

Abstract
Post-traumatic seizures can exacerbate injurious outcomes of severe brain trauma, yet effective treatments are limited owing to the complexity of the pathology underlying the concomitant occurrence of both events. In this study, we tested C‐10068, a novel deuterium-containing analog of (+)-N-methyl-3-ethoxymorphinan, in a rat model of penetrating ballistic-like brain injury (PBBI) and evaluated the effects of C-10068 on PBBI-induced nonconvulsive seizures (NCS), acute neuroinflammation, and neurofunctional outcomes. NCS were detected by electroencephalographic monitoring. Neuroinflammation was evaluated by immunohistochemical markers, for example, glial fibrillary acidic protein and major histocompatibility complex class I, for activation of astrocytes and microglia, respectively. Neurofunction was tested using rotarod and Morris water maze tasks. Three infusion doses of C-10068 (1.0, 2.5, and 5.0 mg/kg/h×72 h) were tested in the antiseizure study. Neuroinflammation and neurofunction were evaluated in animals treated with 5.0 mg/kg/h×72 h C-10068. Compared to vehicle treatment, C-10068 dose dependently reduced PBBI-induced NCS incidence (40–50%), frequency (20–70%), and duration (30–82%). The most effective antiseizure dose of C-10068 (5.0 mg/kg/h×72 h) also significantly attenuated hippocampal astrocyte activation and perilesional microglial reactivity post-PBBI. Within C-10068-treated animals, a positive correlation was observed in reduction in NCS frequency and reduction in hippocampal astrocyte activation. Further, C-10068 treatment significantly attenuated astrocyte activation in seizure-free animals. However, C-10068 failed to improve PBBI-induced motor and cognitive functions with the dosing regimen used in this study. Overall, the results indicating that C-10068 exerts both potent antiseizure and antiinflammatory effects are promising and warrant further investigation.
Key words: : C-10068, dextromethorphan, EEG, neuroinflammation, nonconvulsive seizures, penetrating brain injury
Introduction
Seizures can occur at various stages after severe traumatic brain injury (sTBI), where onset latency ranges from hours/days to months/years postinjury. The early post-traumatic seizures (PTS) occurring within 7 days post-TBI are classified as provoked seizures because they result from the direct effects of the initial brain injury.1,2 Acute postinjury manifestations of PTS have been shown to exacerbate the underlying brain injury through increased metabolic demands, increased intracranial pressure, and increased release of excitatory neurotransmitters.3–7 They may also pose an increased risk for patients to develop late post-traumatic epilepsy.1,8,9 Therefore, early PTS represent an urgent medical concern and should be treated promptly.10,11
Clinical management of PTS in sTBI patients has been challenging with regard to diagnostic capabilities and prophylaxis. Acute PTS may manifest as convulsive or nonconvulsive seizures (NCS). Whereas convulsive seizures can be diagnosed easily and treated promptly, NCS occur without motor manifestation and are often underdiagnosed without electroencephalographic (EEG) monitoring and therefore left untreated. Additionally, the choices of antiepileptic drugs (AEDs) to treat early PTS are limited despite an abundance of AEDs currently available for treating seizure disorders in general. This is partly because most AEDs are designed for oral administration and those AEDs applicable for intravenous (i.v.) administration, (e.g., pentobarbital, benzodiazepines, and propofol) are often known to be associated with adverse effects, such as hypotension, cardiac and/or renal problems, or even increased epileptiform or spike discharge.12 These AED-induced secondary complications can be detrimental to sTBI patients. Currently, phenytoin, which acts as a classic sodium-channel blocker, represents the most commonly used AED for prophylaxis of PTS because it can be administered i.v. as its phosphate prodrug, fosphenytoin, and has shown efficacy in controlling generalized convulsive seizures.13–16 Recently, we have also demonstrated that phenytoin was able to attenuate post-traumatic NCS in a rat model of penetrating brain injury.17 However, the adverse effects of phenytoin on cardiovascular, hepatic, and neurological systems and the narrow therapeutic window owing to its nonlinear pharmacokinetics18 may limit its aggressive use in sTBI patients.
Considering the harmful role of PTS and the lack of safe, effective drugs to mitigate these seizures, efforts to develop new antiseizure drugs aimed at treating early PTS are critically important. Dextromethorphan (DM) is a potent anticonvulsant that acts as an N-methyl-D-aspartate (NMDA) receptor antagonist,19 a voltage-gated calcium-channel antagonist,20 and a sigma-1 receptor agonist.21,22 These activities collectively support the potential of DM to be both a neuroprotective and antiepileptic agent, given that unbalanced excitatory and inhibitory system, iron-channel dysfunction, and sigma-1 receptors are all implicated in the pathological mechanisms of acute brain injuries and seizures as well.23–26 In fact, previous experimental studies have shown that DM treatment protects neuronal damage27–30 and improves motor and cognitive functions27,31,32 in different animal models of brain injury. It also attenuated seizure activities induced by kainite.33,34
However, DM is quickly metabolized to dextrorphan (DX) in vivo by cytochrome P450 2D6, which is commonly present in liver and brain tissue. DX itself is also a potent anticonvulsant, but can cause psychotomimetic symptoms owing to its high affinity to specific PCP sites in the brain.35–37 In order to retain the anticonvulsant properties of DM without in vivo conversion to DX, several attempts have been made to develop DM analogs. These analogs were constructed mainly by modification in positions 3 and 17 of the morphinan ring system, and (+)-N-methyl-3-ethoxymorphinan is one of them, which was referred to as AHN-1-037 in a previous report.38 The structure of (+)-N-methyl-3-ethoxymorphinan differs from DM by having an ethyl ether, rather than a methyl ether, at the 3-position of the morphinan ring. C‐10068 is a deuterium-containing analog of (+)-N-methyl-3-ethoxymorphinan, developed by Concert Pharmaceuticals, Inc. In C-10068, the 3-ethoxy group and the N-methyl group are fully substituted by deuterium. Selective deuterium incorporation results in increased metabolic stability, compared to its parent compound and DM, providing less of the undesirable DX metabolite.39 Studies have shown that C-10068 has similar pharmacology to DM, but binds to the human sigma-1 receptor with ∼6-fold higher affinity.40 C-10068 also demonstrated antiseizure properties in traditional (non-brain-injury related) animal seizure models conducted under the National Institute of Neurological Disorders and Stroke Anticonvulsant Screening Program. Further, in vitro neuroprotection tests (hippocampal slice model) showed that C-10068 inhibited cytotoxicity induced by NMDA and kainic acid.41
The mechanisms of early PTS are likely mingled with the acute pathological cascade of TBI, involving excessive glutamate toxicity and ion-channel dysfunction, which are the primary mechanistic targets of both DM and C-10068. Thus, the primary focus of the current study was to test the dose-response effects of C-10068 against early post-traumatic NCS induced by a penetrating ballistic-like brain injury (PBBI) in rats. Additionally, the potential anti-inflammatory and neuroprotective effects of C-10068 were also evaluated using the most effective antiseizure dose regimen in an attempt to understand the relationship between C-10068's antiseizure and antiinflammation effects, as well as the possible translation of these effects into improvement of neurological functions after the penetrating brain injury.
Methods
Subjects
Adult male Sprague-Dawley rats (275–350 g; Charles River Labs, Raleigh, VA) were used. Rats were housed individually (owing to the presence of indwelling i.v. catheters and EEG head mounts) in a well-ventilated vivarium under a normal 12-h light/dark cycle (lights on 6:00 am–6:00 pm) with free access to food and water before and after brain injury.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Walter Reed Army Institute of Research (WRAIR; Silver Spring, MD). Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals. The animal housing facility was accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Surgeries
During surgical procedures, rats were anesthetized with 2% isoflurane vapor and body temperature was maintained at 37.0°C using a heating blanket (Harvard Apparatus, Holliston, MA).
Electroencephalographic electrode implantation
The epidural EEG electrode was constructed using a 2.5-cm section of insulated Nichrome wire (0.2 mm in diameter, uninsulated at each end; Leico Industries, New York, NY) soldered to a stainless steel screw (0-80×0.32 cm in length; Component Supply Company, Fort Mead, FL). EEG electrode implantation was performed on the anesthetized rat secured on a stereotaxic device. The rat's skull surface was exposed by a mid-line skin incision. Capillary bleeding was cauterized to keep the skull surface dry and clean. Four EEG electrodes (a–d) were symmetrically implanted on the rat skull through tightly fit burr holes over the bilateral frontal and parietal regions of the brain (1 mm anterior and 4 mm posterior to the bregma and±3.5 mm lateral to mid-line) and a reference electrode (R) was placed posterior to the lambda over the transverse sinus (Fig. 1A). The free end of each electrode wire was soldered to a Dale multi-pin connector (March Electronics, West Hempstead, NY) in the sequence of a, b, c, and d, which corresponded to the left frontal, left parietal, right frontal, and right parietal cortical regions, respectively. Each electrode was individually referenced to the electrode R to create a four-channel monoreferential EEG recording montage. Screw electrodes were secured on the skull with tissue adhesive and insulated with a layer of dental acrylic. The whole assembly was then firmly attached to rat skull with more dental acrylic. All rats were allowed at least 5 days of recovery before receiving the PBBI surgery.
