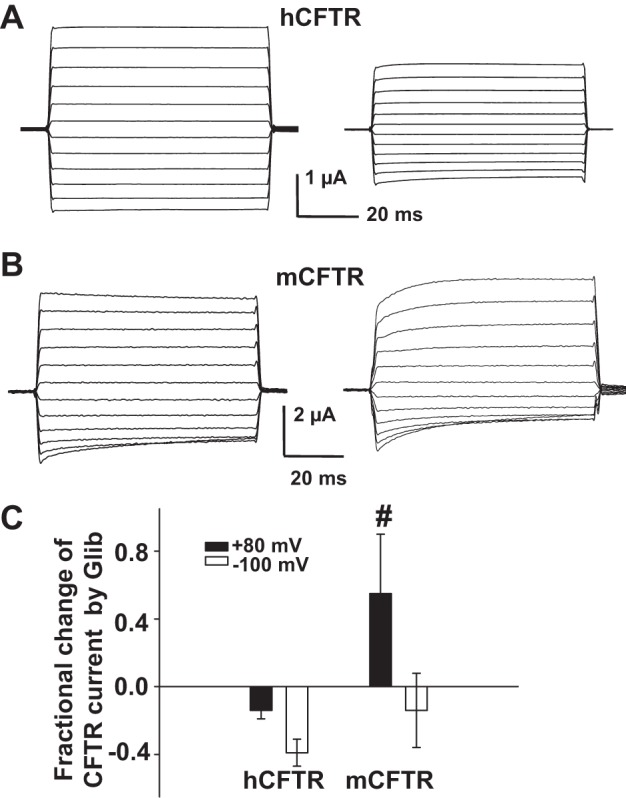
Effects of glibenclamide on hCFTR and mCFTR currents in whole oocytes. Families of TEVC currents in the absence (control, left) and presence (right) of 100 μM extracellular glibenclamide for hCFTR (A) and mCFTR (B). The voltage protocol included a holding potential VM = −30 mV, then membrane potential stepped to potentials ranging from VM = −140 mV to +80 mV in 20-mV increments with a period of 75 ms at each voltage. C: summary of the fractional change in hCFTR and mCFTR current at VM = +80 mV and −100 mV. #P < 0.01 compared with hCFTR at VM = +80 mV; n = 4–5 for each condition.
DPC blocked mCFTR and hCFTR similarly.
Besides GlyH-101, glibenclamide, and VX-770, we also asked whether DPC blocked hCFTR and mCFTR differently and whether DPC could be another tool to differentiate between mCFTR and hCFTR. DPC is a small molecule, fast open pore blocker of hCFTR that blocks in a time-independent manner from the cytoplasmic side (26). We tested the effects of 100 μM DPC on hCFTR and mCFTR utilizing the TEVC technique. As shown in Fig. 12, DPC blocked hCFTR and mCFTR with similar efficacy at positive and negative potentials and did not exhibit potentiation. These data suggest that DPC is not a valuable tool for studying hCFTR compared with mCFTR.
Fig. 12.
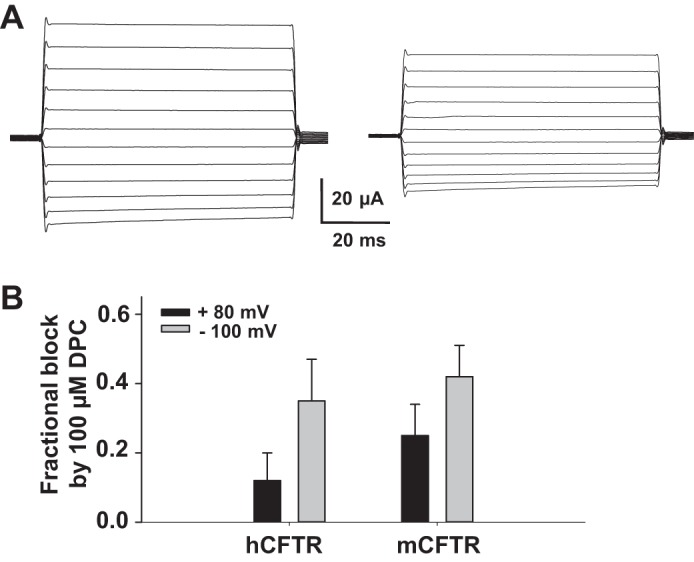
Effects of diphenylamine-2-carboxylate (DPC) on hCFTR and mCFTR in whole oocytes. Representative families of mCFTR currents in the absence (control, left) and presence (right) of 100 μM extracellular DPC. The recording conditions are the same as Fig. 11. Summary of fractional block of hCFTR and mCFTR at VM = +80 mV and −100 mV; n = 4 for each. DPC exhibited only inhibition, without potentiation.
