

Abstract
In the short time since its initial discovery as the cause of rare hypophosphatemic disorders, fibroblast growth factor-23 (FGF-23) has emerged as a major regulator of mineral metabolism and critical component of the bone and mineral adaptation to CKD. However, because elevated FGF-23 levels are also a novel biomarker and possible molecular mediator of increased risks of cardiovascular disease and death in CKD, the initially adaptive response to increase FGF-23 levels to maintain neutral phosphate balance in CKD may ultimately become maladaptive. Incorporating FGF-23 into understanding the complex physiology that governs normal bone and mineral metabolism and its alterations in CKD has filled critical knowledge gaps and opened a new landscape of exciting hypotheses and novel therapeutic strategies to be tested in the continued quest to alleviate the burden of CKD.
Keywords: phosphate, FGF-23, calcium
Introduction
Research of disease mechanisms in the area of CKD can be broadly categorized into two general groups: research on how the kidney is injured and research on how the presence of kidney disease induces injury of other organs (Figure 1). Working in the second space, our group investigates mechanisms of cardiovascular disease in patients with CKD. We emphasize disordered mineral metabolism and, specifically, elevated levels of the bone-derived hormone fibroblast growth factor-23 (FGF-23) as possible novel and modifiable mechanisms of the increased cardiovascular risk that is conferred by CKD.
Figure 1.

Types of research in CKD. Research in the area of CKD can be broadly categorized into studies that investigate mechanisms of how the kidney is injured (left), and mechanisms of how the presence of kidney injury induces systemic damage to other organs (right). Interrupting either set of disease mechanisms could help drive improved clinical outcomes in patients with CKD.
Origin
The origins of FGF-23 investigation in CKD can be traced back to the seminal finding in the late 1990s that higher serum phosphate levels were independently associated with higher risk of death in patients with ESRD undergoing hemodialysis (1). Subsequent studies validated the initial reports and demonstrated qualitatively similar results in patients with earlier stages of CKD and even the general population (2–6). This body of work elevated serum phosphate from the crowd of boxes to be checked on the monthly dialysis rounds report to potential novel risk factor for adverse outcomes in CKD and perhaps beyond.
Bone, parathyroid glands, and gastrointestinal tract contribute to phosphate homeostasis, but the kidney is primarily responsible for regulating the serum phosphate concentration (7). Nonetheless, in the setting of even severe CKD, the serum phosphate is usually maintained within the normal range (8–10). This requires tremendous adaptation. Because phosphate is freely filtered at the glomerulus, the total amount of filtered phosphate decreases in parallel to the reduction in GFR imposed by CKD. Modest increases in serum phosphate levels within the normal range help sustain the filtered load in CKD, but downregulation of phosphate reabsorption by the sodium-phosphate co-transporters, NPT2a and NPT2c, in the apical membrane of the proximal tubule appears to be the most important component of the adaptive response (11,12). Since individuals with normal GFR who consume typical Western diets excrete only 10%–20% of their filtered load of phosphate—the fractional excretion of phosphate—the kidney can readily respond to reduced filtration of phosphate by commensurately decreasing the percentage that NPT2a and NPT2c reabsorb. Indeed, fractional excretion of phosphate often increases from 10%–20% to ≥50% in CKD (9,10,13,14).
Conundrum
How the kidney “knows” to adapt to reduced GFR by excreting a greater proportion of filtered phosphate has long vexed the field. Given its known phosphaturic effects and elevated levels in patients with CKD, parathyroid hormone (PTH) was considered a likely mediator (14). However, creative clinical investigation suggested a more complex mechanism (Figure 2A). Restricting dietary phosphate intake increased circulating levels of 1,25-dihydroxyvitamin D and reduced PTH levels in patients with CKD (15). This demonstrated that 1,25-dihydroxyvitamin D levels are dynamically regulated in CKD and can be modulated by manipulating dietary phosphate intake. Furthermore, this finding cast doubt on PTH as the primary mediator of the phosphaturic response to CKD. If PTH were the primary messenger that modulates renal phosphate reabsorption in response to varying phosphate loads and to CKD, reduced PTH in response to decreased dietary phosphate intake should have been accompanied by a secondary decrease in 1,25-dihydroxyvitamin D levels, the synthesis of which is stimulated by PTH. Instead, restricting dietary phosphate intake increased 1,25-dihydroxyvitamin D levels, suggesting that another unknown factor dynamically regulates renal production of 1,25-dihydroxyvitamin D and is upstream of PTH in phosphate regulation in CKD.
Figure 2.

Critical role of the phosphate–fibroblast growth factor-23 (FGF-23) axis in the dynamic regulation of mineral metabolism in CKD. (A) In patients with CKD, restricting dietary phosphate intake increased levels of 1,25-dihydroxyvitamin D and decreased parathyroid hormone (PTH) levels, whereas phosphate supplementation decreased 1,25-dihydroxyvitamin D levels and increased PTH levels. These data suggested the presence of another, as of then undiscovered, vitamin D–regulating hormone that responds to alterations in dietary phosphate loading. Adapted from reference 15 with permission from the American Society of Clinical Investigation. (B) Neutralizing FGF-23 using monoclonal antibodies in rats with CKD due to anti–glomerular basement membrane disease precipitated severe hyperphosphatemia and fully restored 1,25-dihydroxyvitamin D levels to normal. These data established that FGF-23 was the critical “missing” factor that maintains normal serum phosphate levels and suppresses 1,25-dihydroxyvitamin D production in CKD. Adapted from reference 44 with permission from the Nature Publishing Group.