FIG. 1.
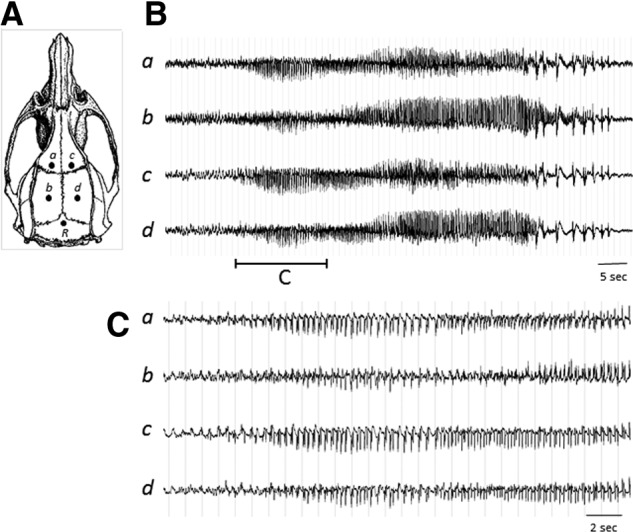
(A) Illustration of EEG electrode configuration. Electrodes a–d were individually referenced to the electrode R to create a four-channel monoreferential EEG recording montage. (B) An example of NCS episode detected by EEG. (C) Seizure initiation shown on an expanded timescale. EEG, electroencephalographic; NCS, nonconvulsive seizures.
Intravenous cannulation
For continuous i.v. infusion of C-10068, each rat was prepared with an indwelling catheter into the jugular vein 2 days after the EEG electrode implantation. The i.v. catheter consisted of a 10.5-cm section of PE-50 tubing coupled with a silastic tip (1 cm) at one end. Under a surgical microscope, the right jugular vein was exposed and isolated. The silastic tip of the catheter was inserted into the jugular vein and secured in position by a piece of 4-0 silk suture. The other end of the catheter was guided subcutaneously to exit through the dorsal neck region. The i.v. catheter was then connected to a piece of infusion tubing, which was attached to the EEG swivel system allowing continuous i.v. infusion in awake and unrestrained animals.
Penetrating ballistic-like brain injury
The PBBI apparatus consisted of a PBBI probe (Kadence Science, Lake Success, NY), a stereotaxic frame equipped with a specially designed probe holder, and a hydraulic pressure-pulse generator (4B080; Mitre Corp, McClean VA). The penetrating probe was made of a 20-G stainless steel tube with fixed perforations along one end, which were sealed by a piece of airtight elastic tubing. The probe was secured on the probe holder with the unperforated end attached to the pulse generator, angled at 50 degrees from the vertical axis and 25 degrees counter clockwise from the midline. During surgery, the head of the anesthetized rat was secured in a stereotaxic device for insertion of the PBBI probe. After a mid-line scalp incision, a right frontal cranial window (diameter=4 mm) was created using a dental drill to expose the right frontal pole of the brain (+4.5 mm anteroposterior [AP], +2 mm mediolateral to bregma). The PBBI probe was advanced through the cranial window into the right hemisphere to a depth of 1.2 cm from the surface of the brain. Once the probe was in place, the pulse generator was activated by a computer to release a pressure pulse calibrated to produce a rapid expansion of the water-filled elastic tubing, forming an elliptical shaped balloon (diameter=0.633 mm; expansion duration, ≤40 ms) to a volume equal to 10% of total brain volume. This rapid inflation/deflation of the balloon mimics the generation of a ballistic force shock wave, thereby creating a temporary cavity in the brain. After the balloon was deflated, the probe was retracted from the brain, the cranial opening was sealed with sterile bone wax, and the skin incision was closed with a piece of 4-0 silk suture.
Pharmacokinetics of C-10068
Material
The chemical compound, C-10068, (+)-(9alpha,13alpha,14alpha)-3-(pentadeuteroethoxy)-17-(trideuteromethyl)morphinan hydrochloride, was developed and synthesized by Concert Pharmaceuticals Inc. and delivered to WRAIR under a Cooperative Research and Development Agreement between the two institutions.
Treatment protocol
Pharmacokinetic studies were performed on naïve rats. The dosing regimen consisted of an i.v. bolus dose using one of the three following doses: 7.5, 15.0, or 20.0 mg/kg (dissolved in saline, 1 mL/kg; N=2–3 per group). Each i.v. bolus dose was immediately followed by an i.v. infusion dose of 8 mg/kg/h for 72 h.
Blood and brain sample collection
Blood samples (200 μL/sample) were collected from the jugular vein immediately after bolus injection and at 1, 4, 24, and 72 h after the initiation of i.v. infusion. At each time point, blood samples were immediately transferred into tubes containing K2EDTA (anticoagulant) and placed on ice. Within 30 min of blood collection, blood samples were centrifuged at 2–8°C for 10 min at 3000 rpm and plasma samples (100–125 μL) were collected and stored at −80°C until analysis. After 72-h infusion of C-10068, each rat was deeply anesthetized with an intramuscular (i.m.) injection of a ketamine (70 mg/kg) and xylazine (6 mg/kg) mixture and euthanized by transcardial (t.c.) exsanguinations with physiological saline. At the end of the saline perfusion, rat brain was removed from the skull, snap-frozen in liquid nitrogen, and stored at −80°C until analysis.
Pharmacokinetic analysis
C-10068 was analyzed using positive ion atmospheric pressure chemical ionization high-performance liquid chromatography with tandem mass spectrometry on an AB Sciex API-4000 tandem mass spectrometer. The m/z ratio for the multiple reaction monitoring transition (294.3-234.2) resulted in a sensitive, selective quantitation of C-10068 in complex biological matrices. Gradient separation was performed by eluting C-10068 on a reverse-phase Xbridge phenyl RP 3.5-um 3.0×100 mm (PN#186003328) analytical column. Samples were processed from matrix by using an acetonitrile protein precipitation extraction followed by vortexing and high-speed centrifugation. The lower limit of quantitation (LLOQ) was determined to be 1 ng/mL using a 20-μL injection volume.
Antiseizure study
Treatment protocol
Three doses of C-10068 (1.0, 2.5, or 5.0 mg/kg/h; N=16–17/dose) were tested in the PBBI-induced NCS model. C-10068 was delivered as continuous i.v. infusion (infusion rate at 0.1 mL/h) for 72 h for each animal. A group of PBBI control animals (N=16) received matching vehicle (normal saline) infusion for 72 h, and sham control animals (N=4) received a 5.0-mg/kg/h C-10068 infusion for 72 h. All treatments were initiated at 30 min post-PBBI or sham procedures and terminated at 72 h postinjury. Animals were euthanized at the end of the treatment for histopathological analysis.
Electroencephalographic recordings
Continuous EEG recordings were performed on awake, freely moving animals housed individually in Plexiglas EEG recording chambers (30L×30W×50H cm) equipped with multi-channel gold contact swivel commutators (Dragonfly Inc., Ridgeley, WV). Each rat was tethered to the swivel commutator by a flexible shielded cable (Plastics One Inc., Roanoke, VA) connected to the Dale connector, which was secured on the rat skull. The swivel commutator was interfaced with an EEG amplifier (Natus Medical Inc., San Carlos, CA). EEG signals were recorded at a sampling rate of 200 Hz and digitized by a computerized data acquisition system (Harmonie software; Natus Medical Inc., San Carlos, CA). EEG recordings with video monitoring were collected from each rat for at least 30 min before PBBI surgery and continued postinjury for 72 h.
Electroencephalographic analysis
An NCS event was defined as rhythmic spike discharges occurring at a frequency of 1–4 Hz for at least 10 sec, with the amplitude distinctively greater than background activities.42 Variations of EEG waveforms also included rhythmic sharp waves (∼1 Hz) or spike waves at irregular intervals of 0.5–2.0 Hz.43 Based on these criteria, the following NCS parameters were included in the statistical analyses: 1) NCS incidence: the number of animals that experienced NCS events within a treatment group, expressed as the percentage of the group; (2) NCS frequency: the number of NCS events experienced by an individual animal; (3) NCS episode duration (sec): the duration of one NCS event; (4) NCS cumulative duration (sec): the sum of NCS episode duration for an individual animal; and (5) NCS onset latency (h): the time lapse between the PBBI and occurrence of the first NCS event. To control for “floor” effects, vehicle-treated animals that displayed zero incidence of post-traumatic NCS and corresponding ranked pairs in each C-10068-treated group were excluded from statistical analyses of NCS frequency duration, as well as latency.17 NCS parameters are expressed as the mean and standard error of the mean (SEM), when appropriate, in the Results section.
