

Abstract
In the present study, we investigated the potential activity of OSI-027, a potent and selective mammalian target of rapamycin (mTOR) complex 1/2 (mTORC1/2) dual inhibitor, against pancreatic cancer cells both in vitro and in vivo. We demonstrated that OSI-027 inhibited survival and growth of both primary and transformed (PANC-1 and MIA PaCa-2 lines) human pancreatic cancer cells. Meanwhile, OSI-027 induced caspase-dependent apoptotic death of the pancreatic cancer cells. On the other hand, caspase inhibitors alleviated cytotoxicity by OSI-027. At the molecular level, OSI-027 treatment blocked mTORC1 and mTORC2 activation simultaneously, without affecting ERK–mitogen-activated protein kinase activation. Importantly, OSI-027 activated cytoprotective autophagy in the above cancer cells. Whereas pharmacological blockage of autophagy or siRNA knockdown of Beclin-1 significantly enhanced the OSI-027-induced activity against pancreatic cancer cells. Specifically, a relatively low dose of OSI-027 sensitized gemcitabine-induced pancreatic cancer cell death in vitro. Further, administration of OSI-027 or together with gemcitabine dramatically inhibited PANC-1 xenograft growth in severe combined immunodeficiency mice, leading to significant mice survival improvement. In summary, the preclinical results of this study suggest that targeting mTORC1/2 synchronously by OSI-027 could be further investigated as a valuable treatment for pancreatic cancer.
Introduction
Pancreatic cancer is arguably one of the most aggressive human tumors (Siegel et al., 2014). The cause of this devastating disease is still debatable (Hart et al., 2008; Hidalgo, 2010). Despite recent advances in medical management of pancreatic cancer, its prognosis and 5-year overall survival are still among the worst of all human malignancies (Schneider et al., 2005; Stathis and Moore, 2010). Pancreatic cancer is characterized by a rapid disease progression without specific clinical symptoms, and many cancers are diagnosed at advanced stages when the surgical resections are not available (Schneider et al., 2005; Stathis and Moore, 2010). Indeed, it is estimated that only 10–20% of pancreatic cancer patients are amenable to surgical interventions (Lockhart et al., 2005; Wray et al., 2005; Gudjonsson, 2009). Postoperative gemcitabine is shown to delay recurrence after complete resection of pancreatic cancer (Eckel et al., 2006; Oettle et al., 2007; Hart et al., 2008). Due to pre-existing or acquired chemoresistance, gemcitabine could only enhance the median survival by less than a few months (Westphal and Kalthoff, 2003; Rivera et al., 2009). Thus, the search for novel and more efficient antipancreatic cancer treatments or gemcitabine adjuvants is undergoing (McKenna and Eatock, 2003; Eckel et al., 2006; Von Hoff et al., 2013). It is also the research focus of our group in recent years (Chen et al., 2013; Tang et al., 2014).
Mammalian target of rapamycin (mTOR) activation is one important contributor of pancreatic cancer cell progression and/or chemoresistance (Ito et al., 2006; Azzariti et al., 2008; Wolpin et al., 2009; Garrido-Laguna et al., 2010). There are at least two multiprotein mTOR complexes identified thus far, including mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Sabatini, 2006; Guertin and Sabatini, 2007). The former mTORC1 is composed of mTOR, Raptor, mLST8, and others, which mediates S6K1 and 4E-BP1 phosphorylation to regulate protein translation and energy metabolism (Sabatini, 2006; Guertin and Sabatini, 2007). mTORC2 is a complex of mTOR, rictor, mSin1, and possible several others, which serves as the upstream kinase of AKT through phosphorylating at the Ser-473 (Sabatini, 2006; Guertin and Sabatini, 2007). Both complexes are important for the cancerous behaviors of pancreatic cancer (Furukawa, 2008; Falasca et al., 2011).