Breakthrough
Investigation of rare causes of vitamin D–resistant rickets provided the breakthrough. Autosomal dominant hypophosphatemic rickets, autosomal recessive hypophosphatemic rickets, X-linked hypophosphatemia, and tumor-induced osteomalacia are a group of rare diseases that share a common biochemical phenotype: Hypophosphatemia caused by renal phosphate wasting; inappropriately low levels of 1,25-dihydroxyvitamin D for the degree of hypophosphatemia; and rickets or osteomalacia, depending on the age of onset (16). Affected patients’ variable levels of PTH, their low levels of 1,25-dihydroxyvitamin D, and the absence of hypercalcemia strongly suggested that PTH was not the offending phosphaturic hormone underlying the phenotype. Investigators reasoned that another circulating factor, a yet to be discovered “phosphatonin,” likely regulates phosphate homeostasis.
Modern genomics opened the field. Studies of families with autosomal dominant hypophosphatemic rickets demonstrated that affected individuals had activating mutations in the gene encoding FGF-23, the last member of the FGF family to be identified (17). Almost simultaneously, microarray analyses revealed markedly increased expression of FGF-23 in tumors that caused tumor-induced osteomalacia (18). Additional research confirmed that the final common pathway in the family of rare hypophosphatemic syndromes is elevated circulating concentrations of FGF-23 (19–21).
Physiology
After the initial discovery of FGF-23 in rare hypophosphatemic disorders, extensive human and animal research rapidly advanced understanding of normal FGF-23 physiology. An endocrine hormone that is secreted by osteocytes and osteoblasts, FGF-23 stimulates renal phosphate excretion by downregulating NPT2a and NPT2c in the proximal tubule (22,23). PTH also stimulates phosphaturia by downregulating these phosphate transporters, but the effects of FGF-23 and PTH on vitamin D regulation diverge (24). Unlike PTH, which stimulates production of 1,25-dihydroxyvitamin D and is suppressed by resulting increases in circulating 1,25-dihydroxyvitamin D concentrations as part of a classic negative endocrine feedback loop, the feedback loop between FGF-23 and 1,25-dihydroxyvitamin D spins in exactly the opposite direction. By downregulating CYP27B1, also known as the renal 1-α hydroxylase, FGF-23 is the primary inhibitor of 1,25-dihydroxyvitamin D production, and 1,25-dihydroxyvitamin D is a major stimulus of FGF-23 production in bone (23,25,26). FGF-23 and PTH also exert opposing effects on CYP24A1, which encodes the 24-hydroxylase that degrades vitamin D metabolites: FGF-23 stimulates CYP24A1, whereas PTH inhibits it (23,26,27).
The main stimuli of FGF-23 production are 1,25-dihydroxyvitamin D and dietary phosphate consumption (28–31). Interestingly, the latter does not appear to be mediated by intermediate changes in serum phosphate, which does not clearly regulate FGF-23 directly (32–34). Indeed, the mechanism of how dietary phosphate intake stimulates bone production of FGF-23 remains unknown. Likewise, how circulating FGF-23 is degraded and removed from the circulation is also unknown. PTH also stimulates FGF-23 as part of a negative endocrine feedback loop they share (35). High serum calcium is another recently appreciated stimulus of FGF-23 (36–38). This makes sense when considered in the broader context of global regulation of mineral metabolism and the parallel in PTH physiology. PTH is primarily the calcium-regulating hormone, but it promotes phosphaturia as a secondary effect. Because PTH-induced bone resorption liberates both calcium and phosphate from bone, PTH-mediated phosphaturia enables pulses of PTH secretion to specifically raise serum calcium without simultaneously raising serum phosphate. Similarly, FGF-23 as phosphate-regulating hormone is secondarily regulated by changes in serum calcium.
The discovery of α-klotho was another recent and major milestone in mineral metabolism research (39). Interruption of the α-klotho gene caused a syndrome of accelerated aging that featured hyperphosphatemia, increased 1,25-dihydroxyvitamin D concentrations, and extensive arterial calcification, similar to FGF-23 deletion (22). Further research demonstrated that α-klotho heterodimerizes with FGF receptors to form a high-affinity receptor for FGF-23 in its classic target cells, including the kidney and parathyroid glands (40). Thus, inactivating mutations in α-klotho or FGF-23 yield similar mineral metabolism phenotypes.
Adaptation
The birth of ELISAs that could precisely measure circulating FGF-23 concentrations was another major breakthrough that propelled the field (21). Armed with this powerful new tool, investigators demonstrated that FGF-23 levels rise as kidney function declines (8,41,42). In our effort to investigate the epidemiology of FGF-23 in CKD, we collaborated with the investigators for the Chronic Renal Insufficiency Cohort (CRIC) study, a prospective observational cohort study that enrolled nearly 4000 patients with intermediate stages of CKD in an effort to identify risk factors for cardiovascular disease, progression of CKD, and death (43).
Working with the CRIC study investigators, we demonstrated that lower eGFR is strongly associated with higher FGF-23 levels (10). Even among individuals with early CKD, marked by modest eGFR reduction, FGF-23 levels are often significantly elevated. Furthermore, FGF-23 elevation appears to antedate significant increases in PTH or serum phosphate levels. Although these data suggested that FGF-23 rises progressively as CKD advances and that FGF-23 elevation precedes other alterations of mineral metabolism in CKD, there are few human data on how changes in mineral metabolites evolve over time within individuals with CKD. In the interim, a seminal animal study provided critical insight into the chronology of disordered mineral metabolism in CKD (44).
