

Abstract
Long overlooked as the virtual compartment and then strictly characterized through descriptive morphologic analysis, the renal interstitium has finally been associated with function. With identification of interstitial renin- and erythropoietin-producing cells, the most prominent endocrine functions of the kidney have now been attributed to the renal interstitium. This article reviews the functional role of renal interstitium.
Keywords: fibrosis, renal interstitium, renin angiotensin system, erythropoietin, fibroblast
Introduction
The physiologic role of the interstitium of the kidney has received comparatively little interest to date. This may partially be attributed to the fact that it was long considered the exclusive domain of descriptive ultrastructural research, which implied that the interstitium was mostly a passive tissue that structurally supported the tubular epithelium (1,2). The renal interstitium received increasing interest in the context of kidney abnormalities, when the role of interstitial fibrosis in progression of CKD became obvious (3–5). Only since transgenic animal studies revealed the physiologic endocrine function of interstitial cells as sources of erythropoietin (Epo) and renin has the physiologic role of the renal interstitium received its due attention. Here we review current knowledge of the form and function of the renal interstitium.
Definition and Ultrastructure of the Renal Interstitium
The renal interstitium is defined as the intertubular, extraglomerular, extravascular space of the kidney. It is bounded on all sides by tubular and vascular basement membranes and is filled with cells, extracellular matrix, and interstitial fluid (1). Its distribution varies within the kidney; it accounts for approximately 8% of the total parenchymal volume in the cortex and up to 40% in the inner medulla (6,7). The term “renal interstitium” is often inadequately used to refer to the peritubular interstitium (the space between tubules, glomeruli, and capillaries); the periarterial connective tissue and the extraglomerular mesangium are considered specialized interstitia (1). It is debated whether microvessels and capillaries, which are located within the peritubular space, are actually part of the renal interstitium or just run through it (1). Furthermore, lymphatics are considered interstitial constituents (1). The tubular interstitium in the cortex and medulla differ with regard to their cellular constituents, extracellular matrix composition, relative volume, and endocrine function, justifying the consideration of cortical and medullary interstitium as separate entities.
The intertubular interstitium harbors dendritic cells, macrophages, lymphocytes, lymphatic endothelial cells (in the cortical intertubular interstitium), and various types of fibroblasts, the hallmark cell type of connective tissues (8). In addition, it is believed that the interstitium plays a role in fluid and electrolyte exchange and insulation (1). Here we focus on fibroblasts and specifically on fibroblasts with endocrine function and their biology in health and disease (other articles have extensively reviewed cellular constituents of the immune system within the renal interstitium [9,10]) (Figure 1).
Figure 1.

Content of the virtual compartment. Left. The photomicrograph displays a cortical section of a paraffin-fixed periodic acid-Schiff–stained mouse kidney. The interstitium, located between tubules, is barely visible. Arrows (black) point to interstitial fibroblasts or to microvessels, respectively (green). Right. Glomerular and tubular cross-sections and contents of the interstitium are illustrated in the schematic. The interstitial compartment contains non–hormone-producing fibroblasts (blue), microvessels (red), perivascular cells (green), renin-producing perivascular cells (orange), juxtaglomerular cells (lilac), and erythropoietin (Epo)-producing fibroblasts (pink). Bottom. Representative drawings are explained.
Renal Fibroblasts
In general, fibroblasts are flattened cells with extended cell processes; in profile they display a fusiform or spindle-like shape and a flattened nucleus (11). Fibroblasts typically are embedded within the fibrillar matrix of connective tissues and are considered prototypical mesenchymal cells. Renal fibroblasts anastomose with each other, forming a continuous network in cortex and medulla (8). Fibroblasts can acquire an activated phenotype with a relatively large oval nucleus with one or two nucleoli, abundant rough endoplasmatic reticulum, and several sets of Golgi apparatus; this reflects their capacity to synthesize substantial amounts of extracellular matrix constituents (12). Under physiologic conditions, however, adult fibroblasts are relatively inactive, the endoplasmatic reticulum is reduced, and the nucleus is flattened and heterochromatic. On the basis of their appearance, it was assumed that the primary function of renal fibroblasts was to provide structural support to nephrons through deposition of extracellular matrix and through direct cell-cell interactions (1). In addition, fibroblasts play an important role in maintaining vascular integrity in close association with vessels (then typically referred to as vascular smooth muscle cells and pericytes) (13,14). Renal fibroblasts are best known for their role in progression of interstitial fibrosis in progressive CKD because they are principal producers of extracellular matrix (4). Finally, fibroblasts have been identified as sources of Epo and renin in the kidney (15,16). It is obvious that such diverse functions are not fulfilled by one cell type but that renal fibroblasts are instead a heterogeneous cell population with distinct functions.
Heterogeneity of Renal Fibroblasts
Our current knowledge of fibroblast heterogeneity evolved from morphologic descriptions to immunophenotyping, and only recently the use of transgenic mouse models provided insights into origins and functions of renal fibroblasts. Ultrastructural analysis first revealed that cortical fibroblasts differ from medullary fibroblasts, as they form a finer network through their radiating cytoplasmatic processes with tubular cells, endothelial cells, and each other. Furthermore, medullary fibroblasts often have a stouter appearance and harbor cytoplasmatic lipid droplets (8). However, such ultrastructural analysis revealed little about distinct functions of fibroblast populations (1,2,8,17). Attempts to link ultrastructural appearance to function were followed by studies that aimed to define renal fibroblast populations through use of fibroblast markers. Such fibroblast markers included the intermediate filament-associated protein vimentin (which is not specific for fibroblasts because it is also present in endothelial cells and neurons) (18); desmin (which is present only in a subset of fibroblasts and in muscle cells) (19,20); collagen receptors, such α1β1 integrin (21) and discoidin domain receptor 2 (which are also present on cells of various other lineages, including endothelial cells) (22); the intracellular calcium-binding protein fibroblast-specific protein 1 (which is present on fibroblasts but also in invasive carcinoma cells) (23); the membrane-bound enzyme ecto-5′-nucleotidase (CD73) (which labels cortical renal fibroblasts but also T-lymphocytes) (24,25); the PDGF receptor PDGF receptor-β (which labels fibroblasts but also macrophages) (26,27); and α-smooth muscle actin (which labels a subset of activated fibroblasts and vascular smooth muscle cells) (28). In addition, the synthesis of collagen types I, III, and V are considered a hallmark feature of fibroblasts, although many other cell types also robustly express these collagens (28). Clearly, all proposed markers had deficiencies, creating an as-yet unresolved dispute in the field regarding identity of fibroblasts and their respective function in kidney health and disease. The confusion is further fueled by assumed different origins of fibroblasts in diseased kidneys (for extensive information, see elsewhere [4,29]). For practical purposes, we here define fibroblasts as the nonvascular, nonepithelial, and noninflammatory cell constituents of the kidney (11). In the following paragraphs we discuss the role of fibroblasts as sources of Epo and renin in kidney health and disease.