Histopathology
At 72 h post-PBBI, EEG recording and treatment were terminated. All rats were deeply anesthetized with an i.m. injection of a ketamine (70 mg/kg) and xylazine (6 mg/kg) mixture and t.c. perfused with 0.1 M of phosphate-buffered saline (PBS), followed by 4% paraformaldehyde. Brains were removed, fixed for 6 h, and then transferred to 20% sucrose solution for cryoprotection. Coronal sections of the brain (40 μm thick) were cut through the entire cerebrum (+4 to −7 mm AP from bregma). The first set of serial brain sections collected at a 960-μm interval from each animal was stained with hematoxylin and eosin (H&E) for histological analysis of lesion volume. Digitized images of H&E-stained brain sections were captured at 0.5× magnification using an Olympus BX61 microscope (Olympus, Center Valley, PA). Lesion area on each affected brain section was defined by tracing the perimeter of the lesion cavity (including injured penumbra), and the lesion volume of each brain was calculated using the Cavalieri formula applied to the sequential brain sections (Inquiry Digital Analysis System; Loats Assoc., Westminster, MD).
Antineuroinflammation study
To evaluate the effects of C-10068 on neuroinflammation and its possible relationship to its effects on PBBI-induced NCS, brain tissues of vehicle-treated animals and those treated with 5.0 mg/kg/h×72 h C-10068 from the above-described antiseizure studies were processed for immunohistochemistry (IHC) of glial fibrillary acidic protein (GFAP) and rat major histocompatibility complex class I (OX-18) markers of activated astrocytes and microglia cells, respectively.
Immunohistochemistry procedure
Two sets of serial brain sections were collected at 960-μm intervals from each animal in vehicle- and C-10068-treated animals mentioned above and stained with GFAP and OX-18. After inactivating endogenous peroxidase activity with hydrogen peroxidase, sections were incubated separately with avidin and biotin solutions (Vector Laboratories, Burlingame, CA) for blocking nonspecific binding of biotin, biotin-binding protein, and lectins. Sections were then incubated free-floating for 1 h in 0.01 M of PBS (pH 7.4) containing 1% normal blocking serum, 0.3% Triton X-100 (Sigma-Aldrich, St. Louis, MO), and one of the specific antibodies (Abs): rabbit anti-GFAP (1:8000; Dako, Carpinteria, CA) and mouse anti-OX-18 (1:6000; AbD Serotec, Raleigh, NC). Subsequently, the immunoreaction product was visualized according to the avidin-biotin complex method with the Vectastin elite ABC kit (Vector Laboratories). All tissue sections were processed simultaneously in batches based on each respective Ab to ensure consistent, uniform staining results. All brain tissue preparation and staining was performed by FD Neurotechnologies, Inc. (Baltimore, MD).
Quantitative analysis of neuroinflammatory profile
For unbiased evaluation of immunohistopathology, quantitative analysis was conducted by an experimenter who was blind to treatment and digital images of GFAP, and OX-18-stained brain sections were captured at uniform criteria for sensitivity and exposure time using the Olympus BX61 microscope. For each set of immunostained brain series, images of all collected sections were taken at 4× magnification for evaluations of gross positive staining. Additionally, high-magnification images were captured at 20× from regions of interest (ROIs), namely, the perilesional regions (GFAP and OX-18) and the hippocampal regions (GFAP) of the most affected brain sections for quantification of GFAP and OX-18 reactivity in these regions. More specifically, for both GFAP and OX-18 immunostains, two 20× images were captured in the perilesional regions (medial and ventral to the lesion) from each of the three brain sections at bregma AP: +2.0, +1.0, and 0.0 mm, yielding a total of six perilesional images for each animal. For GFAP-stained brain series, an additional five 20× images were captured in the hippocampus (cornu ammonis [CA]1–CA3 and the dentate) from each of the two brain sections posterior to the injury core site (bregma AP: −3.0 and −4.0 mm), yielding a total of 10 GFAP-stained hippocampal images for each animal.
Quantification of GFAP and OX-18 reactivity was performed using thresholding analysis provided by ImageJ software (Version 1.44; National Institutes of Health, Bethesda, MD). The threshold value for each stain was determined as the maximal positive staining of the IHC target confined within the 20× image of ROIs with minimal artifacts. The threshold value generated for each specific stain was applied uniformly across all brain sections of each animal in order to ensure objective quantification. Positive staining on a given image was quantified as the number of pixels detected above the selected threshold value and expressed as the ratio to the total pixels of the entire 20× image. The pixel measurement was then automatically converted into area measurement (mm2) according to the image resolution (pixels per mm) and expressed as the percent area (% area) for a given image. The value of positive staining of the specific protein marker was derived by averaging the percent area of positive staining across all analyzed images for the respective ROI for a given animal.
Neuroprotection study
Additional experiments were conducted to evaluate the potential neuroprotective effects of C-10068 on neurobehavioral outcomes. For the neurobehavioral study, the most effective antiseizure dose of C-10068 (5.0 mg/kg/h×72 h) was tested in a separate cohort of animals. These animals were randomly assigned to one of the following three groups: sham (N=12); PBBI+vehicle (N=15); and PBBI+C-10068 (N=15). Motor function was assessed on a rotarod task from 7–10 days post-PBBI and cognitive function was evaluated in the Morris water maze (MWM) task from 13 to 17 days post-PBBI.
Rotarod task
The Rotamex-5 rotarod apparatus (Columbus Instruments, Columbus, OH) was used to measure animals' motor coordination and balance. Before PBBI, all rats were handled and trained to criteria on the rotarod at fixed-speed increments of 5, 10, 15, and 20 rpm using the procedures described previously.44 Baseline measures (2 trials/speed at 10, 15, and 20 rpm and 60 sec per trial) were collected from each animal on the day before PBBI to ensure consistent performance among all animals. At 7 and 10 days postinjury, the ability of animals to remain balanced on the rotating rod was assessed at sequential fixed-speed increments of 10, 15, and 20 rpm for a maximum of 60 sec each trial (2 trials/speed; 60-sec intertrial interval). The outcome measure was defined as the latency (sec) to fall off the rotating rod.
Morris water maze task
Spatial learning and memory abilities were assessed in the MWM test starting 13 days post-PBBI.44 The swimming pool (75 cm deep; 175 cm diameter) was filled with clear water (∼22°C) to a depth of 60 cm with a clear Plexiglas platform being placed at the center of the northwest quadrant (35 cm from the wall of the pool and 1 cm below the water surface). The platform remained in the same position throughout the experiment. Each animal was given four trials per day for 5 consecutive days (30-min intertrial interval) from 13 to 17 days post-PBBI. For each trial, the animal was placed in the MWM at one of four pseudorandomly determined starting points (e.g., north, south, east, and west) and allowed 90 sec to locate the hidden platform. Once the animal located the platform, it was given a 10-sec rest period before being removed to its home cage, which was kept warm by a heating blanket. Animals that could not locate the platform within the allotted time were gently guided to the platform and also allowed to rest for 10 sec on the platform. All trials were recorded with a digital video-tracking system (Noldus EthoVision XT). Primary outcome measures included: 1) latency (sec) to find the hidden platform; 2) swim distance (cm) to the hidden platform; and 3) swim speed (cm/sec).
Statistical analysis
Treatment effects of C-10068 on seizure frequency, duration, and latency were analyzed using Kruskal-Wallis' ranked analysis of variance (ANOVA) and Mann-Whitney's U tests. Fisher's exact test was used for evaluating a trend in proportions of NCS incidence. Student's t-test was used for analyzing immunoreactivity of GFAP and OX-18. Pearson's correlation analysis was used for analyzing the relationship between seizure frequency and immunoreactivity of GFAP and OX-18. Behavioral endpoints were compared by one-way or repeated-measures ANOVA, followed by Tukey's post-hoc analysis, as appropriate, or by using paired t-tests between matched groups. Group differences were considered statistically significant at p<0.05. All data were expressed as mean±SEM, when appropriate.