DISCUSSION
In the present work, we asked whether the clinically useful CFTR potentiator VX-770 and three pore-blocking molecules highly used in CF research have similar impact on wild-type human CFTR and wild-type murine CFTR. The main results are summarized as follows: 1) GlyH-101 both blocked and potentiated mCFTR but only blocked hCFTR. Mutations at some sites important for channel function in hCFTR or mCFTR affected blockade but did not affect potentiation. GlyH-101 was found to increase open probability of single mCFTR channels. 2) Glibenclamide also weakly potentiated mCFTR but blocked both mCFTR and hCFTR. 3) VX-770 potentiated mCFTR strongly but had very little effect on hCFTR. VX-770 increased open probability of single mCFTR channels. VX-770 slowed deactivation of hCFTR but accelerated deactivation of mCFTR after removal of ATP and PKA. VX-770 exhibited potentiation of both hCFTR and mCFTR in an ATP-dependent manner but not in an ATP-independent manner. Hence, in addition to the known effects of VX-770 as a CFTR potentiator, both GlyH-101 and glibenclamide, highly used in CF research for their blocking capabilities, may also provide useful information as potentiators. 4) DPC, as a control, blocked mCFTR and hCFTR similarly, without apparent potentiation.
These results suggest that either mCFTR and hCFTR bind these compounds at different sites or the binding of drug has different consequences in mCFTR and hCFTR. These studies further suggest that chimeras of mCFTR and hCFTR may be useful for identification of the binding sites for CFTR potentiators.
In combination with the homology model of CFTR, the data presented in the present studies predict possible binding pockets in mCFTR. The VX-770 binding pocket is clearly not generated by the consequences of clinical mutations like G551D because several different channel mutants are sensitive to potentiation by this drug (38). There may be a single binding pocket for VX-770 that overcomes the decreased effectiveness of ATP-mediated gating in these mutants. In contrast to its effects as a pure extracellular open pore blocker of hCFTR, GlyH-101 blocked mCFTR from the extracellular side in a manner sensitive to some pore-domain mutations and also potentiated mCFTR from the cytoplasmic side in a manner insensitive to those same mutations, supporting the notion of GlyH-101 binding in two different binding pockets: one that underlies inhibition and one that underlies potentiation. Similar to GlyH-101, glibenclamide most likely blocks mCFTR as well as potentiates mCFTR, via two binding sites on the cytoplasmic side of mCFTR.
The dissimilar sensitivities of mCFTR and hCFTR to potentiators and blockers offer a strong tool for identifying binding sites and functional mechanisms.
It has been suggested that VX-770 potentiates hCFTR mutants by increasing the rate of opening as well as via an ATP-independent manner by acting directly on the NBDs (29, 31). However, recent data indicating that VX-770 potentiates channels bearing multiple disease-causing mutations, spread across CFTR, and that VX-770 potentiates one mutant but not another one in the same domain (for example, VX-770 potentiated TM mutants T338I- and R347H- but not S341P- and E92K-CFTR and potentiated cytoplasmic loop mutants E193K- and K1060T- but not R1066M- and L1065P-CFTR) do not support this conclusion (30, 38). Unlike a previous report indicating that VX-770 did not potentiate mCFTR expressed in FRT cells, measured as forskolin-stimulated transepithelial currents (29), our data indicate that VX-770 potentiated mCFTR expressed in oocytes considerably better than hCFTR. Our data clearly indicate that there are specific, quantitative differences between hCFTR and mCFTR with respect to their response to VX-770.
GlyH-101, originally identified by the Verkman group, is described as highly water soluble and poorly membrane permeant (26). However, GlyH-101 can permeate into intestinal epithelial cells with a half-time of ∼2 h (16). Our initial experiments suggested that GlyH-101 blocks mCFTR considerably more weakly (much higher Kd compared with hCFTR at the same membrane voltage), owing to simultaneous potentiation of mCFTR by GlyH-101 that had diffused to the cytoplasmic side (Kd = 0.60 nM). In fact, we can separate the inhibitory and stimulatory effects of GlyH-101 on mCFTR because the R334A mutation completely abolished the inhibitory effect of GlyH-101 on mCFTR without affecting potentiation. This is not a problem for hCFTR since GlyH-101 does not potentiate hCFTR expressed in Xenopus oocytes when applied to either side of the membrane.