Epo
Epo is an indispensable glycoprotein hormone that is produced by interstitial renal fibroblasts and controls hematopoiesis through promotion of survival, proliferation, and differentiation of erythroid progenitors (30). Epo is best known through its recombinant protein relatives, which are widely used to treat anemia in dialysis patients (a $12 billion market worldwide at its peak in 2006) and its misuse as a performance-enhancing substance in cycling and other sports (31). Insights into the physiologic mechanism that underlies production of endogenous Epo are of interest to understand the molecular basis of Epo deficiency in patients with CKD and potential therapeutic strategies to circumvent lack of Epo. Here we review the origins of endogenous Epo and molecular mechanisms that control Epo production in health and disease.
Identification of Renal Interstitial Fibroblasts as Primary Origin of Epo
In adults, about 90% of Epo is produced by renal interstitial fibroblasts, whereas 10% is contributed by extrarenal sources, primarily liver cells (32). Decreased oxygen tension is the principal stimulus for renal Epo expression, yet Epo production decreases in CKD, which is associated with chronic hypoxia (33,34). When Epo production is impaired in the injured kidney, extrarenal sources of Epo cannot compensate for decreased Epo levels (35). Nevertheless, despite decades of effort, Epo-producing kidney cells have not yet been cultivated, which is why most analyses of Epo production relied on liver-derived hepatoma cells (36). The value of using hepatoma cells to study mechanisms of Epo production is limited, however, because control of Epo expression underlies distinct mechanisms in kidney and liver. To better understand this unusual complexity of endogenous Epo expression, it is worth reviewing the sequence of discoveries that led to the knowledge of Epo expression.
A link between decreased oxygen availability and an adaptive increase in red blood cell count was first suspected by Miescher, who observed increased hemoglobulin levels in patients who entered a high-altitude sanatorium in the Alps (37). Yet it was not until 1953 when Erslev demonstrated that plasma transfer from anemic rabbits caused increased hemoglobin levels (but not white blood cells) in recipient rabbits (38). Goldwasser succeeded in isolating Epo for the first time in 1977, but it took him 17 years and 2500 L of urine from patients with aplastic anemia to achieve this goal (39).
Because of the consistent association between CKD and anemia, the kidney was long suspected as a primary source of Epo. However, Epo expression levels are low, and immunolabeling and in situ hybridization analyses proved to be unreliable; it took extensive studies using transgenic mice to finally, unequivocally identify renal interstitial fibroblasts as primary producers of Epo (16,40–42). Furthermore, analysis of Epo expression in vivo is technically challenging because large regions of noncoding DNA flanking the Epo locus (which contains tissue-specific enhancers and repressors) must be included to adequately reflect endogenous Epo expression. Only recently, use of a 180-kb Epo transgene fused to a green fluorescent protein reporter tag unequivocally located hypoxia-induced Epo expression to fibroblasts in the deep cortex to the outer medulla region (41). The identity of these Epo-producing cells as fibroblasts was confirmed by immunophenotyping with antibodies to CD73.
Regulation of Epo Production
The kidney is the principal source of Epo. Because Epo deficiency is the most important component of the anemia that complicates CKD, cellular origins and molecular mechanisms that control endogenous Epo production are of great clinical relevance (43). Physiologic plasma Epo levels in blood are relatively low at around 100 pg/ml, whereas under hypoxic stress associated with anemia, Epo levels can reach 100,000 pg/ml (44). A drop in tissue PO2 is the principal stimulus for Epo expression, and PO2 levels are consistently higher within the cortical interstitium because there is a constant ratio of blood flow rate and small O2 consumption, relatively independent of cardiac output and renal blood flow (45,46). Hence, a drop in hemoglobin levels and ensuing tissue hypoxia are a prototypical stimulus for Epo expression. Because Epo is not stored and has a relatively short half-life, circulating Epo levels are closely linked to Epo expression in interstitial fibroblasts. The mechanisms that control transcriptional activity have received prominent attention in recent years (summarized in Figure 2) (44). The true value of understanding mechanisms that underlie control of Epo expression may be the possible therapeutic solutions to overcome the transcriptional silencing of Epo, a hallmark of CKD (47). While hypoxia is the principal stimulus for Epo expression under physiologic conditions, Epo transcription is silenced in chronically diseased kidneys, despite the hypoxic conditions that are a hallmark of renal fibrogenesis (34).
Figure 2.

Control of Epo expression. Renal Epo expression largely depends on enhancer regions located between −14 kb and −9.5 kb upstream of the Epo gene, whereas negative regulatory elements located between −6 and −0.4 kb play an important role in preventing ectopic Epo expression. Enhancer regions that control Epo expression in the liver are located at the 3′ end at 0.7 kb downstream of the Epo locus. Under hypoxic conditions hypoxia-inducible factor (HIF)-1α is stabilized, dimerizes with HIF-1β, and translocates to the nucleus, where it can bind to hypoxia response elements within the kidney enhancer region and induce Epo transcriptional activity. Once normoxia is restored, prolyl hydroxylase (PDH1) hydroxylates HIF-1α, causing its association with the von Hippel–Lindau tumor suppressor protein (VHL), ultimately causing ubiquitin-dependent degradation of HIF-1α.
The molecular mechanisms that underlie transcriptional silencing of Epo in fibrotic fibroblasts is only incompletely understood, at least in part because of aforementioned difficulties in the study of Epo expression in cultured renal cells. Two recent studies that used transgenic mice harboring a 180-kD chromosome fragment erythropoietin transgene reported that de novo expression of α-smooth muscle antibody in interstitial fibroblasts correlates negatively with Epo expression, suggesting that additional superimposed intracellular programs render the Epo gene unresponsive to transcriptional stimuli (48,49). In this regard, differential expression of hypoxia-inducible factor (HIF) proteins, of globin transcription factor proteins, or hypermethylation of regulatory elements has been suggested to be involved (48,50). Relevance of such knowledge lies in possible use of these pathways to rescue endogenous Epo production in anemic patients (51).
Impaired Epo Production as Cause of Anemia
Anemia is a common complication of CKD, and the striking effectiveness of recombinant Epo proves that impaired production of endogenous Epo is a major cause of this (52). Decreased Epo levels in patients with CKD are obvious only when anemia is present but cannot be adequately elevated when hematocrit drops, suggesting that part of the Epo production capacity (i.e., through loss of Epo-producing cells) is irreversibly lost (53–55). In general, decreased Epo levels (typically assessed by ELISA in serum) correlate with severity of decreased excretory kidney function. While Epo levels are better preserved in glomerular diseases than in interstitial diseases, suggesting that impaired Epo production is a direct consequence of interstitial disease (56), there is one exception as polycystic kidney disease is often associated with increased Epo production and polycythemia, due to aberrant HIF1α accumulation around cysts due to local hypoxia (57). Nevertheless, circulating Epo levels have been suggested as diagnostic marker of the extent of tubulointerstitial involvement in diabetic nephropathy (58), but due to superimposed regulating factors (including anemia, systemic inflammation, and reduced iron availability), utility of Epo levels as valid diagnostic marker for interstitial fibrosis could not be validated (53). Impaired Epo production as cause of anemia is not limited to CKD but is also increasingly being recognized as a cause of anemia in patients with diabetes mellitus. However, unlike in patients with CKD, Epo production responds adequately in diabetic patients to acute hypoxic stress, suggesting that in principle Epo-producing cells still exist, but that they don’t sense the anemia appropriately (59). While in CKD interstitial cells appear to irreversibly lose capacity to produce Epo, it is conceivable that altered oxygen sensing plays a role in diabetes mellitus. In this regard, in early diabetes renal blood flow is increased, and it is plausible that resulting increased oxygen supply suppresses Epo production. In line with this, blockade of the renin-angiotensin system in rats increased interstitial blood flow (due to decreased intrarenal resistance) and anemia (60), possibly explaining the drop in hematocrit typically observed in the first month of therapy with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers in patients (61).