Results
Pharmacokinetics of C-10068
Blood samples collected immediately after the bolus injections of C-10068 (7.5, 15.0, or 20.0 mg/kg) showed linear dose-dependent increases in plasma concentrations of C-10068 to 1890, 3460, and 5343 (μg/mL), respectively (Fig. 2A). This initial bolus dose-response effect was diminished and no longer differed significantly among groups by 1 h postinjection. Plasma concentrations of C-10068 remained relatively consistent during the chronic infusion protocol, averaging 2683 μg/mL at 1 h, 2700 μg/mL at 4 h, 3474 μg/mL at 24 h, and 3161 μg/mL at 72 h post-treatment initiation (Fig. 2B). At the end of the 72-h infusion period, average brain concentration of C-10068 was 16,862 μg/mL (Fig. 2B), which resulted in a brain/plasma ratio of 5.8 at the termination of the treatment.
FIG. 2.
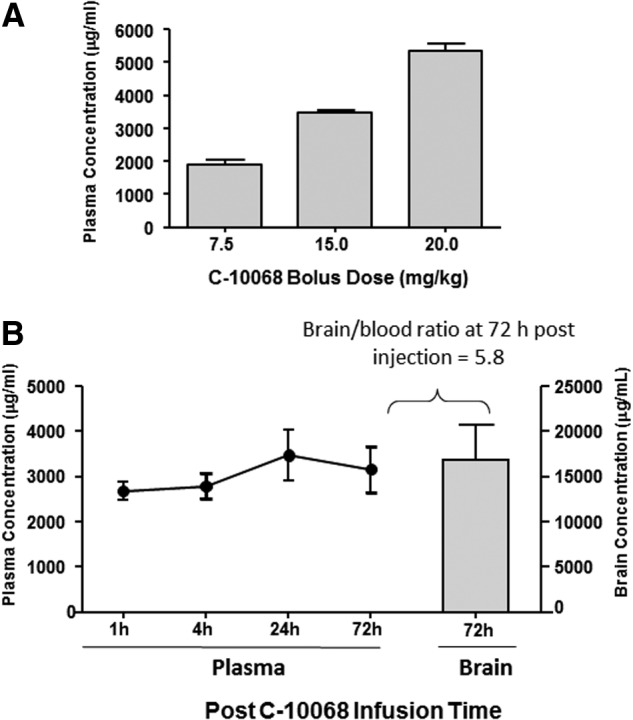
Pharmacokinetic profile of C-10068 in naïve animals. (A) Dose-response profile of C-10068 plasma concentration immediately after a single bolus injection. (B) Steady plasma concentration of C-10068 maintained by the continuous intravenous infusion of C-10068 (8 mg/kg/h×72 h) is shown as the line graph and the brain concentration of C-10068 at the termination of the infusion is shown as the gray bar.
Because brain bioavailability beyond 1 h post-treatment was not influenced by the bolus doses and considering a possible increase in brain bioavailability of C-10068 after penetrating brain injury, the dosing regimen for the antiseizure study was determined at 1.0–5.0 mg/kg/h×72 h infusion without the bolus component.
Antiseizure effects of C-10068
No evidence of NCS was detected in sham control animals treated with C-10068. However, similar to previous findings,17,43 spontaneous NCS occurred in a sporadic fashion post-PBBI detected by 72-h continuous EEG monitoring. No convulsive behavior was observed accompanying EEG ictal discharge based on the review of video clips. EEG ictal discharges manifested as rhythmic spikes or spike/sharp waves with marked augmentation of amplitude, compared to background EEG activities (Fig. 1B,C). The majority of these seizures were generalized NCS occurring in both hemispheres, but, occasionally, focal NCS were also detected from either the injured (electrodes c and d) or the contralateral (electrodes a and b) hemisphere.
In the vehicle-treated group, 75% (12 of 16) of animals experienced NCS with an onset latency ranging between 1 and 48 h post-PBBI (mean onset latency=19.2±4.8 h). On average, each of these rats experienced 8.3±2.4 NCS episodes with a duration of 33±6.5 sec/event, yielding an average cumulative NCS duration of 379.2±153.2 sec/rat. Compared to vehicle treatment, continuous infusion of C-10068 reduced NCS incidence to 41–50% (p>0.05; Fig. 3A) and attenuated NCS episode duration by 7–32% (p>0.05) across all three doses tested, yet lacked significant dose-response effects (data not shown). However, C-10068 treatment dose dependently reduced NCS frequency and cumulative NCS duration, with the most significant effects being achieved at the highest dose tested (5.0 mg/kg/h×72 h). As shown in Figure 3B, compared to the vehicle-treated group, continuous infusion of C-10068 at 1.0, 2.5, and 5.0 mg/kg/h for 72 h reduced NCS frequency to 6.6±2.5 (p>0.05), 4.9±2.6 (p>0.05), and 2.1±0.5 (p<0.05) NCS events per rat, which accounted for 21%, 41%, and 75% reduction in NCS frequency, respectively. Similarly, NCS total duration was reduced by 31%, 40%, and 80% to 263.3±112.0 (p>0.05), 225.8±99.3 (p>0.05), and 69.3±25.5 sec (p<0.05) in the ascending dose order tested (Fig. 3C). Each dose of C-10068 also significantly delayed NCS onset latency to an average of 43.5±8.2–46.7±5.9 h post-PBBI (p<0.05 for each dose; Fig 3D). In vehicle-treated animals, approximately 50% of NCS occurred within the first 24 h postinjury, and, accumulatively, 86% of NCS occurred by 48 h postinjury, whereas in C-10068 treated animals, delay of NCS occurrence after its onset was best evidenced in the highest dose tested (5.0 mg/kg/h×72 h), which showed that only 4% of NCS occurred within the first 24 h postinjury, and, accumulatively, 68% of NCS occurred by 48 h postinjury (Fig. 4).
FIG. 3.

Dose-response effects of C-10068 treatment on PBBI-induced NCS activities: (A) reduction in NCS incidence; (B) reduction in NCS frequency; (C) reduction in cumulative NCS duration; and (D) delay of NCS onset latency. *p<0.05, compared to vehicle-treated group. NCS, nonconvulsive seizures.
FIG. 4.
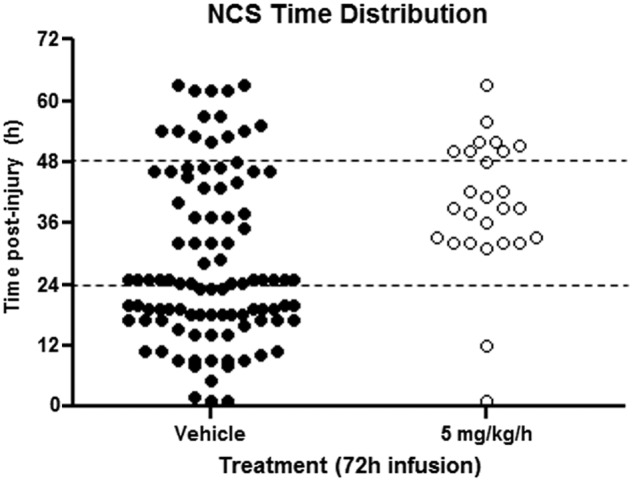
Comparison of time distribution of individual NCS events over a 72-h post-PBBI period between vehicle- (solid black circle) and C-10068-treated (5.0 mg/kg/h×72 h) animals (open circle). Each circle represents a seizure event. NCS, nonconvulsive seizures; PBBI, penetrating ballistic-like brain injury.
Although C-10068 dose dependently attenuated PBBI-induced seizures, it did not reduce brain lesion volume, even at the most effective antiseizure dose tested. Average lesion volume of all treated groups ranged between 73.4±3.5 and 80.1±3.8 mm3 (p>0.05 for all comparisons, data not shown).
Anti-inflammation effects of C-10068
Perilesional region
At 72 h post-PBBI, immunoreactivity of GFAP and OX-18 was markedly increased in the perilesional region of the brain of all vehicle- and C-10068-treated (5.0 mg/kg/h×72 h) animals, whereas in the corresponding regions of the contralateral hemisphere, very few cells with positive GFAP or OX-18 stains were detected (Fig. 5A; OX-18). Compared to vehicle-treated animals, C-10068 treatment significantly attenuated PBBI-induced activation of microglial cells in the perilesional region, evidenced by a 33% (p<0.05) reduction in OX-18 positive staining (Fig. 5B). The effect of C-10068 treatment on GFAP immunoreactivity in the perilesional region resulted in a 14% reduction, which was not statistically significant from that of the vehicle-treated group (p>0.05, data not shown).
FIG. 5.