Traditional mTORC1 inhibitors, including rapamycin and its analogs (rapalogues), have been tested in preclinical pancreatic cancer settings, alone or in combination with other agents (Azzariti et al., 2008; Wolpin et al., 2009). Further, several phase II clinical trials have studied the potential activities of these first-generation mTOR inhibitors in pancreatic cancer patients (Wolpin et al., 2009; Garrido-Laguna et al., 2010; Javle et al., 2010). However, these studies demonstrated only moderated activities by the mTORC1 inhibitors (Wolpin et al., 2009; Garrido-Laguna et al., 2010; Javle et al., 2010). The negative feedback loop activation resulting from mTORC1 inhibition may account for tumor resistance, disease progression, and toxicity in these studies (Wolpin et al., 2009; Garrido-Laguna et al., 2010; Javle et al., 2010). Thus, more efficient strategies should aim for a broader targeting of the mTOR pathway. Developing mTORC1/mTORC2 dual inhibitors could potentially overcome tumor resistance and improve patients' outcome (Wolpin et al., 2009; Javle et al., 2010). Recently, ATP-competitive mTOR inhibitors have been developed (Vilar et al., 2011; Sun, 2013). These inhibitors target the ATP site of the mTOR kinase domain, thereby blocking mTORC1 and mTORC2 simultaneously (Vilar et al., 2011; Sun, 2013). In the current study, we investigated the potential antipancreatic cancer activity of OSI-027, a potent and selective mTORC1/2 dual inhibitor (Bhagwat et al., 2011; Li et al., 2012), in vivo and in vitro.
Materials and Methods
Reagents and chemicals
OSI-027, rapamycin, RAD001, and MEK162 were obtained from Selleck China (Shanghai, China). Gemcitabine, PD98059, U0126, cisplatin, paclitaxel (taxol), chloroquine (Cq), ammonium chloride (NH4Cl), and 3-methyaldenine (3-MA) were obtained from Sigma-Aldrich Co. (St. Louis, MO). The pan caspase inhibitor z-VAD-fmk, the caspase-3 inhibitor z-DVED-fmk, and the caspase-8 inhibitor z-ITED-fmk were purchased from Calbiochem (Darmstadt, Germany). Anti-light chain 3B (LC3B), Beclin-1, p62, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for p-AKT (Ser-473), p-AKT (Thr-308), AKT, p-S6 (Ser-235/236), S6, p-ERK1/2 (Thr-202/Tyr-204), p-S6K1 (Thr-389), and S6K1 were obtained from Cell Signaling Technology (Beverly, MA).
Cell lines
The pancreatic cancer cell lines (PANC-1 and MIA PaCa-2) were purchased from Shanghai Institute of Biological Science, Chinese Academy of Science (Shanghai, China). Cells were cultured in RPMI/DMEM medium (Invitrogen, Shanghai, China), supplemented with 10% fetal bovine serum (FBS; Invitrogen) with antibiotics in a CO2 incubator at 37°C. HPDE6c7, an immortalized but not transformed normal pancreatic epithelial cell line (Bu et al., 2014), was purchased from GuangZhou Jennio Biotech (Guangzhou, China). HPDE6c7 cells were cultivated in DMEM supplemented with 10% FBS.
Culture of primary human pancreatic adenocarcinoma cells
Pancreatic adenocarcinoma tissues from patients with early stage diseases, after informed consent (hospitalized at East Hospital Affiliated to Tongji University in Shanghai, Shanghai, China), were obtained at the time of surgery. Fresh tissues were thoroughly washed in PBS solution (pH 7.4) containing 200 units/mL penicillin–streptomycin and 1 mM DTT (wash buffer). Tissues were then minced by scalpel. Single-cell suspensions were achieved by resuspending cells in 0.05% (w/v) collagenase I dissolved in DMEM. After 1 h, individual cells were pelleted and rinsed twice with DMEM, before resuspending the pellets into cell culture medium (DMEM, 20%-FBS, 2 mM glutamine, 1 mM pyruvate, 10 mM HEPES, 100 units/mL penicillin/streptomycin, 0.1 mg/mL gentamicin, and 2 g/L fungizone) (Min et al., 2014). The study was approved by the Institutional Review Board of Tongji University. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki.
Cell survival assay
As previously reported (Chen et al., 2013; Tang et al., 2014), the number of viable cells after applied treatment was evaluated using CellTiter-Glo (Promega, Shanghai, China) assay, and total luminescence was measured on a Wallac Multilabel Reader (Zaventem, Belgium) (Chen et al., 2013; Tang et al., 2014). The value of the treatment group was normalized to the untreated control group.
Clonogenicity assay of cell proliferation
Cells were seeded and allowed to attach overnight. Next day, cells were treated with indicated concentrations of OSI-027. Following the treatment, the cells were refed with the drug containing medium every 2 days. After 7 days, the colonies were stained with crystal blue and counted. The number of colonies in the treatment group was normalized to that of the untreated control group.