Glibenclamide is a well-known open pore blocker of hCFTR that blocks the channel from the cytoplasmic side (1, 17, 39). We found that glibenclamide blocked mCFTR with a lower Kd compared with hCFTR. When the concentration was dropped to nanomolar levels, glibenclamide lost its blocking effect on mCFTR and we detected that glibenclamide is capable of weakly potentiating mCFTR. In fact, it has been demonstrated previously that glibenclamide can stimulate hCFTR at lower doses (33). Because both glibenclamide-induced inhibition and potentiation of mCFTR occur from the cytoplasmic side, it is hard to separate them completely as is possible with GlyH-101.
CFTR is now known to be “druggable” because VX-770 can target mutants of hCFTR to increase their activity (29). However, both the binding site(s) and mechanisms of action for CFTR potentiators are not known. By taking advantage of the high sequence identity between hCFTR and mCFTR, VX-770 and GlyH-101 binding sites and functional mechanism(s) may be identified by studying first human and murine chimeras followed by point mutants. It is rational to think that the site(s) to which these drugs bind might also be able to be occupied by other drugs that have even greater effect. Combined with computational docking approaches, understanding the mechanism of VX-770 and GlyH-101 will lead to rational drug design to identify better therapeutics for control of CF disease.
GRANTS
The work was supported by National Institutes of Health (NIH) grants (to N. A. McCarty, NIH R01-DK 056481 and R01-DK 075016), a Cystic Fibrosis Foundation research grant (to N. A. McCarty, CFF MCCART14G0), and an EECRSeed Grant (to G. Cui).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.C. and N.A.M. conception and design of research; G.C. performed experiments; G.C. analyzed data; G.C. interpreted results of experiments; G.C. prepared figures; G.C. drafted manuscript; G.C. and N.A.M. edited and revised manuscript; G.C. and N.A.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Becky Kinkead and Stacy Heilman of the Pediatric Grant Editing and Manuscript Support Core at Emory University for editing the manuscript.
REFERENCES
- 1.Budriesi R, Ioan P, Leoni A, Pedemonte N, Locatelli A, Micucci M, Chiarini A, Galietta LJ. Cystic fibrosis: a new target for 4-imidazo[2,1-b]thiazole-1,4-dihydropyridines. J Med Chem 54: 3885–3894, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caci E, Caputo A, Hinzpeter A, Arous N, Fanen P, Sonawane N, Verkman AS, Ravazzolo R, Zegarra-Moran O, Galietta LJ. Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem J 413: 135–142, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Cotten JF, Welsh MJ. Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR. Evidence for disruption of a salt bridge. J Biol Chem 274: 5429–5435, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Cui G, Rahman KS, Infield DT, Kuang C, Prince CZ, McCarty NA. Three charged amino acids in extracellular loop 1 are involved in maintaining the outer pore architecture of CFTR. J Gen Physiol 144: 159–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui G, Song B, Turki HW, McCarty NA. Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflügers Arch 463: 405–418, 2012. [DOI] [PubMed] [Google Scholar]
- 6.Cui G, Zhang ZR, O'Brien AR, Song B, McCarty NA. Mutations at arginine 352 alter the pore architecture of CFTR. J Membr Biol 222: 91–106, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem 287: 36639–36649, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266: 107–109, 1994. [DOI] [PubMed] [Google Scholar]
- 9.Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440: 477–483, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galietta LJ, Moran O. Identification of CFTR activators and inhibitors: chance or design? Curr Opin Pharmacol 4: 497–503, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Grove DE, Rosser MF, Ren HY, Naren AP, Cyr DM. Mechanisms for rescue of correctable-folding defects in CFTRDelta F508. Mol Biol Cell 20: 4059–4069, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol 36: 1–7, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci USA 110: 4404–4409, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lansdell KA, Delaney SJ, Lunn DP, Thomson SA, Sheppard DN, Wainwright BJ. Comparison of the gating behaviour of human and murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in mammalian cells. J Physiol 508: 379–392, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lansdell KA, Kidd JF, Delaney SJ, Wainwright BJ, Sheppard DN. Regulation of murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in Chinese hamster ovary cells. J Physiol 512: 751–764, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levin MH, Kim JK, Hu J, Verkman AS. Potential difference measurements of ocular surface Na+ absorption analyzed using an electrokinetic model. Invest Ophthalmol Vis Sci 47: 306–316, 2006. [DOI] [PubMed] [Google Scholar]