While supplementation with recombinant Epo is highly effective, it is associated with high costs, and its effectiveness can be impaired by antibody formation. Hence, stimulation of endogenous Epo production is an attractive therapeutic strategy. In this regard, inhibition of prolyl-hydroxylase (PHD; the enzyme that makes HIF accessible for Von Hippel–Lindau tumor suppressor protein [VHL] and subsequent proteolytic degradation) has emerged as the primary therapeutic target as several small molecule inhibitors have been developed. The rationale behind PHD inhibition is that conceptually it increases intracellular HIF levels and thus stimulates Epo production. In this regard, the PHD inhibitor FG-4592 is undergoing clinical testing in anemic patients with CKD stages 3 and 4. One possible drawback might be that currently available PHD inhibitors do not display PHD isoform specificity (among the known isoforms PHD1–3, PHD2 is considered the one involved in renal Epo transcriptional control), and adverse effects of enhanced HIF activity (such as angiogenesis as the root of diabetic retinopathy) might be enhanced (62).
Adenosine
One often-underappreciated function of interstitial fibroblasts is their involvement in regulation of renal hemodynamics and microvascular function through generation of extracellular adenosine (63,64). To maintain glomerular filtration within a narrow range, the kidney responds to increased intratubular NaCl levels (upon increased GFR) by vasoconstriction of afferent arterioles (to decrease GFR) (65). A crucial mediator of this tubuloglomerular feedback loop is extracellular adenosine, which is generated through hydrolysis of ATP released by macula densa cells into the interstitium and which causes vasodilation of efferent arterioles (through activation of A2 adenosine receptors) and vasoconstriction of afferent arterioles through activation of A1 receptors, decreasing intraglomerular pressure and GFR (65). Furthermore, adenosine directly inhibits renin release in juxtaglomerular cells. Hence, when GFR and NaCl concentration of tubule fluid increase, adenosine is part of the tubuloglomerular feedback response to lower GFR and NaCl secretion; when GFR and NaCl concentrations decrease, adenosine levels drop under physiologic conditions (66). One enzyme that catalyzes hydrolysis of ATP to adenosine is 5′ectonucleotidase (5′NT, CD73), which is expressed in the kidney predominantly by resident fibroblasts (a characteristic that has made it a popular marker for renal fibroblasts) (63). 5′NT-deficient mice display substantial reduction of extracellular adenosine levels and reduced kidney weight; however, renal blood flow and GFR are unaltered (67), possibly because of intrinsic activity of interstitial ATP on vascular tone (64). In the chronically injured kidney, 5′NT-positive fibroblasts and extracellular adenosine levels accumulate, possibly contributing to the lowered GFR through afferent arteriole constriction, which is typical of CKD (2).
Renin
Renin is another protein that is released by renal cells and indirectly acts systemically (on BP), even though strictly speaking it is more an enzyme than a hormone. With more than 50,000 publications listed in PubMed to date, renin is among the most-studied proteins in biomedical sciences. Renin is a key regulator of the renin-angiotensin-aldosterone system, and plasma renin levels reflect the overall activity of this system (68). The circulating active form of renin cleaves angiotensinogen to form angiotensin I (69). Because angiotensinogen is highly abundant (its concentration is 1000-fold higher than that of angiotensin I), it is the plasma renin activity that determines the rate of angiotensin I formation (68).
Circulating renin is secreted by juxtaglomerular cells (also called granular cells or JGA cells), unique round cells of epithelioid appearance that contain myofilaments and abundant peroxisomes (70). While under physiologic conditions juxtaglomerular cells are located just at the outer edge of the renal interstitium (in the juxtaglomerular interstitium, at the walls of afferent arterioles just at the entrance into glomeruli) additional renin-producing cells are recruited extramurally within the interstitium (discussed in more detail below), and hence they are discussed here as “interstitial cells” for practical purposes (15). Release of renin by juxtaglomerular cells is regulated by hormones such as atrial natriuretic peptide and angiotensin II and by local factors contributed by adjacent tubular epithelial cells, by vascular smooth muscle cells and endothelial cells from afferent arterioles and by sympathetic nerve endings (71).
With regard to local regulatory factors, specialized tubular cells of the cortical thick ascending limb of the loop of Henle act together with macula densa cells to directly translate changes in the tubular NaCl concentration into inverse changes in renin secretion (72). In this system, macula densa cells sense intratubular NaCl concentrations through activity of the Na+-K–2Cl− cotransporter NKCC2 (BSC1) and respond to lower NaCl concentrations by release of prostanoids (generation of active prostaglandin E2 and prostacyclin depends on cyclooxygenases) (71).
Furthermore, renin release is modulated by vascular smooth muscle cells and endothelial cells from afferent arterioles (71). In this system, renin stimulatory factors such as nitric oxide (formed by endothelial nitric oxide synthase) and prostacyclin/prostaglandin E2 are formed in the endothelium adjacent to the granular cells, and inhibitory adenosine (which is converted into ATP) is released by vascular smooth muscle cells (73).
The sympathetic input provided by local nerve endings (in addition to circulating catecholamines) stimulate renin release through the β-adrenergic system (73). The magnitude of sympathetic control of renin release is revealed by double-knockout mice deficient in β1- and β2-receptors, which have 85% reduced plasma renin levels compared with wild-type control mice (74). Hence, renal denervation and baroreceptor stimulation conceptually seemed to be sound antihypertensive strategies. In this regard, failure of renal denervation to significantly lower BP in clinical studies is not yet entirely understood (75). Renal denervation also removes the input of the sympathetic system mediated by α-receptors, including hemodynamic and tubular effects, and systemic catecholamines remain fully functional; these findings suggest that baroreceptor stimulation might be the most promising approach to target the sympathetic nerve system to lower BP (76,77).
Renin secretion is further controlled by various hormones, autocoids, and other factors, including angiotensin II, as atrial natriuretic peptide, vasopressin, oxytocin, aldosterone, glucocorticoids, thyroid hormones, sex steroids, adipokines, dopamine, bradykinin, adrenomedullin, and neuropeptide Y (for further review, see extensive review elsewhere [71,73]). While availability of potent ACE inhibitors and angiotensin receptor blockers (and absence of potent renin blockers) long overshadowed the role of renin in clinical practice, understanding of direct renin-receptor mediated effects and availability of aliskiren revived interest in renin biology (78).