Effects of C-10068 treatment (5.0 mg/kg/h×72 h) on PBBI-induced activation of microglia cells (A and B) and astrocytes (C and D). (A) Images of OX-18-stained brain section taken at the perilesional region (a: A 4× image of the brain section; b: A 20× image captured from an area marked in a). (B) C-10068 treatment significantly reduced OX-18-stained activation of microglial cells in the perilesional region of the brain. (C) Images of GFAP-stained hippocampus (a: A 4× image of the hippocampus; b: A 20× image captured from CA1; c: A 20× image captured from the dentate). (D) C-10068 treatment significantly reduced GFAP-stained activation of astrocytes in the hippocampal region of the brain. *p<0.05, compared to vehicle-treated group. PBBI, penetrating ballistic-like brain injury; GFAP, glial fibrillary acidic protein; CA1, cornu ammonis 1.
Hippocampal region
Increased GFAP immunoreactivity was observed in the ipsilateral hippocampus, compared to the contralateral hippocampus, at 72 h post-PBBI (Fig. 5C). Treatment with C-10068 significantly reduced PBBI-induced activation of hippocampal astrocytes, evidenced by a 36% (p<0.05) reduction in GFAP-positive staining in the ipsilateral hippocampus, compared to that in the matching ROI of vehicle-treated animals (Fig. 5D). PBBI did not alter OX-18 immunoreactivity in the ipsilateral hippocampus, compared to that of the contralateral hippocampus, in both vehicle- and C-10068-treated animals at 72 h postinjury.
Correlations between nonconvulsive seizure activities and neuroinflammation
The relationship between NCS activities and the degree of neuroinflammation were further examined in order to understand the possible mechanistic basis for the therapeutic effects of C-10068 treatment. For this purpose, the relationship between NCS frequency and neuroinflammation (GFAP and OX-18 immunoreactivity) as well as the effects of C-10068 treatment on such relationship were analyzed separately.
The relationship between NCS frequency and the activation of astrocytes and microglial cells was first evaluated independently of the treatments by pooling all animals together and then reclassifying them based on their NCS status, such that one group included animals that exhibited NCS (S+ group) and the other group included animals that did not exhibit NCS (S− group). The analysis of GFAP immunoreactivity (the average value of positive GFAP staining in both hippocampus and perilesional region) showed that activation of astrocytes was significantly stronger in S+ animals than that in S− animals, showing a 44% difference in GFAP-positive staining in favor of S+ animals (p<0.05; Fig. 6A). Furthermore, there was a moderate, but significant, positive relationship between GFAP immunoreactivity and NCS frequency across all individual animals (Pearson's r=0.49; p<0.05; Fig. 6B). In contrast, activation of microglia cells in the perilesional region of the brain and NCS activities appeared to be independent of each other. OX-18-positive staining in this region was very similar between S− and S+ animals (p=0.41; Fig. 6C), and no correlative relationship between OX-18 immunoreactivity and NCS frequency was detected across all individual animals (Pearson's r=0.06; p>0.05; Fig. 6D).
FIG. 6.

Correlation analysis of NCS frequency and GFAP immunoreactivity based on seizure status of animals (independent of treatments). (A) Increased GFAP immunoreactivity in pooled S+ animals, compared to pooled S− animals. (B) Positive correlations between GFAP immunoreactivity and NCS frequency. (C) Similar OX-18 immunoreactivity in pooled S+ and pooled S− animals. (D) Lack of correlation between OX-18 immunoreactivity and NCS frequency. *p<0.05, compared to S− animals. NCS, nonconvulsive seizures; GFAP, glial fibrillary acidic protein.
The relationship between NCS activities and neuroinflammation was further examined with respect to treatment effects of C-10068. For this purpose, the correlation analysis focused only on hippocampal GFAP immunoreactivity because this appeared to provide the most sensitive measure of increased GFAP immunoreactivity relative to NCS activity. Similar to the approach described above, animals within vehicle- and C-10068-treated groups were compartmentalized into S− and S+ subgroups. The effects of C-10068 treatment on GFAP-positive staining was compared to that of vehicle treatment based on the matched NCS status. As shown in Figure 7A, significant reductions (42%) in hippocampal GFAP immunoreactivity were detected in C-10068-treated S− animals, compared to vehicle-treated S− animals (p<0.05). A similar trend was observed in S+ animals, where C-10068 treatment reduced hippocampal GFAP-positive staining by 21% in S+ animals, compared to that of vehicle-treated S+ animals, but this magnitude of reduction failed to reach statistical significance (p>0.05). Overall, C-10068-treated animals showed a moderate, but statistically significant, positive correlation between NCS frequency and hippocampal GFAP immunoreactivity (Pearson's r=0.55; p<0.05; Fig. 7B)
FIG. 7.
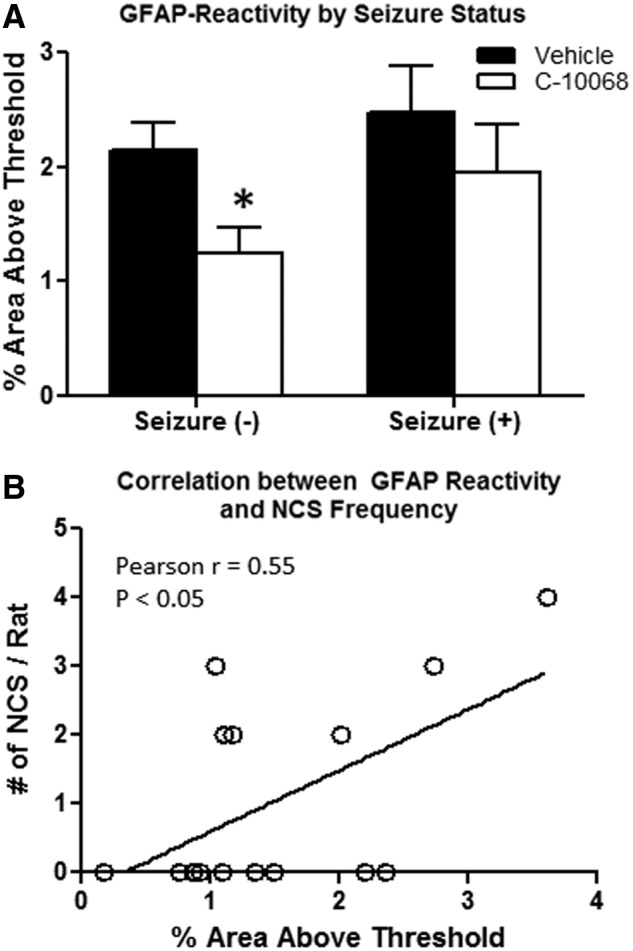
Effects of C-10068 treatment on hippocampal GFAP immunoreactivity based on seizure status. (A) Significant effects of C-10068 treatment on reducing hippocampal GFAP immunoreactivity in the S− animals. (B) Significant positive correlation between GFAP reactivity and NCS frequency. *p<0.05, compared to vehicle-treated S− animals. GFAP, glial fibrillary acidic protein; NCS, nonconvulsive seizures.
Neuroprotective effects of C-10068
Rotarod test
Before PBBI, all rats achieved criteria performance on the rotarod task. Compared to sham controls, rats sustaining PBBI displayed significant difficulty maintaining balance on the rotating rod at 7 and 10 days postinjury, compared to sham controls. C-10068 treatment had no effect on improving animals' motor deficits. As shown in Figure 8A, the latency to fall off the rotating rod (average measures of the 2 testing days) was significantly decreased by 43% in vehicle- and 64% in C-10068-treated groups, respectively (p<0.05 for each group, compared to the sham group). However, the 21% reduction in the C-10068 group was not statistically significant, compared to vehicle-treated group (p=0.092).
FIG. 8.
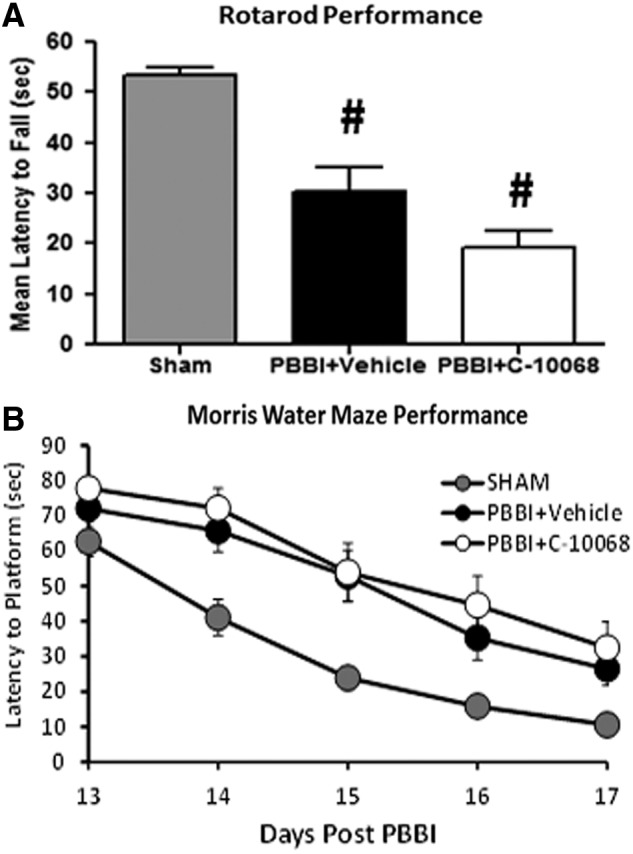
Lack of neuroprotective effects of C-10068 on rotarod performance (A) and Morris water maze performance (B). #p<0.05, compared to sham controls. PBBI, penetrating ballistic-like brain injury.