Caspase-3 activity assay
After treatment, cytosolic proteins from approximately 2–3×106 cells were extracted in hypotonic cell lysis buffer described in (Zhu et al., 2014). Twenty microgram of cytosolic extracts were added to caspase assay buffer (312.5 mM HEPES, pH 7.5, 31.25% sucrose, 0.3125% CHAPS) with Ac-DEVD-AFC (15 μg/mL) (Calbiochem) as the substrate. After incubation at 37°C for 1 h, the amount of AFC liberated was measured using a spectrofluorometer (Thermo Labsystems, Helsinki, Finland) with excitation of 380 nm and emission wavelength of 460 nm.
Terminal deoxynucleotidyl transferase dUTP nick end labeling staining
The pancreatic cancer cell apoptosis was detected by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) In Situ Cell Death Detection Kit (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer's instructions. Cells were also stained with 4′,6′-diamino-2-phenylindole (DAPI, blue fluorescence; Molecular Probes) to visualize the cell nuclei. Cell apoptosis rate was calculated by the TUNEL percentage (TUNEL vs. DAPI) detected under a fluorescence microscope (Zeiss).
Western blotting
Protein samples were prepared in lysis buffer [5 mM MgCl2, 137 mM KCl, 1 mM EDTA, 1 mM EGTA, 1% CHAPS, 10 mM HEPES (pH 7.5)], normalized using NanoDrop measurement (Thermo Scientific, Nanjing, China), and boiled in sodium dodecyl sulfate (SDS) sample buffer. Samples were then loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels (14% for detection of LC3B), and transferred onto polyvinylidene difluoride membranes, and then labeled with indicated primary and secondary antibodies. Antibody binding was detected by the ECL Detection Kit (Amersham, Shanghai, China). The intensity of each band was quantified using ImageJ software (National Institutes of Health, Baltimore, Maryland).
Beclin-1 siRNA
Pancreatic cancer cells were transfected with the commercially available siRNA designed to knockdown Beclin-1 (Cell Signaling Tech), or with a scramble control siRNA (Santa Cruz Biotechnology). SiRNA (100 nM) transfection was performed through Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The transfection took 36 h. After transfection, expression of Beclin-1 and loading (GAPDH) was tested by western blots.
Mice tumor xenograft
Severe combined immunodeficiency (SCID) mice (6–8 weeks old) were inoculated by intraperitoneal injection (i.p.). with 1×107 PANC-1 pancreatic cells, and treatment began 4 weeks (Humbert et al., 2010) post tumor implant with i.p. of gemcitabine (25 mg/kg, daily) with/without p.o. daily of OSI-027 (25 mg/kg) (Bhagwat et al., 2011), for a total of 21 days. Tumor volume (in cm3) was recorded every week for a total of 4 weeks. Mice survival at week 7 was also recorded. The survival data were plotted as the Kaplan–Meier graph using SPSS 16.0 software, and p-value was calculated. The animal experiments were approved by the Institutional Animal Care and Use Committee.
Statistical analyses
The data presented are mean±standard error. Statistical differences were analyzed by one-way ANOVA followed by multiple comparisons performed with post hoc Bonferroni test (SPSS version 18.0, Chicago, CA). Values of p<0.05 were considered statistically significant. The durations of treatment and concentrations of drugs were selected based on references or pre-experiment results.
Results
OSI-07 inhibits pancreatic cancer cell survival and growth
As shown in Figure 1A, OSI-027 dose-dependently inhibited the survival of PANC-1 pancreatic cancer cells. The activity of OSI-027 was most potent at 500 nM, but was inefficient at 10 nM (Fig. 1A). Meanwhile, the anti-PANC-1 activity of OSI-027 was also time dependent, and was most evident at 72 h of treatment (Fig. 1A). Similar titration- and time-dependent activity of OSI-027 was also observed in MIA PaCa-2 pancreatic cancer cells (Fig. 1B). OSI-027 was more potent than the same concentration (100 nM) of traditional mTORC1 inhibitors (rapamycin and RAD001) in inhibiting PANC-1/MIA PaCa-2 cell survival (Fig. 1C). The effect of OSI-027 on primary human pancreatic cancer cells was also tested. As demonstrated, OSI-027 (100 nM, 72 h) treatment dramatically suppressed primary cell survival (Fig. 1D). Notably, HPDE6c7 (an immortalized, but not transformed pancreatic epithelial cell line, noncancerous) cells were insensitive to the same OSI-027 treatment (Fig. 1D). Clonogenicity assay results showed that OSI-027 at the concentrations between 50–500 nM potently inhibited growth of PANC-1 (Fig. 1E) and MIA PaCa-2 (Fig. 1F) cells.
FIG. 1.