After inducing CKD in rats by administering anti–glomerular basement membrane antibodies, the investigators demonstrated significantly elevated FGF-23 coincident with the earliest, most subtle increase in serum creatinine. Only later, after the FGF-23 was dramatically elevated, did they observe progressive 1,25-dihydroxyvitamin D deficiency that was accompanied by elevated PTH. Still later, they finally observed increases in serum phosphate, but this occurred only after kidney disease was severe, serum creatinine was obviously elevated, and FGF-23 levels were increased 10- to 100-fold.
In addition to unraveling temporal aspects of the pathophysiologic cascade, this study reported two other landmark findings. To help deduce the effects of FGF-23 in CKD, the investigators administered neutralizing monoclonal antibodies against FGF-23 to the rats with established CKD. The FGF-23 neutralizing antibodies increased serum phosphate levels rapidly and severely (Figure 2B). This indicated that FGF-23 is critical for maintaining normal serum phosphate levels in CKD. Furthermore, neutralizing FGF-23 completely normalized 1,25-dihydroxyvitamin D levels, despite no improvement in kidney function (Figure 2B). This finding powerfully contradicted the prevailing pathophysiologic paradigm that emphasized the kidney’s inability to make adequate amounts of 1,25-dihydroxyvitamin D as a central mechanism of disordered mineral metabolism in CKD. In actuality, the kidney retains that ability in CKD, as was shown earlier in humans fed low-phosphate diets (Figure 2A) (15). Instead, the primary mechanism of 1,25-dihydroxyvitamin D deficiency in CKD is inhibition of its production, and the messenger of that inhibitory signal is elevated FGF-23.
The discovery of FGF-23 has forced us to revisit fundamental aspects of how CKD alters bone and mineral metabolism (45). In the updated, post–FGF-23 paradigm, the earliest alteration of mineral metabolism in CKD appears to be increased circulating FGF-23 levels, which suppresses renal production of 1,25-dihydroxyvitamin D and accelerates its degradation (Figure 3). Once 1,25-dihydroxyvitamin D deficiency becomes severe enough to impede gastrointestinal calcium absorption and exert inadequate feedback inhibition of PTH production, secondary hyperparathyroidism ensues. Perhaps most important from a clinical perspective, all of these adaptations become firmly entrenched behind a shroud of normal serum phosphate levels because hyperphosphatemia occurs only after patients reach advanced CKD or even ESRD (9,10).
Figure 3.

Cascade of disordered mineral metabolism in CKD in the “post–fibroblast growth factor-23 [FGF-23]” era. Increased FGF-23 is currently the earliest detectable alteration in mineral metabolism in CKD. Gradually increasing FGF-23 levels suppress circulating concentrations of 1,25-dihydroxyvitamin D leading to secondary hyperparathyroidism. Elevated levels of FGF-23 and parathyroid hormone combine with reduced 1,25-dihydroxyvitamin D levels to maintain normal serum phosphate levels. Hyperphosphatemia results only after these adaptive responses are no longer able to overcome critical reductions in GFR, which usually does not occur until severely advanced CKD or even ESRD. Adapted from reference 45 with permission from the American Society of Nephrology.
Opportunity
Recognizing that phosphate homeostasis is severely deranged before the serum phosphate levels are overtly elevated presents an important opportunity to enhance clinical management of patients with CKD. The current approach to managing abnormal phosphate homeostasis in CKD is to treat patients with phosphate-lowering therapies only after they manifest overt hyperphosphatemia (46,47). This means that treatment candidacy can begin only after the eGFR declines to <15–20 ml/min per 1.73 m2 and often, not until patients reach ESRD (Figure 3). In the future, perhaps we will use FGF-23 testing to identify which patients with CKD and normal serum phosphate levels actually have abnormal phosphate homeostasis that should justify their candidacy for early treatment. Using elevated FGF-23 as an early warning system could translate into years of earlier delivery of treatment compared with the current approach. This strategy should not be foreign to nephrologists. Most patients with established secondary hyperparathyroidism have normal serum calcium levels. Yet, we often prescribe them PTH-lowering agents to prevent and treat complications, such as high-turnover bone disease, because we recognize that high PTH reflects disordered calcium homeostasis regardless of the normal serum calcium. More important, we understand that the serum calcium is often normal because of the secondary increases in PTH. We should test whether it would be beneficial to apply a similar approach to disordered phosphate homeostasis by beginning treatment when FGF-23 levels are elevated while serum phosphate levels are still normal.
Biomarker
To generate support for using FGF-23 testing to guide earlier delivery of treatment of disordered phosphate homeostasis, we tested whether elevated FGF-23 can serve as a biomarker of risks of cardiovascular disease and death in CKD. In our first exploration, we investigated FGF-23 levels and risk of death among incident hemodialysis patients in the Accelerated Mortality on Renal Replacement (ArMORR) study (48). In a prospective nested case-control study of 200 patients who died during the first year after initiating hemodialysis and 200 patients who survived the first year, higher FGF-23 levels, measured on the first day of outpatient hemodialysis, were independently associated with higher risk of death (Figure 4A). The risk of death increased in a stepwise pattern with stepwise increases in FGF-23 levels, and the magnitude of the FGF-23 effect was substantially larger than the analogous results for serum phosphate (Figure 4B).