Morris water maze test
Similarly, compared to sham controls, PBBI produced significant impairment on the cognitive function in all injured animals, evidenced by significantly longer latency (F2,39=8.09; p<0.005; Fig. 8B) and longer swim distance (F2,39=7.58; p<0.005; data not shown) to locate the hidden platform in the MWM task. There were no between-group differences in swim speeds, indicating that cognitive impairment was not confounded by animals' motor deficits (data not shown). Overall, treatment with C-10068 had no effect on MWM performance (Fig. 8B).
Discussion
In the current study, we demonstrated that treatment with C-10068 provided significant dose-dependent protection against acute post-traumatic NCS in the rat PBBI model. The most efficacious antiseizure dose of C-10068 also significantly attenuated PBBI-induced neuroinflammation by reducing activated astrocytes in the hippocampus and reactive microglia in the perilesional region of the brain. Correlation analysis revealed that severity of NCS activity was positively associated with degree of neuroinflammation across all animals, regardless of treatments. The positive correlation between the degree of hippocampal GFAP immunoreactivity and the number of NCS events was also evident within the C-10068-treated cohort, although the anti-inflammation effects of C-10068 appeared to be relatively more prominent in animals lacking NCS activities. However, the dual antiseizure and antiinflammation effects of C-10068 did not translate to neuroprotection against injury-induced functional deficits.
Most AEDs are designed to modulate specific mechanisms underlying seizure activities. The putative antiepileptic targets include imbalance of excitatory and inhibitory neurotransmitters (e.g., glutamate antagonist and gamma-aminobutyric acid agonist) and/or dysfunction of ion channel (e.g., sodium- or calcium-channels blockers). As mentioned above in the Introduction, C-10068 shares similar pharmacological functions of DM as an NMDA receptor antagonist, a voltage-gated calcium-channel antagonist, as well as a sigma-1 receptor ligand. All of these properties are likely to contribute to its antiseizure properties. Whereas deuterium modification improves metabolic stability of C-10068, it does not alter its pharmacological function and also avoid being converted to DX. Earlier studies have shown that some neuroprotective actions may be attributable to DM's primary metabolite, DX, which is a potent noncompetitive NMDA antagonist20,45,46; however, DM has clear neuroprotective properties in its own right and its metabolism to DX is not necessary for these effects.47 This is evidenced in reports that dimemorfan, another DM analog, which cannot be metabolized to DX, demonstrates equivalent anticonvulsant activity to DM in a kainate-induced seizure model.34 Shin and colleagues also demonstrated that pretreatment with dimemorfan and DM were equally effective in preventing hippocampal cell loss. Dimemorfan exhibits protective effects against ischemic stroke, likely through sigma-1 receptor modulation, resulting in reduced glutamate accumulation.48 These results suggest that metabolic formation of DX may not be required for the observed neuroprotective effects of the morphinan compounds.
One of the major pharmacological properties of DM and C-10068 that distinguishes them from most approved AEDs is that the morphinans have a high affinity for sigma-1 receptors, which have been implicated in seizure protection.33,49 Further, evidence suggests that sigma-1 receptors play a major role in suppressing excess calcium influx associated with glutamate-receptor–mediated excitotoxicity.22,50,51 Thus, in the event of seizures, sigma-1 receptors might be involved in balancing the dysfunction of the glutamatergic system and ion channels to ameliorate neuronal hyperactivities. In support of this theory, previous studies have shown that pretreatment with a sigma-1 receptor antagonist, BD1047, dose dependently counteracts the antiseizure effects of sigma-1 receptor ligands, such as DM, carbetapentane, and dimemorfan.33,34 In this regard, the antiseizure effects of C-10068 observed in this study might also be partly mediated by its agonistic action at the sigma-1 receptor-binding site, representing a promising antiseizure property different from most currently used AEDs.
The mechanisms underlying the protective effects of C-10068 against PBBI-induced post-traumatic NCS may not be limited to its pharmacological attributes, but might also result from the concatenation of its effects pertaining to both seizures and TBI. This is evidenced by its dual therapeutic effects against PBBI-induced NCS and neuroinflammation. Activation of microglia and astrocytes have been considered the hallmark of neuroinflammation after various brain injuries.52 Previously, we reported that marked activation of both astrocytes and microglia cells were observed in the brain post-PBBI.53–57 Although the exact anatomical origin(s) of NCS initiation post-PBBI could not be identified in this study, ample studies have shown that gliosis-mediated inflammation induced by brain insults plays an important role in the pathogenesis of seizures and epilepsy.58–62 Therefore, the protective effects of C-10068 against PBBI-induced gliosis may be equally important to its antiseizure properties in this brain injury model. The current results suggest that the complexity of PTS may require drugs with multiple mechanisms of action or combination therapy to achieve desired therapeutic benefits.
The dual antiseizure and antiinflammatory effects of C-10068 are intriguing because seizures and neuroinflammation often coexist and negatively influence each other.58 Thus, their concomitant occurrence post-sTBI may worsen the injury outcomes. However, causal relationships between seizures and neuroinflammation in the context of a sTBI are difficult to determine, because the brain injury itself plays a major role in development of both events. Thus, the mechanism(s) that underlies the dual antiseizure/anti-inflammation therapeutic effects of C-10068 observed in this study may be difficult to dissect. Nevertheless, the current findings showing a significant positive correlation between GFAP up-regulation and seizure activity indicate a complementary relationship between the antiseizure and antiinflammatory effects of C-10068. Further, two lines of evidence showing 1) stronger attenuation of GFAP immunoreactivity in the seizure free (S−) animals as a result of C-10068 treatment (Fig. 7A) and 2) the lack of correlation between C-10068's effects on perilesional OX-18 immunoreactivity and NCS activities (Fig. 6D) may suggest that, to some degree, the anti-inflammatory properties of C-10068 might be independent of its antiseizure properties.
In TBI patients, early PTS occur in a sporadic and unpredicted nature, such that the timing of treatments can be difficult to determine. Although deuterium substitution has improved the metabolic stability of C-10068, the half-life of C-10068 (∼2 h in rats) is still relatively short for effectively treating this type of seizure on an intermittent interval. In this study, the PBBI-induced NCS model closely mimics the spontaneous, sporadic nature of clinical post-traumatic NCS; therefore, the continuous i.v. infusion regimen of C-10068 might have offered a pharmacokinetic advantage to maintain its sustained exposure, which may, in part, be responsible for its success in achieving timely therapeutic effects.
Although treatment with C-10068 achieved the intended antiseizure effects and showed potent anti-inflammatory effects, the failure to observe a neuroprotection benefits in either the rotarod test or the MWM test was disappointing. The lack of an observed functional benefit in the current study was unexpected given that the pharmacology of C-10068 is closely related to DM, which has previously demonstrated neuroprotective properties in various experimental models of brain injury, including the PBBI model used in this study.26,32 One explanation for these differences may be owing to the timing and modality of neurobehavioral measures used to evaluate C-10068 versus DM post-PBBI. More specifically, the previous study limited the evaluation of DM to acute (≤7 days) postinjury outcome measures using a balance beam task and a novel object recognition task. In contrast, the current study evaluated motor performance using the rotarod at 7 and 10 days postinjury and cognitive abilities on the MWM task from 13 to 17 days postinjury. The extent to which the differences between the testing paradigms used in the two studies influenced the results is beyond the scope of this discussion. However, the neuroprotective potential of C-10068 may have been diminished owing to the increased interval between treatment cessation and initiation of behavioral testing. In keeping with this, it is possible that extended treatment durations with C-10068 may be needed to confer significant therapeutic benefit on more delayed neurobehavioral outcome measures.