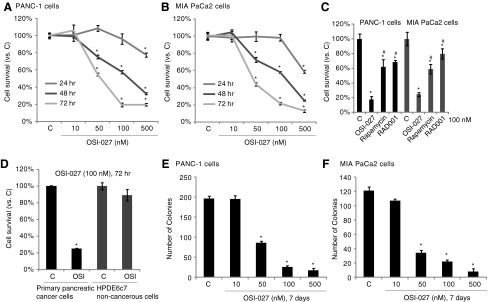
OSI-07 inhibits human pancreatic cancer cell survival and growth in vitro. Transformed pancreatic cancer cells (PANC-1 or MIA PaCa-2 lines), the primary cancer cells or the noncancerous HPDE6c7 pancreatic epithelial cells, were either left untreated (“C”) or stimulated with applied concentrations of OSI-027, rapamycin, or RAD001 for an indicated time, cell survival was analyzed by the CellTiter-Glo assay (A–D), and cell proliferation was analyzed by the clonogenicity assay (E, F). Data are expressed as mean±standard error (SE), experiments were repeated five times. n=5 for each assay. *p<0.05 versus “C” group. #p<0.05 versus OSI-027 group (for C).
OSI-027 induces apoptotic death of pancreatic cancer cells
The potential role of OSI-027 on cell apoptosis was also tested. As demonstrated, OSI-027 dose-dependently increased the percentage of TUNEL-positive pancreatic cancer cells (PANC-1 and MIA PaCa-2 lines) (Fig. 2A, B), indicating apoptosis induction in these cells. Again, OSI-027 is a more efficient apoptosis inducer than the same concentration (100 nM) of traditional mTORC1 inhibitors—rapamycin and RAD001 (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/dna). The caspase-3 activity of OSI-027-treated pancreatic cancer cells was also increased (Fig. 2C, D), further confirming apoptosis activation. Caspase inhibitors, including the caspase-3-specific inhibitor z-DVED-fmk, the caspase-8-specific inhibitor z-ITED-fmk, and the pan-caspase inhibitor z-VAD-fmk were applied. All these inhibitors suppressed OSI-027-induced cytotoxicity against PANC-1 cells (Fig. 2E) and MIA PaCa-2cells (data not shown). The above caspase inhibitors also alleviated cell death by OSI-027 in primary human pancreatic cancer cells (Fig. 2F). Together, the results suggest that OSI-07 induces caspase-dependent apoptotic death of pancreatic cancer cells.
FIG. 2.
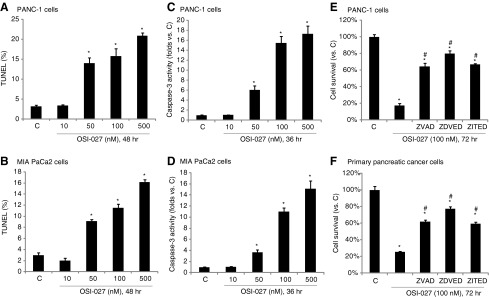
OSI-027 induces human pancreatic cancer cell apoptosis in vitro. Human pancreatic cancer lines (PANC-1 or MIA PaCa-2) were either left untreated (“C”), or stimulated with applied concentration of OSI-027, cells were then cultured for an indicated time, and cell apoptosis was analyzed by staining assay (A, B) and caspase-3 activity assay (C, D). PANC-1 or primary human pancreatic cancer cells, pretreated with the caspase-3 inhibitor z-DVED-fmk (ZDVED, 30 μM), the caspase-8 inhibitor z-ITED-fmk (ITED, 30 μM), or the pan caspase inhibitor z-VAD-fmk (ZVAD, 30 μM) for 1 h, were stimulated with OSI-027 (100 nM) for 72 h, cell survival was analyzed by CellTiter-Glo assay (E, F). Data are expressed as mean±SE, experiments were repeated five times. n=5 for each assay. *p<0.05 versus “C” group. #p<0.05 versus OSI-027 only group (E, F).