Figure 4.
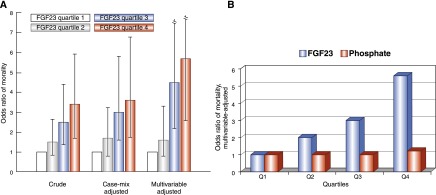
Elevated fibroblast growth factor-23 (FGF-23) is a powerful risk factor for death that surpasses elevated serum phosphate. (A) In a prospective cohort study, higher levels of FGF-23 on the first day of long-term outpatient hemodialysis were independently associated with increased risk of 1-year mortality on dialysis. The results were noteworthy for the large magnitude of effect, the monotonic graded effect across the range of baseline FGF-23 levels, and the minimal amount of confounding that is indicated by qualitatively similar results across the three different multivariable modeling strategies. Adapted from reference 48 with permission from the Massachusetts Medical Society. (B) As a biomarker of risk of death on dialysis, the strong monotonic effect of elevated FGF-23 vastly overshadowed the effect of higher serum phosphate, as shown by the corresponding odds ratios for each that were derived from the fully adjusted multivariable model. Adapted from reference 48 with permission from the Massachusetts Medical Society.
The results were especially noteworthy for their striking lack of confounding. Regardless of which covariates we included in the multivariable-adjusted models, the estimates of risk of death according to baseline FGF-23 levels were qualitatively unchanged (Figure 4A). This is unusual for epidemiologic studies in general but especially unusual for dialysis studies because so many factors influence survival on dialysis. When investigators account for these confounding factors in multivariable models, the relationship between the exposure and outcome of interest usually changes substantially and, in some cases, can reverse completely. For example, in the observational studies that established elevated serum phosphate as an independent risk factor for death in patents with ESRD, higher phosphate was actually associated with lower risk of death in unadjusted analyses (2). Only after multivariable adjustment did the now-familiar link between hyperphosphatemia and increased mortality emerge.
Our finding of a large-magnitude, monotonic association between FGF-23 and mortality that was minimally confounded by other survival factors elicited an important pivot in our thinking. We initially embarked on our analysis of the ArMORR study aiming to demonstrate that elevated FGF-23 could serve as a superior biomarker of phosphate-related risk of death relative to the serum phosphate itself. However, we emerged from the ArMORR study wondering whether elevated FGF-23 may instead be a contributing culprit in CKD-associated death rather than simply being a predictive biomarker of it. For the first time, we considered that some of the risk of death that had been attributed to hyperphosphatemia in the pre–FGF-23 era might be truly attributable to underlying, previously undiscovered and thus unmeasured FGF-23 elevations.
As was the case for phosphate, once FGF-23 was identified and validated as a risk factor for death in ESRD, subsequent studies investigated it in earlier stages of CKD. Among the nearly 4000 patients with CKD stage 2–4 in the CRIC study, we reported that higher baseline FGF-23 at enrollment was independently associated with significantly increased risk of death during the subsequent 5 years of follow-up (49). Similar to the ESRD data, the results demonstrated a minimally confounded, monotonically graded association between elevated FGF-23 levels and higher mortality risk. Other studies reported similar results (50,51).
Because cardiovascular disease is the leading cause of death in CKD, we postulated that higher cardiovascular risk was likely an important underlying mechanism to explain the strong link between elevated FGF-23 and mortality. Several prospective cohort studies demonstrated that elevated FGF-23 levels are associated with higher risk of cardiovascular events, but the strongest link was usually with congestive heart failure (52–54). Heart failure is a common cardiovascular complication of CKD with a multifactorial and complex pathophysiology, but one important mechanism is left ventricular hypertrophy (LVH), which is known to be a leading pattern of cardiovascular injury in CKD (55). Presence of LVH is consistently associated with mortality in diverse populations, including CKD, and likely contributes mechanistically to death by promoting diastolic heart failure through hypertrophy and eventually dysfunction of cardiac myocytes, and by increasing risk of arrhythmogenic sudden death due to microvascular ischemia and intramyocardial fibrosis that interferes with normal cardiac conduction (56–58).
Maladaptation
Stimulated by our initial finding that higher FGF-23 was independently associated with greater left ventricular mass in a small cross-sectional study of patients with CKD (59), and by studies that implicated FGF2 in the molecular pathogenesis of LVH (60–62), we investigated whether FGF-23 might coopt similar FGF receptor signaling pathways to contribute mechanistically to development of LVH in CKD. In an analysis of the 3000 CRIC study participants who underwent echocardiography, we found that higher FGF-23 was independently associated with greater left ventricular mass and higher prevalence of LVH (63). Several other cross-sectional studies similarly reported strong associations between elevated FGF-23 and LVH (64,65). We extended these results by demonstrating that higher FGF-23 levels were also prospectively associated with increased risk of developing new-onset LVH in the subgroup of CRIC Study participants who demonstrated no evidence of LVH at baseline and underwent a second echocardiographic examination approximately 3 years later (63).
To test whether a direct effect of FGF-23 on cardiac myocytes could explain these associative human data, we cultured neonatal rat ventricular myocytes in the presence of increasing concentrations of FGF-23, using FGF2 as the positive control. Similar to FGF-2, FGF-23 induced hypertrophy of the cardiac myocytes. We could block the effect by co-incubating the cells with a pan–FGF receptor inhibitor, indicating that the prohypertrophic effects of FGF-23 on cardiac myocytes were FGF receptor-dependent (63).