In summary, we demonstrated, in this study, that treatment with C-10068, a novel deuterium-substituted morphinan, significantly attenuated post-traumatic NCS and neuroinflammation in a rat model of penetrating brain injury. Although it is not possible to ascertain the causative relationship between seizures and inflammation post-PBBI, and thereby the mechanistic actions of C-10068 on both events remain elusive, the observed positive correlations of C-10068's effects on PBBI-induced NCS and neuroinflammation suggest dual therapeutic functions for this compound. Considering the difficulties in treating post-traumatic NCS in sTBI patients owing to the refractory nature of these seizures and the concerns of inherent adverse effects of most AEDs used clinically, the dual therapeutic properties of C-10068 are promising and warrant further investigation of this compound as a novel antiseizure drug candidate for treating seizures caused by severe brain trauma.
Acknowledgments
This work was supported by funding provided by the U.S. Army Medical Research and Material Command, Combat Casualty Care Research Program and Concert Pharmaceuticals Inc.
The authors thank Ms. Ying Cao, Ms. Rebecca Pedersen, Mr. Justin Sun, and Mr. Zhilin Liao for their excellent technical support.
This material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author and are not to be construed as official or as reflecting true views of the Department of the Army or the Department of Defense. Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals (NRC Publication, 2011 edition).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Asikainen I., Kaste M., and Sarna S. (1999). Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia 40, 584–589 [DOI] [PubMed] [Google Scholar]
- 2.Beghi E. (2003). Overview of studies to prevent posttraumatic epilepsy. Epilepsia 44, Suppl. 10, 21–26 [DOI] [PubMed] [Google Scholar]
- 3.Jordan K.G. (1995). Neurophysiologic monitoring in the neuroscience intensive care unit. Neurol. Clin. 13, 579–626 [PubMed] [Google Scholar]
- 4.Vespa P., Prins M., Ronne-Engstrom E., Caron M., Shalmon E., Hovda D.A., Martin N.A., and Becker D.P. (1998). Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: a microdialysis study. J. Neurosurg. 89, 971–982 [DOI] [PubMed] [Google Scholar]
- 5.Vespa P.M., Miller C., McArthur D., Eliseo M., Etchepare M., Hirt D., Glenn T.C., Martin N., and Hovda D. (2007). Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit. Care Med. 35, 2830–2836 [PMC free article] [PubMed] [Google Scholar]
- 6.Vespa P.M., O'Phelan K., Shah M., Mirabelli J., Starkman S., Kidwell C., Saver J., Nuwer M.R., Frazee J.G., McArthur D.A., and Martin N.A. (2003). Acute seizures after intracerebral hemorrhage: a factor in progressive midline shift and outcome. Neurology 60, 1441–1446 [DOI] [PubMed] [Google Scholar]
- 7.Young G.B., and Jordan K.G. (1998). Do nonconvulsive seizures damage the brain?–Yes. Archives of neurology 55, 117–119 [DOI] [PubMed] [Google Scholar]
- 8.Bushnik T., Englander J., and Duong T. (2004). Medical and social issues related to posttraumatic seizures in persons with traumatic brain injury. J. Head Trauma Rehabil. 19, 296–304 [DOI] [PubMed] [Google Scholar]
- 9.Englander J., Bushnik T., Duong T.T., Cifu D.X., Zafonte R., Wright J., Hughes R., and Bergman W. (2003). Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 84, 365–373 [DOI] [PubMed] [Google Scholar]
- 10.Agrawal A., Timothy J., Pandit L., and Manju M. (2006). Post-traumatic epilepsy: an overview. Clin. Neurol. Neurosurg. 108, 433–439 [DOI] [PubMed] [Google Scholar]
- 11.Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons; Joint Section on Neurotrauma and Critical Care, AANS/CNS, Bratton S.L., Chestnut R.M., Ghajar J., McConnell Hammond F.F., Harris O.A., Hartl R., Manley G.T., Nemecek A., Newell D.W., Rosenthal G., Schouten J., Shutter L., Timmons S.D., Ullman J.S., Videtta W., Wilberger J.E., and Wright D.W. (2007). Guidelines for the management of severe traumatic brain injury. XIII. Antiseizure prophylaxis. J. Neurotrauma 24, Suppl. 1, S83–S86 [DOI] [PubMed] [Google Scholar]
- 12.Jirsch J., and Hirsch L.J. (2007). Nonconvulsive seizures: developing a rational approach to the diagnosis and management in the critically ill population. Clin. Neurophysiol. 118, 1660–1670 [DOI] [PubMed] [Google Scholar]
- 13.Hirsch L.J. (2008). Nonconvulsive seizures in traumatic brain injury: what you don't see can hurt you. Epilepsy Curr. 8, 97–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Temkin N.R., Dikmen S.S., Anderson G.D., Wilensky A.J., Holmes M.D., Cohen W., Newell D.W., Nelson P., Awan A., and Winn H.R. (1999). Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J. Neurosurg. 91, 593–600 [DOI] [PubMed] [Google Scholar]
- 15.Temkin N.R., Dikmen S.S., Wilensky A.J., Keihm J., Chabal S., and Winn H.R. (1990). A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N. Engl. J. Med. 323, 497–502 [DOI] [PubMed] [Google Scholar]
- 16.Wohns R.N., and Wyler A.R. (1979). Prophylactic phenytoin in severe head injuries. J. Neurosurg. 51, 507–509 [DOI] [PubMed] [Google Scholar]
- 17.Mountney A., Shear D.A., Potter B., Marcsisin S.R., Sousa J., Melendez V., Tortella F.C., and Lu X.C. (2013). Ethosuximide and phenytoin dose-dependently attenuate acute nonconvulsive seizures after traumatic brain injury in rats. J. Neurotrauma 30, 1973–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCluggage L.K., Voils S.A., and Bullock M.R. (2009). Phenytoin toxicity due to genetic polymorphism. Neurocrit. Care 10, 222–224 [DOI] [PubMed] [Google Scholar]
- 19.Tortella F.C., Pellicano M., and Bowery N.G. (1989). Dextromethorphan and neuromodulation: old drug coughs up new activities. Trends Pharmacol. Sci. 10, 501–507 [DOI] [PubMed] [Google Scholar]
- 20.Carpenter C.L., Marks S.S., Watson D.L., and Greenberg D.A. (1988). Dextromethorphan and dextrorphan as calcium channel antagonists. Brain Res. 439, 372–375 [DOI] [PubMed] [Google Scholar]
- 21.Klette K.L., DeCoster M.A., Moreton J.E., and Tortella F.C. (1995). Role of calcium in sigma-mediated neuroprotection in rat primary cortical neurons. Brain Res. 704, 31–41 [DOI] [PubMed] [Google Scholar]
- 22.Klette K.L., Lin Y., Clapp L.E., DeCoster M.A., Moreton J.E., and Tortella F.C. (1997). Neuroprotective sigma ligands attenuate NMDA and trans-ACPD-induced calcium signaling in rat primary neurons. Brain Res. 756, 231–240 [DOI] [PubMed] [Google Scholar]
- 23.Hatton J. (2001). Pharmacological treatment of traumatic brain injury: a review of agents in development. CNS Drugs 15, 553–581 [DOI] [PubMed] [Google Scholar]
- 24.Lerche H., Shah M., Beck H., Noebels J., Johnston D., and Vincent A. (2013). Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 591, 753–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer G.C. (2001). Neuroprotection by NMDA receptor antagonists in a variety of neuropathologies. Curr. Drug Targets 2, 241–271 [DOI] [PubMed] [Google Scholar]
- 26.Werling L.L., Lauterbach E.C., and Calef U. (2007). Dextromethorphan as a potential neuroprotective agent with unique mechanisms of action. Neurologist 13, 272–293 [DOI] [PubMed] [Google Scholar]
- 27.Block F., and Schwarz M. (1996). Dextromethorphan reduces functional deficits and neuronal damage after global ischemia in rats. Brain Res. 741, 153–159 [DOI] [PubMed] [Google Scholar]
- 28.Britton P., Lu X.C., Laskosky M.S., and Tortella F.C. (1997). Dextromethorphan protects against cerebral injury following transient, but not permanent, focal ischemia in rats. Life Sci. 60, 1729–1740 [DOI] [PubMed] [Google Scholar]
- 29.Schmid-Elsaesser R., Zausinger S., Hungerhuber E., Baethmann A., and Reulen H.J. (1998). Monotherapy with dextromethorphan or tirilazad—but not a combination of both—improves outcome after transient focal cerebral ischemia in rats. Exp. Brain Res. 122, 121–127 [DOI] [PubMed] [Google Scholar]