OSI-027 blocks mTORC1/2 activation, while inducing cytoprotective autophagy in cultured pancreatic cancer cells
OSI-027 is a potent mTORC1/2 dual inhibitor (Bhagwat et al., 2011), its activity on mTORC1/2 activation in cultured pancreatic cancer cells was tested. Western blot results showed that phosphorylation of S6K1 and S6, the indicators of mTORC1 activation, as well as phosphorylation of AKT at Ser-473, the indicator of mTORC2 activation, were almost completely blocked by OSI-027 in PANC-1 cells (Fig. 3A) and MIA PaCa2 cells (Fig. 3B). Phosphorylation of AKT at Thr-308 and phosphorylation of ERK1/2, on the other hand, were intact in OSI-027-treated cells (Fig. 3A, B). Thus, OSI-027 blocked mTORC1/2 activation in pancreatic cancer cells. AKT activity was partially inhibited (Ser-473). Significantly, MEK/ERK inhibitors, including PD98059, U0126, and MEK162 (Ascierto et al., 2013; Kusters-Vandevelde et al., 2014), sensitized the OSI-027-induced lethality in both PANC-1 (Fig. 3C) and MIA PaCa-2cells (Fig. 3D). As expected, the above inhibitors blocked MEK/ERK activation in the pancreatic cancer cells (data not shown).
FIG. 3.

Signaling changes by OSI-027 in pancreatic cancer cells. Human pancreatic cancer cell lines (PANC-1 or MIA PaCa-2) were either left untreated (“C”), or treated with OSI-027 (50 nM) for an indicated time (6/24 h), expression of listed proteins was tested by western blots (A, B, and E). PANC-1 (C) or MIA PaCa-2 (D) cells were treated with OSI-027 (50 nM), with or without MEK/ERK inhibitor PD98059 (500 nM), U0126 (500 nM), or MEK162 (100 nM) for 72 h, cell survival was tested by CellTiter-Glo assay. PANC-1 or primary pancreatic cancer cells were preadded with 3-methyladenine (3-MA, 0.5 mM), chloroquine (Cq, 10 μM), or ammonium chloride (NH4Cl, 10 mM) for 1 h, followed by OSI-027 (50 nM) treatment, cell survival was tested by CellTiter-Glo assay 72 h after OSI-027 treatment (F, G). PANC-1 cells, transfected with scramble control siRNA (“sc-RNAi”) or Beclin-1 siRNA (100 nM each, 36 h), were either left untreated (“C”), or treated with OSI-027 (50 nM) for 72 h, expression of Beclin-1 and GAPDH was tested by western blots (H), cell survival was also tested (I). Phosphorylation or expression of indicated proteins were quantified, and was normalized to the nonphosphorylated kinase or the loading control (GAPDH). LC3-II intensity was normalized to that of LC3-I to calculate the LC3-I/II conversion. For (C, D, F, G, and I): Data are expressed as mean±SE, experiments were repeated five times. n=5 for each assay.*p<0.05 versus “C” group. #p<0.05 versus OSI-027 only group (C, D, F, G). #p<0.05 versus “sc-RNAi” group (I).
Notably, in both PANC-1 and MIA PaCa-2cells, OSI-027 treatment induced autophagy activation, which was evidenced by p62 degradation, Beclin-1 upregulation, and LC3-I to LC3-II conversion (Fig. 3E). Significantly, autophagy inhibitors, including chloroquine (Cq), ammonium chloride (NH4Cl), and 3-methylaldenine (3-MA), all enhanced OSI-027 (50 nM)-induced activity in transformed (PANC-1, Fig. 3F) and primary (Fig. 3G) pancreatic cancer cells. These autophagy inhibitors alone had no significant effect on the survival of tested pancreatic cancer cells (data not shown). To further study the role of autophagy in OSI-027's activity, siRNA strategy was applied to knockdown the key autophagy protein Beclin-1 (Fu et al., 2013). Beclin-1 siRNA significantly downregulated Beclin-1 expression in PANC-1 cells (Fig. 3H). Importantly, OSI-027-induced cytotoxicity was enhanced by Beclin-1 silencing (Fig. 3I). Similar results were also observed in MIA PaCa-2 cells (Supplementary Fig. S2). These results suggest that autophagy activation by OSI-027 exerted a cytoprotective role, counteracting OSI-027-induced antipancreatic cancer activity.
OSI-027 sensitizes gemcitabine-induced antipancreatic cancer activity in vitro and in vivo
We also tested the potential role of OSI-027 on gemcitabine. As shown, in both transformed [PANC-1 and MIA PaCa2, both gemcitabine-resistant lines (Humbert et al., 2010)] and primary pancreatic cancer cells, gemcitabine (100 μM, 72 h) only exerted weak activity on cell survival (Fig. 4A–C). However, in the presence of a relatively low concentration of OSI-027 (50 nM, see Fig. 1), its activity was dramatically increased (Fig. 4A–C). OSI-027 alone only slightly inhibited cell survival (Fig. 4A, C). In the meantime, OSI-027 (50 nM) also sensitized the antipancreatic cancer activity by paclitaxel (Taxol), but not cisplatin (Supplementary Fig. S3A,B). Although the Taxol sensitization activity by OSI-027 appeared weaker as compared to the activity of gemcitabine.