Besides supporting a direct effect of FGF-23 on cardiac myocytes, this experiment was important because it demonstrated that FGF-23 could have direct effects on cells that do not express α-klotho, such as cardiac myocytes. This challenged the ingrained dogma at the time that α-klotho was an obligate FGF-23 co-receptor without which FGF-23 could not exert effects. However, certain effects of FGF-23 may have escaped detection in the studies that established this view. When defining the tissue distribution of FGF-23 actions, investigators injected FGF-23 and measured activation of Ras-mitogen–activated protein kinase (MAPK) signaling as the read-out of a positive FGF-23 response because this pathway was known to mediate the α-klotho–dependent actions of FGF-23 in the kidney (40). However, focusing exclusively on Ras-MAPK activation to infer FGF-23 activity would fail to detect activation of other signaling pathways by FGF-23. Indeed, we reported that the α-klotho–independent effects of FGF-23 on cardiac myocytes were primarily mediated by activation of the phospholipase Cγ-calcineurin-nuclear factor of activated T cells (NFAT) cascade rather than the Ras-MAPK cascade (63). Importantly, activation of the PLCγ-calcineurin-NFAT signaling in cardiac myocytes is a known mechanism of pathologic cardiac hypertrophy due to other stimuli (66).
To test whether FGF-23 could induce LVH in vivo, we conducted a series of experiments in rodents in which we raised FGF-23 levels through genetic or pharmacologic approaches (63). In each case, LVH resulted. Most relevant to CKD, we investigated the 5/6 nephrectomy rat model of CKD, which is known to develop severe hypertension and LVH along with markedly elevated FGF-23 levels. When we injected 5/6 nephrectomy rats with a pan–FGF receptor inhibitor, beginning at the time of nephrectomy, we prevented LVH. Furthermore, administering the pan–FGF receptor inhibitor to 5/6 nephrectomy rats after they had already developed LVH reversed established LVH and reduced intramyocardial fibrosis (67). These data support FGF receptor activation as an important mechanism of LVH in 5/6 nephrectomy rats but do not provide direct evidence that the FGF receptor inhibitor specifically prevented FGF-23–induced cardiac hypertrophy. Additional validation studies are needed.
If elevated FGF-23 does contribute directly to the pathogenesis of cardiac hypertrophy and heart failure in CKD, one logical therapeutic approach to prevent these adverse effects would be to target FGF-23 itself to eliminate its effects, for example, using monoclonal neutralizing antibodies. Unfortunately, this approach increased mortality in 5/6 nephrectomy rats, which developed severe hyperphosphatemia and arterial calcification, which is another leading pattern of cardiovascular injury in patients with CKD that is strongly associated with increased risk of death (68,69).
The role of FGF-23 in arterial calcification is uncertain. Some clinical studies suggest an association and others found none (59,70). Variable study designs, sample sizes, and approaches to adjusting for confounding likely underlie this heterogeneity. We found no association between FGF-23 and coronary artery calcification in the 1500 CRIC study participants who underwent computed tomography of the coronary arteries (71). In complementary laboratory experiments, FGF-23 treatment, alone or in combination with exogenous α-klotho, could not induce calcification of isolated vascular smooth muscle cells or explanted aortic rings. In contrast, higher serum phosphate, even within the normal range, was independently associated with greater prevalence of coronary artery calcification, and higher phosphate concentrations dose-dependently induced ex vivo vascular calcification.
The procalcification effects of phosphate have been reported previously and are well known to the CKD research and clinical communities (72–75). However, this was the first study of phosphate and calcification that adjusted for FGF-23 and demonstrated that the phosphate effects were clearly independent of FGF-23 (71). Previously, it was unclear whether the association of higher serum phosphate with calcification would “hold up” to adjustment for FGF-23 given that many previously reported associations of phosphate with adverse outcomes, notably mortality and cardiovascular events, were substantially attenuated when FGF-23 was incorporated into the analyses (Figure 4B).
Pivot
Our calcification results stimulated yet another shift in our view of how disordered phosphate homeostasis influences cardiovascular disease in CKD. Whereas we initially hypothesized that FGF-23 could be a superior biomarker of toxicity that is caused directly by high serum phosphate, we later veered to the other end of the spectrum to propose that elevated FGF-23 might be the true villain causing much of the toxicity that was perhaps erroneously attributed to phosphate in the pre-FGF-23 era. Now, we shifted to an intermediate position between these extremes: FGF-23 directly promotes cardiac injury, and hyperphosphatemia directly promotes arterial injury. Together, these nonoverlapping and perhaps synergistic toxicities contribute causally to the high rates of cardiovascular disease in CKD. This construct helps to explain the lethality of the monoclonal FGF-23 antibodies in 5/6 nephrectomy rats (68), which completely abrogated all FGF-23 effects, causing severe hyperphosphatemia, malignant calcification, and death. Like other hormones, both severe FGF-23 deficiency and severe FGF-23 excess are pathologic, and what constitutes an initial adaptation in CKD can eventually deteriorate into maladaptation (Figure 5).
Figure 5.