Under physiologic conditions, secretion and activation of prorenin by juxtaglomerular cells are sufficient for maintenance of GFR (71). During pathologic conditions, however, additional perivascular fibroblasts (i.e., pericytes, vascular smooth muscle cells) of afferent arterioles express prorenin, which has led to increased interest in the ontogeny of renin-producing cells (79). Under basal conditions, renin produced by juxtaglomerular cells is sufficient to main BP and intravascular volume (71). Upon prolonged threat to volume homeostasis (i.e., through exposing mice to a low salt diet and ACE inhibitors) additional cells along afferent arterioles express renin, in angiotensinogen-deficient mice renin is even expressed by fibroblasts (pericytes) all through the kidney cortex (80,81). Models of CKD are also associated with recruitment of additional renin-producing cells. Because increased renin expression is often at the root of hypertension, and because higher on-treatment plasma renin activity contributes to progression of CKD and increased cardiovascular risk, mechanisms that cause such inopportune increase in renin release and particularly recruitment of renin producers are of imminent interest (82,83).
Interplay of Renin-Angiotensin and Epo Systems
Clinicians know that patients with renovascular hypertension and kidney transplant recipients with transplant renal artery stenosis have higher hematocrit values compared with patients who have normal renal arteries (84). The underlying pathomechanism of this clinical observation is that ensuing renal hypoperfusion activates the renin-angiotensin system (as described in detail above) and that angiotensin II stimulates erythropoiesis through stimulating Epo secretion and through its directs stimulating effect on erythropoiegenesis in the bone marrow (85). This mechanisms explains why effective blockage of the renin-angiotensin system through ACE inhibitors and/or angiotensin receptor blockers is often associated with a decrease in hematocrit (86,87) and that ACE inhibitors are effective in normalizing post-transplant erythrocytosis (88). ACE inhibitors even affect hematocrit in hemodialysis patients, who require recombinant Epo substitution because of impaired endogenous Epo production (as discussed in more detail above). This occurs because treatment with ACE inhibitors is associated with higher recombinant human Epo (rhEpo) requirements (whereas withdrawal of ACE inhibitors decreases rhEpo doses), reflecting the direct effect of angiotensin II on erythropoiegenesis (89). Exceptions to the link of renin-angiotensin system activation and increased hematocrit are often cases in which increment of red blood cells mass are masked by volume expansion (i.e., in patients with severe heart failure) (90).
Hypertension is the most relevant side-effect of rhEpo therapy (91). However, the increase in BP that is often observed in dialysis patients receiving rhEpo therapy is independent of the renin-angiotensin system; several clinical studies did not demonstrate renin-angiotensin system activation upon rhEpo therapy (92). Instead, the increased BP observed with rhEpo therapy is most likely a direct systemic vasoconstriction (91). Nevertheless, rhEpo-induced hypertension is correctable with the typical antihypertensive therapeutic regimen, including ACE/angiotensin receptor blocker inhibition (91).
Origins of Hormone-Producing Interstitial Cells
If we are going to develop specific therapeutic interventions, we need to understand the origins and development of fibroblast subpopulations, including those that produce Epo, those that produce renin, and those that proliferate and lay down the extracellular matrix in fibrosis. Such studies are done by crossing mice in which cre-recombinase is expressed under control of promoters specific for a certain lineage (such as myelin protein zero for neural crest or FoxD1 for mesenchyme) or indicating Epo or renin expression (epo-cre or renin-cre) to mice that harbor a floxed reporter gene, irreversibly tagging them even when expression of the original marker is lost upon further cell differentiation.
Published data mostly suggest that renin- and Epo-producing cells are of two distinct lineages: Renin-producing cells are considered to be derivates of FoxD1 mesenchymal cell progenitor cells, whereas Epo-producing cells have been suggested to be of neural crest origin, although existing data to support this are limited (Figure 3). Evidence for neural crest origin of Epo-producing cells stems from one study that analyzed P0-Cre;R26tdRFP;Epo-GFP triple transgenic mice (49). In this system, Cre-recombinase expression is driven by the myelin protein zero promoter, which is expressed by neural crest cells in early embryonic development (93), tagging deriving cells through recombination of the RFP reporter allele (94). These mice were crossed with reporter mice in which GFP is fused to a 180-kD chromosomal fragment containing the mouse Epo coding region and all necessary regulatory elements and displays specific Epo expression within kidney interstitial cells (95). On the basis of the observation that >75% of Epo-GFP–positive cells were also positive for RFP, it was assumed that most Epo-producing cells are of neural crest origin (information on percentage of RFP-positive cells that were also positive for GFP was not provided) (49). This study also analyzed kidneys of P0-Cre;R26ECFP double transgenic mice (in these mice, derivate cells of myelin protein zero-positive cells are tagged through expression of ECFP) and found that almost all interstitial cells in the adult kidney that expressed PDGF receptor-β and/or CD73 were also ECFP positive. The researchers concluded that almost all interstitial fibroblasts were of neural crest origin. In summary, this study suggested that Epo-producing interstitial cells are derivates of neural crest, just like all interstitial fibroblasts (Figure 3). The authors do not indicate whether Epo-producing cells are a specific subpopulation of fibroblasts or whether all fibroblasts can potentially produce Epo (49). This study somewhat contradicts a school of thought suggesting that all resident interstitial fibroblasts in adult kidneys are derivates of mesenchymal FoxD1-positive progenitor cells (27).
Figure 3.
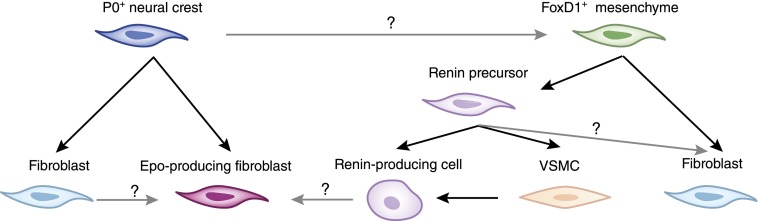
Origins of Epo- and renin-producing renal cells. Epo-producing fibroblasts are presumed to derive from myelin protein zero–positive (P0+) neural crest precursor cells. Whether all fibroblasts have the capacity to produce Epo is not yet known. Another school of thought suggests that almost all fibroblasts derive from FoxD1-positive mesenchymal progenitor cells. Renin precursor cells have been suggested to originate from FoxD1-positive precursors also. It is not yet known whether renin precursors give rise to generic fibroblasts. One study suggests the possibility that renin-producing cells can convert to Epo-producing fibroblasts. VSMC, vascular smooth muscle cell.