FIG. 4.
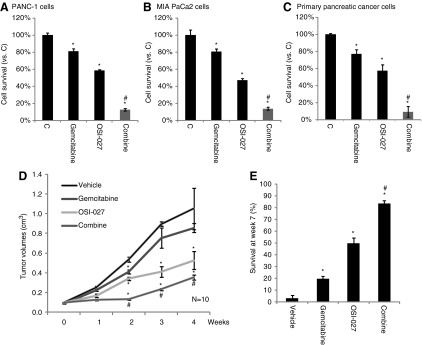
OSI-027 and gemcitabine synergism against pancreatic cancer cells. Transformed (PANC-1 or MIA PaCa-2lines) (A, B) or primary (C) pancreatic cancer cells were either left untreated (“C”), or stimulated with OSI-027 (50 nM) and/or gemcitabine (100 μM) for 72 h, cell survival was analyzed by the CellTiter-Glo assay. Severe combined immunodeficiency mice were ear tagged and randomized into four different groups (n=10 for each group): vehicle control, OSI-027 (25 mg/kg, p.o.), gemcitabine (25 mg/kg, i.p.), and OSI-027 plus gemcitabine. Treatments are mentioned in Materials and Methods. Tumor volume (in cm3) (D) was recorded every week for a total of 4 weeks. Mice survival at week 7 was also presented (E, three individual experiments). Data are presented as mean±SE. *p<0.05 versus “C”/vehicle group, #p<0.05 versus gemcitabine only group.
To test the activity of OSI-027 and/or gemcitabine in vivo, SCID mice bearing PANC-1 xenograft model was applied (See Materials and Methods). As demonstrated, daily gemcitabine (25 mg/kg, i.p.) administration only slightly inhibited PANC-1 xenograft growth in SCID mice (Fig. 4D). On the other hand, oral administration of OSI-027 (25 mg/kg, daily) in SCID mice significantly repressed PANC-1 cell growth in vivo (Fig. 4D). When the OSI-027 and gemcitabine were coadministered, PANC-1 xenograft growth was further inhibited, although the difference between combo and OSI-027 alone was not that dramatic (Fig. 4D). Interestingly, the mice survival was significantly improved after the coadministration (Fig. 4E), and the combo activity was significantly more potent than either agent alone (Fig. 4E). Therefore, mice survival beneficial by the coadministration may not be solely dependent on tumor volume regression; it could be due to other unidentified mechanisms, which warrant further investigations. Mice treated with the above regimens did not show any signs of wasting, and the body weights were not significantly affected (data not shown).
Discussions
mTORC1 and mTORC2 are formed and regulated by different proteins and are also driven by multiple different compensatory feedback loops (Vilar et al., 2011). The first generation of mTOR inhibitors only target mTORC1. Thus the antiproliferative property of these inhibitors is relatively weak (Vilar et al., 2011). Meanwhile, mTORC1 inhibition often results in feedback activations of the prosurvival signalings, including PI3K–AKT and ERK–mitogen-activated protein kinase signalings (Chen et al., 2010; Vilar et al., 2011). OSI-027 is novel and potent mTORC1/2 dual inhibitor. Previous studies have shown that OSI-027 exerted robust antitumor activity in several different human xenograft models (Bhagwat et al., 2011). Furthermore, it showed superior efficacy compared with rapamycin (Bhagwat et al., 2011). In this study, our results showed that targeting mTORC1/2 simultaneously by OSI-027 significantly inhibited pancreatic cancer cell growth both in vivo and in vitro. Further, AKT Ser-473 was also dramatically inhibited by OSI-027 in tested cancer cells, where ERK1/2 activation was not affected. Thus, the concurrent suppression of mTORC2 in addition to mTORC1 is likely to be a more effective strategy in the treatment of pancreatic cancer. As a matter of fact, we showed that OSI-027 was more potent than the same concentration of rapamycin or RAD001 in inhibiting pancreatic cancer cell survival, and inducing cell apoptosis.