Mineral metabolism in CKD: adaptation devolves into maladaptation. (A) Adaptation: High dietary phosphate intake and reduced phosphate excretory capacity due to CKD stimulate bone production of fibroblast growth factor-23 (FGF-23). Increased FGF-23 levels stimulate phosphaturia and reduce circulating concentrations of 1,25-dihydroxyvitamin D, which decreases the efficiency of intestinal calcium and phosphate absorption. Secondary increases in parathyroid hormone levels in response to impaired calcium absorption further enhance phosphaturia and stimulate bone resorption. Collectively, these adaptations result in normal serum calcium and phosphate levels. (B) Maladaptation: Continuous activation of these initially adaptive pathways due to unrelenting CKD may lead ultimately to maladaptive adverse effects in several end organs, including cardiac hypertrophy that promotes heart failure, progression of CKD to ESRD, and renal osteodystrophy that increases risk of fracture.
Intervention
An updated construct that elevates both FGF-23 and phosphate excess as mediators of cardiovascular toxicity in CKD has important therapeutic implications. Rather than embracing approaches that target reduction of FGF-23 or phosphate at the expense of elevated levels of the other, we need to develop therapeutic strategies that address both phosphate and FGF-23 excess simultaneously (76). Ideally, optimal interventions will also increase renal expression of α-klotho, deficiency of which is also implicated in the pathogenesis of both arterial calcification and LVH (77,78).
Because dietary phosphate consumption is a leading stimulus of FGF-23 production, reducing phosphate intake through public policy might be one approach to simultaneously attenuate FGF-23 and phosphate toxicity. With no mandate in the United States for companies to list specific phosphate-based ingredients or total phosphate content on food labels, individuals can unknowingly consume large amounts of readily absorbable inorganic phosphate in their diets because of the frequent use of phosphate-based additives in the food supply (79). This may be especially problematic in lower-socioeconomic communities; these communities may be prone to consume larger amounts of processed foods that contain the most added phosphate because these items are cheaper than fresh, unprocessed foods (80). Public policy approaches might focus on regulating use of phosphate-based additives or trying to alleviate the negative dietary impact of “food deserts.” These are neighborhoods with limited access to fresh healthy foods that are disproportionately populated by minorities and the socioeconomically disadvantaged, which are the same populations with the highest rates of CKD.
Among patients with established CKD, we should test whether earlier delivery of dietary interventions and treatments that block phosphate absorption, for example, low-phosphate diet, phosphate binders, niacin derivatives, and newer experimental therapies, can reduce the load of phosphate that must be excreted and thereby reduce FGF-23 levels without raising serum phosphate. To date, small studies suggest that diets containing higher proportions of vegetarian- versus meat-based protein sources may lower FGF-23 levels in CKD (81), but pilot clinical trials aimed at lowering FGF-23 in CKD using phosphate binders have yielded inconsistent and often disappointing results (82,83).
Finally, we should consider other approaches that leverage novel molecular targets defined by cutting-edge research. One potentially promising approach is selective blockade of the culprit FGF receptor that mediates the deleterious cardiac effects of FGF-23. The mammalian genome encodes four FGF receptor isoforms (84). FGF receptor 1 is thought to be the most important isoform that accounts for the α-klotho–dependent effects of FGF-23 in the kidney (40). On the basis of the different signaling pathways activated by FGF-23 in the heart versus the kidney, we hypothesize that a different FGF receptor isoform likely mediates the α-klotho–independent effects of FGF-23 on the heart. If so, it might be possible to develop therapies that selectively block the FGF receptor isoform that mediates the undesirable effects of FGF-23 without sacrificing the desirable effects of FGF-23 to maintain normal serum phosphate levels in the face of advancing CKD.
Disclosures
M.W. has received research support, served as a consultant, or received honoraria from Amgen, Astra Zeneca, DiaSorin, Keryx, Luitpold, Opko, Pfizer, Sanofi, and Shire.
Acknowledgments
I thank the American Society of Nephrology and the American Heart Association for this award. Along with the National Institutes of Health, the American Society of Nephrology and the American Heart Association were instrumental in supporting my research endeavors and career development through generous grant support.
Many investigators in the broad areas of mineral metabolism paved the road we travelled to study FGF-23 in CKD. My contributions are simply a natural extension of theirs. During the address at Renal Week 2014, I acknowledged in pictures many, but unfortunately, not possibly all of the scientists whose seminal findings were the foundation of my team’s efforts.
Like never before, science is a team effort. All of the data I presented from our group represent the fruits of numerous and invaluable local and worldwide collaborations.
I owe much of my personal success to spectacular mentorship by Ravi Thadhani, and to many individuals whose leadership helped support my career development across three outstanding and nurturing academic medical centers, the Massachusetts General Hospital, the University of Miami Miller School of Medicine, and the Feinberg School of Medicine at Northwestern University.