- 17.McCarty NA. Permeation through the CFTR chloride channel. J Exp Biol 203: 1947–1962, 2000. [DOI] [PubMed] [Google Scholar]
- 18.McCarty NA, Zhang ZR. Identification of a region of strong discrimination in the pore of CFTR. Am J Physiol Lung Cell Mol Physiol 281: L852–L867, 2001. [DOI] [PubMed] [Google Scholar]
- 19.McDonough S, Davidson N, Lester HA, McCarty NA. Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron 13: 623–634, 1994. [DOI] [PubMed] [Google Scholar]
- 20.Mense M, Vergani P, White DM, Altberg G, Nairn AC, Gadsby DC. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J 25: 4728–4739, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuberger T, Burton B, Clark H, Van Goor F. Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol Biol 741: 39–54, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Ostedgaard LS, Rogers CS, Dong Q, Randak CO, Vermeer DW, Rokhlina T, Karp PH, Welsh MJ. Processing and function of CFTR-DeltaF508 are species-dependent. Proc Natl Acad Sci USA 104: 15370–15375, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penmatsa H, Frederick CA, Nekkalapu S, Conoley VG, Zhang W, Li C, Kappes J, Stokes DC, Naren AP. Clinical and molecular characterization of S1118F-CFTR. Pediatr Pulmonol 44: 1003–1009, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott-Ward TS, Cai Z, Dawson ES, Doherty A, Da Paula AC, Davidson H, Porteous DJ, Wainwright BJ, Amaral MD, Sheppard DN, Boyd AC. Chimeric constructs endow the human CFTR Cl− channel with the gating behavior of murine CFTR. Proc Natl Acad Sci USA 104: 16365–16370, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sondo E, Tomati V, Caci E, Esposito AI, Pfeffer U, Pedemonte N, Galietta LJ. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am J Physiol Cell Physiol 301: C872–C885, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Y, Sonawane ND, Salinas D, Qian L, Pedemonte N, Galietta LJ, Verkman AS. Evidence against the rescue of defective DeltaF508-CFTR cellular processing by curcumin in cell culture and mouse models. J Biol Chem 279: 40629–40633, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Stahl M, Stahl K, Brubacher MB, Forrest JN Jr. Divergent CFTR orthologs respond differently to the channel inhibitors CFTRinh-172, glibenclamide, and GlyH-101. Am J Physiol Cell Physiol 302: C67–C76, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan LP, Wallace DP, Grantham JJ. Epithelial transport in polycystic kidney disease. Physiol Rev 78: 1165–1191, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106: 18825–18830, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 13: 29–36, 2014. [DOI] [PubMed] [Google Scholar]
- 31.Venglarik CJ, Schultz BD, DeRoos AD, Singh AK, Bridges RJ. Tolbutamide causes open channel blockade of cystic fibrosis transmembrane conductance regulator Cl− channels. Biophys J 70: 2696–2703, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walters R, Welsh M. Mechanism by which calcium phosphate coprecipitation enhances adenovirus-mediated gene transfer. Gene Ther 6: 1845–1850, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Li G, Clancy JP, Kirk KL. Activating cystic fibrosis transmembrane conductance regulator channels with pore blocker analogs. J Biol Chem 280: 23622–23630, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Wang W, Roessler BC, Kirk KL. An electrostatic interaction at the tetrahelix bundle promotes phosphorylation-dependent cystic fibrosis transmembrane conductance regulator (CFTR) channel opening. J Biol Chem 289: 30364–30378, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Wu J, Bernard K, Li G, Wang G, Bevensee MO, Kirk KL. ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc Natl Acad Sci USA 107: 3888–3893, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, Bot A, Jorna H, de Jonge HR, Scholte BJ. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros 10, Suppl 2: S152–S171, 2011. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Yu M, Jan YN, Jan LY. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proc Natl Acad Sci USA 94: 1568–1572, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP Jr, Urrutia A, Joubran J, Seepersaud S, Sussky K, Hoffman BJ, Van Goor F. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 11: 237–245, 2012. [DOI] [PubMed] [Google Scholar]
- 39.Zhang ZR, Cui G, Zeltwanger S, McCarty NA. Time-dependent interactions of glibenclamide with CFTR: kinetically complex block of macroscopic currents. J Membr Biol 201: 139–155, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Zhang ZR, Song B, McCarty NA. State-dependent chemical reactivity of an engineered cysteine reveals conformational changes in the outer vestibule of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 280: 41997–42003, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Zhang ZR, Zeltwanger S, McCarty NA. Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J Membr Biol 175: 35–52, 2000. [DOI] [PubMed] [Google Scholar]