One important finding of this study is the autophagy activation by OSI-027, which exerts prosurvival roles in pancreatic cancer cells. Pharmacological blockage of autophagy significantly enhanced OSI-027-induced antipancreatic cancer activity. Of the tested autophagy inhibitors, Cq is the lysosomotropic drug that disrupts autophagic protein degradation (Glaumann and Ahlberg, 1987). 3-MA inhibits LC3B-I to LC3B-II conversion, and prevents autophagosome formation (Seglen and Gordon, 1982). NH4Cl increases intralysosomal pH thus preventing autophagic protein degradations. All these inhibitors enhanced OSI-027-induced cytotoxicity against primary and transformed pancreatic cancer cells. Further, siRNA-mediated knockdown of Beclin-1, a key protein for autophagosome formation (Fu et al., 2013), also facilitated OSI-027-induced pancreatic cancer cell death. Thus, autophagy intervention could be an important strategy to further increase the activity of OSI-027.
The advanced or metastatic pancreatic cancer is an extremely aggressive human malignancy with a median overall survival of only several months (Hidalgo, 2010; Siegel et al., 2014). There are several reasons of the dismal prognosis of this disease, including late diagnosis, absence of diagnostic serum markers, resistance to the conventional chemotherapy (i.e., gemcitabine), and the high metastatic potential (Costello and Neoptolemos, 2011). Gemcitabine is thus far the only approved single chemoagent for the treatment of pancreatic cancer, and its benefit in increasing patients' overall survival has been documented (Heinemann et al., 2008). However, the activity of gemcitabine is generally gentle, and the combination strategies have been widely tested. As a result of chemoresistance, the majority of phase III trials exploring gemcitabine-based combinations have failed to improve the overall survival, the only expectances are erlotinib (Moore et al., 2007) and nab-paclitaxel (Von Hoff et al., 2013). Our in vivo and in vitro results demonstrated that adding a low concentration of OSI-027 could potentially increase gemcitabine's activity both in vivo and in vitro.
Conclusions
A first-in-human phase I clinical trial of OSI-027 is ongoing, and already presented with preliminary evidence of OSI-027's pharmacologic activity. The results showed that OSI-027 was well tolerated at tested doses, although the maximum tolerated dose has yet to be defined (Tan et al., Journal of Clinical Oncology, 2010 ASCO Annual Meeting Abstracts). The preclinical results of this study suggest that targeting mTORC1/2 synchronously by OSI-027 could be possibly further investigated as a valuable treatment for pancreatic cancer.
Supplementary Material
Acknowledgment
This work is funded by the research startup funds of the East Hospital Affiliated to Tongji University in Shanghai.
Disclosure Statement
No competing financial interests exist.
References
- Ascierto P.A., et al. (2013). MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol 14, 249–256 [DOI] [PubMed] [Google Scholar]
- Azzariti A., et al. (2008). Synergic antiproliferative and antiangiogenic effects of EGFR and mTor inhibitors on pancreatic cancer cells. Biochem Pharmacol 75, 1035–1044 [DOI] [PubMed] [Google Scholar]
- Bhagwat S.V., et al. (2011). Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther 10, 1394–1406 [DOI] [PubMed] [Google Scholar]
- Bu H.Q., et al. (2014). Oridonin induces apoptosis in SW1990 pancreatic cancer cells via p53- and caspase-dependent induction of p38 MAPK. Oncol Rep 31, 975–982 [DOI] [PubMed] [Google Scholar]
- Chen B., et al. (2013). Cisplatin-induced non-apoptotic death of pancreatic cancer cells requires mitochondrial cyclophilin-D-p53 signaling. Biochem Biophys Res Commun 437, 526–531 [DOI] [PubMed] [Google Scholar]
- Chen X.G., et al. (2010). Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol Carcinog 49, 603–610 [DOI] [PubMed] [Google Scholar]
- Costello E., and Neoptolemos J.P. (2011). Pancreatic cancer in 2010: new insights for early intervention and detection. Nat Rev Gastroenterol Hepatol 8, 71–73 [DOI] [PubMed] [Google Scholar]
- Eckel F., et al. (2006). Pancreatic cancer: a review of recent advances. Expert Opin Investig Drugs 15, 1395–1410 [DOI] [PubMed] [Google Scholar]
- Falasca M., et al. (2011). Targeting phosphoinositide 3-kinase pathways in pancreatic cancer—from molecular signalling to clinical trials. Anticancer Agents Med Chem 11, 455–463 [DOI] [PubMed] [Google Scholar]
- Fu L.L., et al. (2013). Beclin-1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol 45, 921–924 [DOI] [PubMed] [Google Scholar]
- Furukawa T. (2008). Molecular targeting therapy for pancreatic cancer: current knowledge and perspectives from bench to bedside. J Gastroenterol 43, 905–911 [DOI] [PubMed] [Google Scholar]