Finally, but most importantly, this award would have remained a fantasy were it not for the great support of my wonderful immediate and extended families.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Block GA, Hulbert-Shearon TE, Levin NW, Port FK: Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am J Kidney Dis 31: 607–617, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM: Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 15: 2208–2218, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, Sherrard DJ, Andress DL: Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 16: 520–528, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G, Cholesterol And Recurrent Events Trial Investigators : Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 112: 2627–2633, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Kalantar-Zadeh K, Kuwae N, Regidor DL, Kovesdy CP, Kilpatrick RD, Shinaberger CS, McAllister CJ, Budoff MJ, Salusky IB, Kopple JD: Survival predictability of time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int 70: 771–780, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Dhingra R, Sullivan LM, Fox CS, Wang TJ, D’Agostino RB, Sr, Gaziano JM, Vasan RS: Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 167: 879–885, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Murer H, Hernando N, Forster I, Biber J: Proximal tubular phosphate reabsorption: Molecular mechanisms. Physiol Rev 80: 1373–1409, 2000 [DOI] [PubMed] [Google Scholar]
- 8.Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Jüppner H, Wolf M: Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 16: 2205–2215, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Craver L, Marco MP, Martínez I, Rue M, Borràs M, Martín ML, Sarró F, Valdivielso JM, Fernández E: Mineral metabolism parameters throughout chronic kidney disease stages 1-5—achievement of K/DOQI target ranges. Nephrol Dial Transplant 22: 1171–1176, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M: Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79: 1370–1378, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slatopolsky E, Gradowska L, Kashemsant C, Keltner R, Manley C, Bricker NS: The control of phosphate excretion in uremia. J Clin Invest 45: 672–677, 1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prié D, Ureña Torres P, Friedlander G: Latest findings in phosphate homeostasis. Kidney Int 75: 882–889, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Slatopolsky E, Robson AM, Elkan I, Bricker NS: Control of phosphate excretion in uremic man. J Clin Invest 47: 1865–1874, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slatopolsky E, Caglar S, Gradowska L, Canterbury J, Reiss E, Bricker NS: On the prevention of secondary hyperparathyroidism in experimental chronic renal disease using “proportional reduction” of dietary phosphorus intake. Kidney Int 2: 147–151, 1972 [DOI] [PubMed] [Google Scholar]
- 15.Portale AA, Booth BE, Halloran BP, Morris RC, Jr: Effect of dietary phosphorus on circulating concentrations of 1,25-dihydroxyvitamin D and immunoreactive parathyroid hormone in children with moderate renal insufficiency. J Clin Invest 73: 1580–1589, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carpenter TO: The expanding family of hypophosphatemic syndromes. J Bone Miner Metab 30: 1–9, 2012 [DOI] [PubMed] [Google Scholar]
- 17.ADHR Consortium : Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26: 345–348, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T: Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 98: 6500–6505, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Jüppner H, Econs MJ: The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab 86: 497–500, 2001 [DOI] [PubMed] [Google Scholar]
- 20.De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY, Kumar R, Levine MA, Schiavi SC: Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res 17: 1102–1110, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Jüppner H: Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348: 1656–1663, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T: Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113: 561–568, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T: FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19: 429–435, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Kempson SA, Lötscher M, Kaissling B, Biber J, Murer H, Levi M: Parathyroid hormone action on phosphate transporter mRNA and protein in rat renal proximal tubules. Am J Physiol 268: F784–F791, 1995 [DOI] [PubMed] [Google Scholar]
- 25.Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N: Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 280: 2543–2549, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Bai XY, Miao D, Goltzman D, Karaplis AC: The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem 278: 9843–9849, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Shigematsu T, Horiuchi N, Ogura Y, Miyahara T, Suda T: Human parathyroid hormone inhibits renal 24-hydroxylase activity of 25-hydroxyvitamin D3 by a mechanism involving adenosine 3′,5′-monophosphate in rats. Endocrinology 118: 1583–1589, 1986 [DOI] [PubMed] [Google Scholar]
- 28.Ferrari SL, Bonjour J-P, Rizzoli R: Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 90: 1519–1524, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Burnett SM, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS: Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21: 1187–1196, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Antoniucci DM, Yamashita T, Portale AA: Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab 91: 3144–3149, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Wesseling-Perry K, Pereira RC, Sahney S, Gales B, Wang HJ, Elashoff R, Jüppner H, Salusky IB: Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int 79: 112–119, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, Shuto E, Nashiki K, Arai H, Yamamoto H, Takeda E: Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int 70: 2141–2147, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Isakova T, Gutierrez O, Shah A, Castaldo L, Holmes J, Lee H, Wolf M: Postprandial mineral metabolism and secondary hyperparathyroidism in early CKD. J Am Soc Nephrol 19: 615–623, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scanni R, vonRotz M, Jehle S, Hulter HN, Krapf R: The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol 25: 2730–2739, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T: PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am J Physiol Renal Physiol 299: F882–F889, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, Ono K, Kakitani M, Tomizuka K, Fujita T, Fukumoto S, Yamashita T: Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol 289: F1088–F1095, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Ortiz ME, Lopez I, Muñoz-Castañeda JR, Martinez-Moreno JM, Ramírez AP, Pineda C, Canalejo A, Jaeger P, Aguilera-Tejero E, Rodriguez M, Felsenfeld A, Almaden Y: Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol 23: 1190–1197, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.David V, Dai B, Martin A, Huang J, Han X, Quarles LD: Calcium regulates FGF-23 expression in bone. Endocrinology 154: 4469–4482, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI: Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390: 45–51, 1997 [DOI] [PubMed] [Google Scholar]