This thinking is based on a study that used FoxD1-GFP-Cre;R26STOPlacZ double transgenic mice and found that almost all interstitial cells in the adult kidney expressing PDGF receptor-β and/or CD73 were also LacZ positive. Those investigators concluded that almost all interstitial fibroblasts were derivates of FoxD1-positive mesenchymal progenitor cells (albeit only 20% of fibroblasts were LacZ positive when inducible FoxD1-CreER;R26STOPlacZ transgenic mice were used) (27). Neither study conclusively determined whether FoxD1 mesenchymal progenitors are of extrarenal origin (Figure 3). In this context, there is ample evidence that juxtaglomerular renin-producing cells are progeny of renin-positive precursor cells (cells that also give rise to perivascular fibroblast-like cells, which can be recruited to produce renin when needed). This concept is mainly based on studies that analyzed kidneys of Ren-1d-Cre;R26STOPlacZ and Ren-1d-Cre;Z/EG double transgenic mice as well as in Ren-1d-Cre;R26STOPlacZ ;ATGF−/− composite mice (in these mice, angiotensinogen deficiency causes additional recruitment of renin-producing cells [96]), which documented common lineage for perivascular fibroblasts and renin-producing juxtaglomerular cells (70,79). These aforementioned renin precursor cells are presumably progeny of FoxD1-positive progenitor cells, but this information has been disclosed only as “unpublished data” (Figure 3) (97). A recent study by the Kurtz group demonstrated pro-collagen production by renin precursor cells, hinting at a common lineage of renin cells and non–renin-producing fibroblasts (98).
To explore the role of hypoxia as possible stimulus for renin production, Kurtz and coworkers ablated VHL in renin-producing cells (Renin-1d-Cre;Vhllox/lox) based on the concept that absence of VHL causes HIF accumulation (due to insufficient proteolytic degradation), mimicking hypoxic signaling. Unexpected findings were that renin-producing cells were decreased (it had been widely presumed that hypoxia stimulates renin production) and that VHL-deficient juxtaglomerular cells produced Epo instead of renin (99). Although it is unclear whether such clamping of HIF at abnormally high levels (and resulting Epo overproduction and renin suppression) ever happens under in non–genetically modified conditions, this observation raises several conceptual questions. For example, future studies should determine whether renin and Epo production are interchangeable functions of a specialized interstitial cell type, whether all renal fibroblasts have potential to produce Epo or renin, or, in a broader view, what renin- and Epo-producing cells truly are.
Pathophysiology of the Renal Interstitium
Just as the renal interstitium plays an integral role in physiologic kidney function, it is a central determinant of the fate of diseased kidneys. Chronic progressive kidney injury is unequivocally associated with both quantitative and qualitative changes of the renal interstitium, which are commonly referred to as “interstitial fibrosis.” The fibrotic interstitium is no longer invisible because it substantially expands (although in normal kidneys the relative volume taken up by the interstitium is no higher than 5%–10%, it often occupies >60% of kidney tissue in a severely diseased kidney). Such expansion is predominantly caused by accumulation of extracellular matrix (ECM; fibers), ECM-producing fibroblasts, and mononuclear cells. While ECM-producing fibroblasts (and often also renin-producing cells) accumulate in the fibrosing interstitium, Epo-producing fibroblasts are lost, raising the question of whether Epo-producing fibroblasts convert into non–Epo-producing fibroblasts in CKD and whether renin-producing cells could be therapeutically converted into Epo producers (Figure 4). While numerous cell types, including resident non–hormone-producing fibroblasts, endothelial cell, epithelial cells, and bone marrow–derived fibrocytes contribute to accumulation of ECM-producing fibroblasts in kidney fibrosis (29), the contribution of hormone-producing cells to accumulation of activated fibroblasts in CKD is yet unknown. Of note, there is a glaring disconnect between groups of different research interests: Most studies to elucidate origins of fibroblasts in fibrotic kidneys, including from our groups, did not analyze possible Epo or renin expression of fibroblasts. Studies from groups with an interest in Epo- and/or renin-expressing cells reported that these cells could contribute to collagen production in CKD but did not use transgenic fate mapping systems, which are established in the fibrosis field. Impaired hormone production, however, is not the cardinal feature of the fibrosed interstitium in progressive CKD, as excretory kidney function is also affected. In fact, the extent of tubulointerstitial fibrosis is the single best determinant for requirement of RRT, even in primary glomerular nephropathies (100–102) because the expansion of the interstitium, paired with the overall kidney contraction, causes functional nephron loss through ensuing hypoxia (caused by microvascular rarefaction and hindered diffusion of oxygen and nutrients), and stenosis of glomerulotubular necks and tubular segments through physical pressure (103–105) (for in-depth information regarding the molecular events which underlie renal fibrogenesis we refer to extensive reviews elsewhere [4,105–108]).
Figure 4.

From physiology to pathophysiology of the renal interstitium. Pathologic involvement of the interstitium, so-called fibrosis, determines progression of CKD. The schematic illustrates quantitative and qualitative changes that define the switch from physiologic (left) to fibrotic (pathophysiologic) interstitium (right). The interstitium not only increases in volume due to accumulation of fibrotic extracellular matrix, but fibroblasts capable of producing collagen accumulate whereas Epo-producing fibroblasts are diminished. Renin-producing perivascular cells accumulate within the interstitium.
Outlook
Epo and renin production are two of the kidney’s most prominent functions, and these functions are localized to the fibroblasts of the renal interstitium. Thus, understanding of fibroblast origins and phenotypic plasticity is as relevant as ever. Future work will reveal whether reprogramming of fibroblasts may not only circumvent progressive deterioration of excretory kidney function but also limit relative Epo deficiency and renin overproduction in the future.
Disclosures
None.