- Garrido-Laguna I., et al. (2010). Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br J Cancer 103, 649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaumann H., and Ahlberg J. (1987). Comparison of different autophagic vacuoles with regard to ultrastructure, enzymatic composition, and degradation capacity—formation of crinosomes. Exp Mol Pathol 47, 346–362 [DOI] [PubMed] [Google Scholar]
- Gudjonsson B. (2009). Pancreatic cancer: survival, errors and evidence. Eur J Gastroenterol Hepatol 21, 1379–1382 [DOI] [PubMed] [Google Scholar]
- Guertin D.A., and Sabatini D.M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 [DOI] [PubMed] [Google Scholar]
- Hart A.R., et al. (2008). Pancreatic cancer: a review of the evidence on causation. Clin Gastroenterol Hepatol 6, 275–282 [DOI] [PubMed] [Google Scholar]
- Heinemann V., et al. (2008). Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer 8, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M. (2010). Pancreatic cancer. N Engl J Med 362, 1605–1617 [DOI] [PubMed] [Google Scholar]
- Humbert M., et al. (2010). Masitinib combined with standard gemcitabine chemotherapy: in vitro and in vivo studies in human pancreatic tumour cell lines and ectopic mouse model. PLoS One 5, e9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D., et al. (2006). In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int J Cancer 118, 2337–2343 [DOI] [PubMed] [Google Scholar]
- Javle M.M., et al. (2010). Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer 10, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters-Vandevelde H.V., et al. (2014). Experimental treatment of NRAS-mutated neurocutaneous melanocytosis with MEK162, a MEK-inhibitor. Acta Neuropathol Commun 2, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. (2012). Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res Treat 134, 1057–1066 [DOI] [PubMed] [Google Scholar]
- Lockhart A.C., et al. (2005). Treatment for pancreatic cancer: current therapy and continued progress. Gastroenterology 128, 1642–1654 [DOI] [PubMed] [Google Scholar]
- McKenna S., and Eatock M. (2003). The medical management of pancreatic cancer: a review. Oncologist 8, 149–160 [DOI] [PubMed] [Google Scholar]
- Min H., et al. (2014). Bortezomib induces protective autophagy through AMP-activated protein kinase activation in cultured pancreatic and colorectal cancer cells. Cancer Chemother Pharmacol 74, 167–176 [DOI] [PubMed] [Google Scholar]
- Moore M.J., et al. (2007). Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25, 1960–1966 [DOI] [PubMed] [Google Scholar]
- Oettle H., et al. (2007). Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 297, 267–277 [DOI] [PubMed] [Google Scholar]
- Rivera F., et al. (2009). Treatment of advanced pancreatic cancer: from gemcitabine single agent to combinations and targeted therapy. Cancer Treat Rev 35, 335–339 [DOI] [PubMed] [Google Scholar]
- Sabatini D.M. (2006). mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 6, 729–734 [DOI] [PubMed] [Google Scholar]
- Schneider G., et al. (2005). Pancreatic cancer: basic and clinical aspects. Gastroenterology 128, 1606–1625 [DOI] [PubMed] [Google Scholar]
- Seglen P.O., and Gordon P.B. (1982). 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 79, 1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R., et al. (2014). Cancer statistics, 2014. CA Cancer J Clin 64, 9–29 [DOI] [PubMed] [Google Scholar]
- Stathis A., and Moore M.J. (2010). Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol 7, 163–172 [DOI] [PubMed] [Google Scholar]
- Sun S.Y. (2013). mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 340, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.Y., et al. (2014). GDC-0980-induced apoptosis is enhanced by autophagy inhibition in human pancreatic cancer cells. Biochem Biophys Res Commun 453, 533–538 [DOI] [PubMed] [Google Scholar]
- Vilar E., et al. (2011). Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther 10, 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D.D., et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369, 1691–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal S., and Kalthoff H. (2003). Apoptosis: targets in pancreatic cancer. Mol Cancer 2, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpin B.M., et al. (2009). Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol 27, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray C.J., et al. (2005). Surgery for pancreatic cancer: recent controversies and current practice. Gastroenterology 128, 1626–1641 [DOI] [PubMed] [Google Scholar]
- Zhu Y.R., et al. (2014). Bufotalin-induced apoptosis in osteoblastoma cells is associated with endoplasmic reticulum stress activation. Biochem Biophys Res Commun 451, 112–118 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.