- 30.Tortella F.C., Britton P., Williams A., Lu X.C., and Newman A.H. (1999). Neuroprotection (focal ischemia) and neurotoxicity (electroencephalographic) studies in rats with AHN649, a 3-amino analog of dextromethorphan and low-affinity N-methyl-D-aspartate antagonist. J. Pharmacol. Exp. Ther. 291, 399–408 [PubMed] [Google Scholar]
- 31.Block F., and Schwarz M. (1998). Global ischemic neuronal damage relates to behavioural deficits: a pharmacological approach. Neuroscience 82, 791–803 [DOI] [PubMed] [Google Scholar]
- 32.Shear D.A., Williams A.J., Sharrow K., Lu X.C., and Tortella F.C. (2009). Neuroprotective profile of dextromethorphan in an experimental model of penetrating ballistic-like brain injury. Pharmacol. Biochem. Behav. 94, 56–62 [DOI] [PubMed] [Google Scholar]
- 33.Kim H.C., Jhoo W.K., Kim W.K., Shin E.J., Cheon M.A., Shin C.Y., and Ko K.H. (2001). Carbetapentane attenuates kainate-induced seizures via sigma-1 receptor modulation. Life Sci. 69, 915–922 [DOI] [PubMed] [Google Scholar]
- 34.Shin E.J., Nah S.Y., Kim W.K., Ko K.H., Jhoo W.K., Lim Y.K., Cha J.Y., Chen C.F., and Kim H.C. (2005). The dextromethorphan analog dimemorfan attenuates kainate-induced seizures via sigma1 receptor activation: comparison with the effects of dextromethorphan. Br. J. Pharmacol. 144, 908–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aram J.A., Martin D., Tomczyk M., Zeman S., Millar J., Pohler G., and Lodge D. (1989). Neocortical epileptogenesis in vitro: studies with N-methyl-D-aspartate, phencyclidine, sigma and dextromethorphan receptor ligands. J. Pharmacol. Exp. Ther. 248, 320–328 [PubMed] [Google Scholar]
- 36.Chapman A.G., and Meldrum B.S. (1989). Non-competitive N-methyl-D-aspartate antagonists protect against sound-induced seizures in DBA/2 mice. Eur. J. Pharmacol. 166, 201–211 [DOI] [PubMed] [Google Scholar]
- 37.Tortella F.C., Ferkany J.W., and Pontecorvo M.J. (1988). Anticonvulsant effects of dextrorphan in rats: possible involvement in dextromethorphan-induced seizure protection. Life Sci. 42, 2509–2514 [DOI] [PubMed] [Google Scholar]
- 38.Tortella F.C., Robles L., Witkin J.M., and Newman A.H. (1994). Novel anticonvulsant analogs of dextromethorphan: improved efficacy, potency, duration and side-effect profile. J. Pharmacol. Exp. Ther. 268, 727–733 [PubMed] [Google Scholar]
- 39.Uttamsingh V., Nguyen S., Bridson G., Morgan A., and Graham P. (2013). C-10068: a novel sigma agonist morphinan with a superior metabolism and pharmacokinetic profile. Presented at the 10th ISSX International Meeting September 29–October 3, 2013, Toronto, Ontario, Canda [Google Scholar]
- 40.Graham P., Aslanian A., Nguyen S., Uttamsingh V., Morgan A., Bridson G., Sabounjian L., and Shipley S. (2012). C-10068: novel morphinan with a unique pharmacological profile. Presented at Epilepsy Pipeline Meeting, February 2–4, 2012, San Francisco, CA [Google Scholar]
- 41.Graham P. (2011). C-10068: novel sigma-1 agonist for epilepsy and neuroprotection. Epilepsy Pipeline Meeting, April 27–29, 2011, Aventara, FL [Google Scholar]
- 42.Hartings J.A., Williams A.J., and Tortella F.C. (2003). Occurrence of nonconvulsive seizures, periodic epileptiform discharges, and intermittent rhythmic delta activity in rat focal ischemia. Exp. Neurol. 179, 139–149 [DOI] [PubMed] [Google Scholar]
- 43.Lu X.C., Mountney A., Chen Z., Wei G., Cao Y., Leung L.Y., Khatri V., Cunningham T., and Tortella F.C. (2013). Similarities and differences of acute nonconvulsive seizures and other epileptic activities following penetrating and ischemic brain injuries in rats. J. Neurotrauma 30, 580–590 [DOI] [PubMed] [Google Scholar]
- 44.Shear D.A., Lu X.C., Bombard M.C., Pedersen R., Chen Z., Davis A., and Tortella F.C. (2010). Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. J. Neurotrauma 27, 1911–1923 [DOI] [PubMed] [Google Scholar]
- 45.Goldberg M.P., Pham P.C., and Choi D.W. (1987). Dextrorphan and dextromethorphan attenuate hypoxic injury in neuronal culture. Neurosci. Lett. 80, 11–15 [DOI] [PubMed] [Google Scholar]
- 46.Choi D.W. (1987). Dextrorphan and dextromethorphan attenuate glutamate neurotoxicity. Brain Res. 403, 333–336 [DOI] [PubMed] [Google Scholar]
- 47.Kim H.C., Bing G., Jhoo W.K., Kim W.K., Shin E.J., Im D.H., Kang K.S., and Ko K.H. (2003). Metabolism to dextrorphan is not essential for dextromethorphan's anticonvulsant activity against kainate in mice. Life Sci. 72, 769–783 [DOI] [PubMed] [Google Scholar]
- 48.Shen Y.C., Wang Y.H., Chou Y.C., Liou K.T., Yen J.C., Wang W.Y., and Liao J.F. (2008). Dimemorfan protects rats against ischemic stroke through activation of sigma-1 receptor-mediated mechanisms by decreasing glutamate accumulation. J. Neurochem. 104, 558–572 [DOI] [PubMed] [Google Scholar]
- 49.Thurgur C., and Church J. (1998). The anticonvulsant actions of sigma receptor ligands in the Mg2+-free model of epileptiform activity in rat hippocampal slices. Br. J. Pharmacol. 124, 917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cobos E.J., Entrena J.M., Nieto F.R., Cendan C.M., and Del Pozo E. (2008). Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr. Neuropharmacol. 6, 344–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Debonnel G., and de Montigny C. (1996). Modulation of NMDA and dopaminergic neurotransmissions by sigma ligands: possible implications for the treatment of psychiatric disorders. Life Sci. 58, 721–734 [DOI] [PubMed] [Google Scholar]
- 52.Seifert G., Carmignoto G., and Steinhauser C. (2010). Astrocyte dysfunction in epilepsy. Brain Res. Rev. 63, 212–221 [DOI] [PubMed] [Google Scholar]
- 53.Cunningham T.L., Cartagena C.M., Lu X.C., Konopko M., Dave J.R., Tortella F.C., and Shear D.A. (2014). Correlations between blood-brain barrier disruption and neuroinflammation in an experimental model of penetrating ballistic-like brain injury. J. Neurotrauma 31, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu X.C., Si Y., Williams A.J., Hartings J.A., Gryder D., and Tortella F.C. (2009). NNZ-2566, a glypromate analog, attenuates brain ischemia-induced non-convulsive seizures in rats. J. Cereb. Blood Flow Metab. 29, 1924–1932 [DOI] [PubMed] [Google Scholar]
- 55.Williams A.J., Hartings J.A., Lu X.C., Rolli M.L., Dave J.R., and Tortella F.C. (2005). Characterization of a new rat model of penetrating ballistic brain injury. J. Neurotrauma 22, 313–331 [DOI] [PubMed] [Google Scholar]
- 56.Williams A.J., Hartings J.A., Lu X.C., Rolli M.L., and Tortella F.C. (2006). Penetrating ballistic-like brain injury in the rat: differential time courses of hemorrhage, cell death, inflammation, and remote degeneration. J. Neurotrauma 23, 1828–1846 [DOI] [PubMed] [Google Scholar]
- 57.Williams A.J., Wei H.H., Dave J.R., and Tortella F.C. (2007). Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J. Neuroinflammation 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi J., and Koh S. (2008). Role of brain inflammation in epileptogenesis. Yonsei Med. J. 49, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D'Ambrosio R. (2004). The role of glial membrane ion channels in seizures and epileptogenesis. Pharmacol. Ther. 103, 95–108 [DOI] [PubMed] [Google Scholar]
- 60.Devinsky O., Vezzani A., Najjar S., De Lanerolle N.C., and Rogawski M.A. (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184 [DOI] [PubMed] [Google Scholar]
- 61.Tian G.F., Azmi H., Takano T., Xu Q., Peng W., Lin J., Oberheim N., Lou N., Wang X., Zielke H.R., Kang J., and Nedergaard M. (2005). An astrocytic basis of epilepsy. Nat. Med. 11, 973–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vezzani A., Aronica E., Mazarati A., and Pittman Q.J. (2013). Epilepsy and brain inflammation. Exp. Neurol. 244, 11–21 [DOI] [PubMed] [Google Scholar]