Acknowledgments
M.Z. is supported by DFG grant ZE523/2-1. R.K. is supported by National Institutes of Health grant DK081576 and by the Cancer Prevention and Research Institute of Texas and the Cancer Metastasis Research Center.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Lemley KV, Kriz W: Anatomy of the renal interstitium. Kidney Int 39: 370–381, 1991 [DOI] [PubMed] [Google Scholar]
- 2.Kaissling B, Le Hir M: The renal cortical interstitium: Morphological and functional aspects. Histochem Cell Biol 130: 247–262, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuncio GS, Neilson EG, Haverty T: Mechanisms of tubulointerstitial fibrosis. Kidney Int 39: 550–556, 1991 [DOI] [PubMed] [Google Scholar]
- 4.Zeisberg M, Neilson EG: Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 21: 1819–1834, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Eddy AA: Molecular insights into renal interstitial fibrosis [editorial]. J Am Soc Nephrol 7: 2495–2508, 1996 [DOI] [PubMed] [Google Scholar]
- 6.Kriz W, Napiwotzky P: Structural and functional aspects of the renal interstitium. Contrib Nephrol 16: 104–108, 1979 [DOI] [PubMed] [Google Scholar]
- 7.Knepper MA, Danielson RA, Saidel GM, Post RS: Quantitative analysis of renal medullary anatomy in rats and rabbits. Kidney Int 12: 313–323, 1977 [DOI] [PubMed] [Google Scholar]
- 8.Takahashi-Iwanaga H: The three-dimensional cytoarchitecture of the interstitial tissue in the rat kidney. Cell Tissue Res 264: 269–281, 1991 [DOI] [PubMed] [Google Scholar]
- 9.Teteris SA, Engel DR, Kurts C: Homeostatic and pathogenic role of renal dendritic cells. Kidney Int 80: 139–145, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J: The renal mononuclear phagocytic system. J Am Soc Nephrol 23: 194–203, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalluri R, Zeisberg M: Fibroblasts in cancer. Nat Rev Cancer 6: 392–401, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Zeisberg M, Strutz F, Müller GA: Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol 13[Suppl 3]: S111–S120, 2000 [PubMed] [Google Scholar]
- 13.Enge M, Bjarnegård M, Gerhardt H, Gustafsson E, Kalén M, Asker N, Hammes HP, Shani M, Fässler R, Betsholtz C: Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J 21: 4307–4316, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abramsson A, Lindblom P, Betsholtz C: Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112: 1142–1151, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurtz A: Renin release: Sites, mechanisms, and control. Annu Rev Physiol 73: 377–399, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Kurtz A, Eckardt KU, Neumann R, Kaissling B, Le Hir M, Bauer C: Site of erythropoietin formation. Contrib Nephrol 76: 14–20, discussion 21–23, 1989 [DOI] [PubMed] [Google Scholar]
- 17.Komuro T: Re-evaluation of fibroblasts and fibroblast-like cells. Anat Embryol (Berl) 182: 103–112, 1990 [DOI] [PubMed] [Google Scholar]
- 18.Franke WW, Schmid E, Osborn M, Weber K: Different intermediate-sized filaments distinguished by immunofluorescence microscopy. Proc Natl Acad Sci U S A 75: 5034–5038, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazarides E, Balzer DR, Jr: Specificity of desmin to avian and mammalian muscle cells. Cell 14: 429–438, 1978 [DOI] [PubMed] [Google Scholar]
- 20.Tuszynski GP, Frank ED, Damsky CH, Buck CA, Warren L: The detection of smooth muscle desmin-like protein in BHK21/C13 fibroblasts. J Biol Chem 254: 6138–6143, 1979 [PubMed] [Google Scholar]
- 21.Gardner H, Kreidberg J, Koteliansky V, Jaenisch R: Deletion of integrin alpha 1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol 175: 301–313, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Vogel W, Gish GD, Alves F, Pawson T: The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell 1: 13–23, 1997 [DOI] [PubMed] [Google Scholar]
- 23.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG: Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130: 393–405, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marxer-Meier A, Hegyi I, Loffing J, Kaissling B: Postnatal maturation of renal cortical peritubular fibroblasts in the rat. Anat Embryol (Berl) 197: 143–153, 1998 [DOI] [PubMed] [Google Scholar]
- 25.Thompson LF, Ruedi JM, Glass A, Low MG, Lucas AH: Antibodies to 5′-nucleotidase (CD73), a glycosyl-phosphatidylinositol-anchored protein, cause human peripheral blood T cells to proliferate. J Immunol 143: 1815–1821, 1989 [PubMed] [Google Scholar]
- 26.Fellström B, Klareskog L, Heldin CH, Larsson E, Rönnstrand L, Terracio L, Tufveson G, Wahlberg J, Rubin K: Platelet-derived growth factor receptors in the kidney—upregulated expression in inflammation. Kidney Int 36: 1099–1102, 1989 [DOI] [PubMed] [Google Scholar]
- 27.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS: Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabbiani G, Kapanci Y, Barazzone P, Franke WW: Immunochemical identification of intermediate-sized filaments in human neoplastic cells. A diagnostic aid for the surgical pathologist. Am J Pathol 104: 206–216, 1981 [PMC free article] [PubMed] [Google Scholar]
- 29.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R: Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacombe C, Mayeux P: Biology of erythropoietin. Haematologica 83: 724–732, 1998 [PubMed] [Google Scholar]
- 31.Jelkmann W: Physiology and pharmacology of erythropoietin. Transfus Med Hemother 40: 302–309, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fried W, Kilbridge T, Krantz S, McDonald TP, Lange RD: Studies on extrarenal erythropoietin. J Lab Clin Med 73: 244–248, 1969 [PubMed] [Google Scholar]
- 33.Cahan C, Hoekje PL, Goldwasser E, Decker MJ, Strohl KP: Assessing the characteristic between length of hypoxic exposure and serum erythropoietin levels. Am J Physiol 258: R1016–R1021, 1990 [DOI] [PubMed] [Google Scholar]
- 34.Nangaku M, Eckardt KU: Hypoxia and the HIF system in kidney disease. J Mol Med (Berl) 85: 1325–1330, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Weidemann A, Johnson RS: Nonrenal regulation of EPO synthesis. Kidney Int 75: 682–688, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Chiang CK, Nangaku M, Tanaka T, Iwawaki T, Inagi R: Endoplasmic reticulum stress signal impairs erythropoietin production: a role for ATF4. Am J Physiol Cell Physiol 304: C342–C353, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Miescher F: Über die Beziehungen zwischen Meereshöhe und Beschaffenheit des Blutes. Korrespbl Schweiz Arz 23: 809–830, 1893 [Google Scholar]
- 38.Erslev A: Humoral regulation of red cell production. Blood 8: 349–357, 1953 [PubMed] [Google Scholar]
- 39.Wojchowski D: Eugene Goldwasser (1922-2010). Nature 470: 40, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Eckardt KU, Koury ST, Tan CC, Schuster SJ, Kaissling B, Ratcliffe PJ, Kurtz A: Distribution of erythropoietin producing cells in rat kidneys during hypoxic hypoxia. Kidney Int 43: 815–823, 1993 [DOI] [PubMed] [Google Scholar]
- 41.Pan X, Suzuki N, Hirano I, Yamazaki S, Minegishi N, Yamamoto M: Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice. PLoS ONE 6: e25839, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maxwell AP, Lappin TR, Johnston CF, Bridges JM, McGeown MG: Erythropoietin production in kidney tubular cells. Br J Haematol 74: 535–539, 1990 [DOI] [PubMed] [Google Scholar]
- 43.Unger EF, Thompson AM, Blank MJ, Temple R: Erythropoiesis-stimulating agents—time for a reevaluation. N Engl J Med 362: 189–192, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Macdougall IC: Optimizing the use of erythropoietic agents—pharmacokinetic and pharmacodynamic considerations. Nephrol Dial Transplant 17[Suppl 5]: 66–70, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Pagel H, Jelkmann W, Weiss C: O2-supply to the kidneys and the production of erythropoietin. Respir Physiol 77: 111–117, 1989 [DOI] [PubMed] [Google Scholar]
- 46.Jelkmann W: Regulation of erythropoietin production. J Physiol 589: 1251–1258, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh AK: Anemia of chronic kidney disease. Clin J Am Soc Nephrol 3: 3–6, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Dewi FR, Fatchiyah F: Methylation impact analysis of erythropoietin (EPO) gene to hypoxia inducible factor-1α (HIF-1α) activity. Bioinformation 9: 782–787, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, Yamamura K, Nagoshi N, Shibata S, Rao TN, Fehling HJ, Fukatsu A, Minegishi N, Kita T, Kimura T, Okano H, Yamamoto M, Yanagita M: Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 121: 3981–3990, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Souma T, Yamazaki S, Moriguchi T, Suzuki N, Hirano I, Pan X, Minegishi N, Abe M, Kiyomoto H, Ito S, Yamamoto M: Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol 24: 1599–1616, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernhardt WM, Wiesener MS, Scigalla P, Chou J, Schmieder RE, Günzler V, Eckardt KU: Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 21: 2151–2156, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Artunc F, Risler T: Serum erythropoietin concentrations and responses to anaemia in patients with or without chronic kidney disease. Nephrol Dial Transplant 22: 2900–2908, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Mercadal L, Metzger M, Casadevall N, Haymann JP, Karras A, Boffa JJ, Flamant M, Vrtovsnik F, Stengel B, Froissart M; NephroTest Study Group: Timing and determinants of erythropoietin deficiency in chronic kidney disease. Clin J Am Soc Nephrol 7: 35–42, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandra M, Clemons GK, McVicar MI: Relation of serum erythropoietin levels to renal excretory function: Evidence for lowered set point for erythropoietin production in chronic renal failure. J Pediatr 113: 1015–1021, 1988 [DOI] [PubMed] [Google Scholar]
- 55.Thomas M, Tsalamandris C, MacIsaac R, Jerums G: Anaemia in diabetes: An emerging complication of microvascular disease. Curr Diabetes Rev 1: 107–126, 2005 [DOI] [PubMed] [Google Scholar]
- 56.de Klerk G, Wilmink JM, Rosengarten PC, Vet RJ, Goudsmit R: Serum erythropoietin (ESF) titers in anemia of chronic renal failure. J Lab Clin Med 100: 720–734, 1982 [PubMed] [Google Scholar]
- 57.Eckardt KU, Möllmann M, Neumann R, Brunkhorst R, Burger HU, Lonnemann G, Scholz H, Keusch G, Buchholz B, Frei U: Erythropoietin in polycystic kidneys. J Clin Invest 84: 1160–1166, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inomata S, Itoh M, Imai H, Sato T: Serum levels of erythropoietin as a novel marker reflecting the severity of diabetic nephropathy. Nephron 75: 426–430, 1997 [DOI] [PubMed] [Google Scholar]
- 59.Bosman DR, Osborne CA, Marsden JT, Macdougall IC, Gardner WN, Watkins PJ: Erythropoietin response to hypoxia in patients with diabetic autonomic neuropathy and non-diabetic chronic renal failure. Diabet Med 19: 65–69, 2002 [DOI] [PubMed] [Google Scholar]
- 60.Naeshiro I, Sato K, Chatani F, Sato S: Possible mechanism for the anemia induced by candesartan cilexetil (TCV-116), an angiotensin II receptor antagonist, in rats. Eur J Pharmacol 354: 179–187, 1998 [DOI] [PubMed] [Google Scholar]
- 61.Ertürk S, Ateş K, Duman N, Karatan O, Erbay B, Ertuğ E: Unresponsiveness to recombinant human erythropoietin in haemodialysis patients: Possible implications of angiotensin-converting enzyme inhibitors. Nephrol Dial Transplant 11: 396–397, 1996 [DOI] [PubMed] [Google Scholar]
- 62.Franke K, Gassmann M, Wielockx B: Erythrocytosis: The HIF pathway in control. Blood 122: 1122–1128, 2013 [DOI] [PubMed] [Google Scholar]
- 63.Le Hir M, Kaissling B: Distribution and regulation of renal ecto-5′-nucleotidase: Implications for physiological functions of adenosine. Am J Physiol 264: F377–F387, 1993 [DOI] [PubMed] [Google Scholar]
- 64.Nishiyama A, Rahman M, Inscho EW: Role of interstitial ATP and adenosine in the regulation of renal hemodynamics and microvascular function. Hypertens Res 27: 791–804, 2004 [DOI] [PubMed] [Google Scholar]
- 65.Li L, Mizel D, Huang Y, Eisner C, Hoerl M, Thiel M, Schnermann J: Tubuloglomerular feedback and renal function in mice with targeted deletion of the type 1 equilibrative nucleoside transporter. Am J Physiol Renal Physiol 304: F382–F389, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vallon V, Mühlbauer B, Osswald H: Adenosine and kidney function. Physiol Rev 86: 901–940, 2006 [DOI] [PubMed] [Google Scholar]
- 67.Ozüyaman B, Ding Z, Buchheiser A, Koszalka P, Braun N, Gödecke A, Decking UK, Zimmermann H, Schrader J: Adenosine produced via the CD73/ecto-5′-nucleotidase pathway has no impact on erythropoietin production but is associated with reduced kidney weight. Pflugers Arch 452: 324–331, 2006 [DOI] [PubMed] [Google Scholar]
- 68.Kobori H, Nangaku M, Navar LG, Nishiyama A: The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007 [DOI] [PubMed] [Google Scholar]
- 69.Baylis C, Engels K, Hymel A, Navar LG: Plasma renin activity and metabolic clearance rate of angiotensin II in the unstressed aging rat. Mech Ageing Dev 97: 163–172, 1997 [DOI] [PubMed] [Google Scholar]
- 70.Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA: Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol 281: F345–F356, 2001 [DOI] [PubMed] [Google Scholar]
- 71.Castrop H, Höcherl K, Kurtz A, Schweda F, Todorov V, Wagner C: Physiology of kidney renin. Physiol Rev 90: 607–673, 2010 [DOI] [PubMed] [Google Scholar]
- 72.Skøtt O, Briggs JP: Direct demonstration of macula densa-mediated renin secretion. Science 237: 1618–1620, 1987 [DOI] [PubMed] [Google Scholar]
- 73.Hackenthal E, Paul M, Ganten D, Taugner R: Morphology, physiology, and molecular biology of renin secretion. Physiol Rev 70: 1067–1116, 1990 [DOI] [PubMed] [Google Scholar]
- 74.Kim SM, Chen L, Faulhaber-Walter R, Oppermann M, Huang Y, Mizel D, Briggs JP, Schnermann J: Regulation of renin secretion and expression in mice deficient in beta1- and beta2-adrenergic receptors. Hypertension 50: 103–109, 2007 [DOI] [PubMed] [Google Scholar]
- 75.Bhatt DL, Kandzari DE, O’Neill WW, D’Agostino R, Flack JM, Katzen BT, Leon MB, Liu M, Mauri L, Negoita M, Cohen SA, Oparil S, Rocha-Singh K, Townsend RR, Bakris GL; SYMPLICITY HTN-3 Investigators: A controlled trial of renal denervation for resistant hypertension. N Engl J Med 370: 1393–1401, 2014 [DOI] [PubMed] [Google Scholar]
- 76.Fisher JP, Paton JF: The sympathetic nervous system and blood pressure in humans: Implications for hypertension. J Hum Hypertens 26: 463–475, 2012 [DOI] [PubMed] [Google Scholar]
- 77.Davidson AC, Bisognano JD: Interventional approaches for resistant hypertension. Curr Opin Nephrol Hypertens 21: 475–480, 2012 [DOI] [PubMed] [Google Scholar]
- 78.Zeisberg M, Müller GA: Mechanistic insights into the antifibrotic activity of aliskiren in the kidney. Hypertens Res 35: 266–268, 2012 [DOI] [PubMed] [Google Scholar]
- 79.Sequeira López ML, Pentz ES, Nomasa T, Smithies O, Gomez RA: Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 6: 719–728, 2004 [DOI] [PubMed] [Google Scholar]
- 80.Ayan S, Roth JA, Freeman MR, Bride SH, Peters CA: Partial ureteral obstruction dysregulates the renal renin-angiotensin system in the fetal sheep kidney. Urology 58: 301–306, 2001 [DOI] [PubMed] [Google Scholar]
- 81.Mimura I, Nangaku M: The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 6: 667–678, 2010 [DOI] [PubMed] [Google Scholar]
- 82.Sealey JE, Alderman MH, Furberg CD, Laragh JH: Renin-angiotensin system blockers may create more risk than reward for sodium-depleted cardiovascular patients with high plasma renin levels. Am J Hypertens 26: 727–738, 2013 [DOI] [PubMed] [Google Scholar]
- 83.Huby AC, Kavvadas P, Alfieri C, Abed A, Toubas J, Rastaldi MP, Dussaule JC, Chatziantoniou C, Chadjichristos CE: The RenTg mice: A powerful tool to study renin-dependent chronic kidney disease. PLoS ONE 7: e52362, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tarazi RC, Frohlich ED, Dustan HP, Gifford RW, Jr, Page IH: Hypertension and high hematocrit. Another clue to renal arterial disease. Am J Cardiol 18: 855–858, 1966 [DOI] [PubMed] [Google Scholar]
- 85.Vlahakos DV, Marathias KP, Madias NE: The role of the renin-angiotensin system in the regulation of erythropoiesis. Am J Kidney Dis 56: 558–565, 2010 [DOI] [PubMed] [Google Scholar]
- 86.Kuriyama S, Tomonari H, Tokudome G, Horiguchi M, Hayashi H, Kobayashi H, Ishikawa M, Hosoya T: Antiproteinuric effects of combined antihypertensive therapies in patients with overt type 2 diabetic nephropathy. Hypertens Res 25: 849–855, 2002 [DOI] [PubMed] [Google Scholar]
- 87.Jacobsen P, Andersen S, Jensen BR, Parving HH: Additive effect of ACE inhibition and angiotensin II receptor blockade in type I diabetic patients with diabetic nephropathy. J Am Soc Nephrol 14: 992–999, 2003 [DOI] [PubMed] [Google Scholar]
- 88.Vlahakos DV, Marathias KP, Agroyannis B, Madias NE: Posttransplant erythrocytosis. Kidney Int 63: 1187–1194, 2003 [DOI] [PubMed] [Google Scholar]
- 89.Ertürk S, Nergizoğlu G, Ateş K, Duman N, Erbay B, Karatan O, Ertuğ AE: The impact of withdrawing ACE inhibitors on erythropoietin responsiveness and left ventricular hypertrophy in haemodialysis patients. Nephrol Dial Transplant 14: 1912–1916, 1999 [DOI] [PubMed] [Google Scholar]
- 90.Volpe M, Tritto C, Testa U, Rao MA, Martucci R, Mirante A, Enea I, Russo R, Rubattu S, Condorelli GL, Cangianiello S, Trimarco B, Peschle C, Condorelli M: Blood levels of erythropoietin in congestive heart failure and correlation with clinical, hemodynamic, and hormonal profiles. Am J Cardiol 74: 468–473, 1994 [DOI] [PubMed] [Google Scholar]
- 91.Eckardt KU: Cardiovascular consequences of renal anaemia and erythropoietin therapy. Nephrol Dial Transplant 14: 1317–1323, 1999 [DOI] [PubMed] [Google Scholar]
- 92.Rosario R, Epstein M: Relationship between erythropoietin administration and alterations of renin-angiotensin-aldosterone. J Renin Angiotensin Aldosterone Syst 7: 135–138, 2006 [DOI] [PubMed] [Google Scholar]
- 93.Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K: A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol 212: 191–203, 1999 [DOI] [PubMed] [Google Scholar]
- 94.Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ: Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol 37: 43–53, 2007 [DOI] [PubMed] [Google Scholar]
- 95.Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M: Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 111: 5223–5232, 2008 [DOI] [PubMed] [Google Scholar]
- 96.Kim HS, Maeda N, Oh GT, Fernandez LG, Gomez RA, Smithies O: Homeostasis in mice with genetically decreased angiotensinogen is primarily by an increased number of renin-producing cells. J Biol Chem 274: 14210–14217, 1999 [DOI] [PubMed] [Google Scholar]
- 97.Sequeira Lopez ML, Gomez RA: Development of the renal arterioles. J Am Soc Nephrol 22: 2156–2165, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Karger C, Kurtz F, Steppan D, Schwarzensteiner I, Machura K, Angel P, Banas B, Risteli J, Kurtz A: Procollagen I-expressing renin cell precursors. Am J Physiol Renal Physiol 305: F355–F361, 2013 [DOI] [PubMed] [Google Scholar]
- 99.Kurt B, Paliege A, Willam C, Schwarzensteiner I, Schucht K, Neymeyer H, Sequeira-Lopez ML, Bachmann S, Gomez RA, Eckardt KU, Kurtz A: Deletion of von Hippel-Lindau protein converts renin-producing cells into erythropoietin-producing cells. J Am Soc Nephrol 24: 433–444, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bohle A, Bader R, Grund KE, Mackensen S, Neunhoeffer J: Serum creatinine concentration and renal interstitial volume. Analysis of correlations in endocapillary (acute) glomerulonephritis and in moderately severe mesangioproliferative glomerulonephritis. Virchows Arch A Pathol Anat Histol 375: 87–96, 1977 [DOI] [PubMed] [Google Scholar]
- 101.Bohle A, Christ H, Grund KE, Mackensen S: The role of the interstitium of the renal cortex in renal disease. Contrib Nephrol 16: 109–114, 1979 [DOI] [PubMed] [Google Scholar]
- 102.Risdon RA, Sloper JC, De Wardener HE: Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet 2: 363–366, 1968 [DOI] [PubMed] [Google Scholar]
- 103.Cohen EP: Fibrosis causes progressive kidney failure. Med Hypotheses 45: 459–462, 1995 [DOI] [PubMed] [Google Scholar]
- 104.Cohen EP, Regner K, Fish BL, Moulder JE: Stenotic glomerulotubular necks in radiation nephropathy. J Pathol 190: 484–488, 2000 [DOI] [PubMed] [Google Scholar]
- 105.Becker GJ, Hewitson TD: The role of tubulointerstitial injury in chronic renal failure. Curr Opin Nephrol Hypertens 9: 133–138, 2000 [DOI] [PubMed] [Google Scholar]
- 106.Zeisberg M, Kalluri R: Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 304: C216–C225, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eddy AA: Progression in chronic kidney disease. Adv Chronic Kidney Dis 12: 353–365, 2005 [DOI] [PubMed] [Google Scholar]
- 108.Remuzzi G, Bertani T: Pathophysiology of progressive nephropathies. N Engl J Med 339: 1448–1456, 1998 [DOI] [PubMed] [Google Scholar]