- 40.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T: Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770–774, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Larsson T, Nisbeth U, Ljunggren O, Jüppner H, Jonsson KB: Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64: 2272–2279, 2003 [DOI] [PubMed] [Google Scholar]
- 42.Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, Fukagawa M: Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis 44: 250–256, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Feldman HI, Appel LJ, Chertow GM, Cifelli D, Cizman B, Daugirdas J, Fink JC, Franklin-Becker ED, Go AS, Hamm LL, He J, Hostetter T, Hsu CY, Jamerson K, Joffe M, Kusek JW, Landis JR, Lash JP, Miller ER, Mohler ER, 3rd, Muntner P, Ojo AO, Rahman M, Townsend RR, Wright JT, Chronic Renal Insufficiency Cohort (CRIC) Study Investigators : The Chronic Renal Insufficiency Cohort (CRIC) study: design and methods. J Am Soc Nephrol 14[Suppl 2]: S148–S153, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, Yamashita T, Fukumoto S, Shimada T: Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 78: 975–980, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Wolf M: Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol 21: 1427–1435, 2010 [DOI] [PubMed] [Google Scholar]
- 46.National Kidney Foundation : K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 42[Suppl 3]: S1–S201, 2003 [PubMed] [Google Scholar]
- 47.Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group : KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl 113: S1–S130, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M: Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 359: 584–592, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutiérrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M, Chronic Renal Insufficiency Cohort (CRIC) Study Group : Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305: 2432–2439, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kendrick J, Cheung AK, Kaufman JS, Greene T, Roberts WL, Smits G, Chonchol M, HOST Investigators : FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 22: 1913–1922, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandenburg VM, Kleber ME, Vervloet MG, Tomaschitz A, Pilz S, Stojakovic T, Delgado G, Grammer TB, Marx N, März W, Scharnagl H: Fibroblast growth factor 23 (FGF23) and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis 237: 53–59, 2014 [DOI] [PubMed] [Google Scholar]
- 52.Ix JH, Katz R, Kestenbaum BR, de Boer IH, Chonchol M, Mukamal KJ, Rifkin D, Siscovick DS, Sarnak MJ, Shlipak MG: Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol 60: 200–207, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, Zhang X, Nessel L, Hamano T, Grunwald JE, Raj DS, Yang W, He J, Lash JP, Go AS, Kusek JW, Feldman H, Wolf M, Chronic Renal Insufficiency Cohort (CRIC) Study Investigators : Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol 25: 349–360, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seiler S, Rogacev KS, Roth HJ, Shafein P, Emrich I, Neuhaus S, Floege J, Fliser D, Heine GH: Associations of FGF-23 and sKlotho with cardiovascular outcomes among patients with CKD stages 2-4. Clin J Am Soc Nephrol 9: 1049–1058, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glassock RJ, Pecoits-Filho R, Barberato SH: Left ventricular mass in chronic kidney disease and ESRD. Clin J Am Soc Nephrol 4[Suppl 1]: S79–S91, 2009 [DOI] [PubMed] [Google Scholar]
- 56.Silberberg JS, Barre PE, Prichard SS, Sniderman AD: Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int 36: 286–290, 1989 [DOI] [PubMed] [Google Scholar]
- 57.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP: Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322: 1561–1566, 1990 [DOI] [PubMed] [Google Scholar]
- 58.Drazner MH, Rame JE, Marino EK, Gottdiener JS, Kitzman DW, Gardin JM, Manolio TA, Dries DL, Siscovick DS: Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: The Cardiovascular Health Study. J Am Coll Cardiol 43: 2207–2215, 2004 [DOI] [PubMed] [Google Scholar]
- 59.Gutiérrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, Sarwar A, Hoffmann U, Coglianese E, Christenson R, Wang TJ, deFilippi C, Wolf M: Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 119: 2545–2552, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corda S, Mebazaa A, Gandolfini MP, Fitting C, Marotte F, Peynet J, Charlemagne D, Cavaillon JM, Payen D, Rappaport L, Samuel JL: Trophic effect of human pericardial fluid on adult cardiac myocytes. Differential role of fibroblast growth factor-2 and factors related to ventricular hypertrophy. Circ Res 81: 679–687, 1997 [DOI] [PubMed] [Google Scholar]
- 61.Parker TG, Packer SE, Schneider MD: Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest 85: 507–514, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scheinowitz M, Kotlyar A, Zimand S, Ohad D, Leibovitz I, Bloom N, Goldberg I, Nass D, Engelberg S, Savion N, Eldar M: Basic fibroblast growth factor induces myocardial hypertrophy following acute infarction in rats. Exp Physiol 83: 585–593, 1998 [DOI] [PubMed] [Google Scholar]
- 63.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-O M, Kusek JW, Keane MG, Wolf M: FGF23 induces left ventricular hypertrophy. J Clin Invest 121: 4393–4408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE: Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 207: 546–551, 2009 [DOI] [PubMed] [Google Scholar]
- 65.Jovanovich A, Ix JH, Gottdiener J, McFann K, Katz R, Kestenbaum B, de Boer IH, Sarnak M, Shlipak MG, Mukamal KJ, Siscovick D, Chonchol M: Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis 231: 114–119, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frey N, Olson EN: Cardiac hypertrophy: The good, the bad, and the ugly. Annu Rev Physiol 65: 45–79, 2003 [DOI] [PubMed] [Google Scholar]
- 67.Di Marco GS, Reuter S, Kentrup D, Grabner A, Amaral AP, Fobker M, Stypmann J, Pavenstädt H, Wolf M, Faul C, Brand M: Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol Dial Transplant 29: 2028–2035, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, Tsai MM, Cattley RC, Wronski TJ, Xia X, Li X, Henley C, Eschenberg M, Richards WG: FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 122: 2543–2553, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sigrist MK, Taal MW, Bungay P, McIntyre CW: Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol 2: 1241–1248, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, Choukroun G, Drueke TB, Massy ZA, European Uremic Toxin (EUTox) Work Group : FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int 23: 2017–2025, 2012 [DOI] [PubMed] [Google Scholar]
- 71.Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, Chavkin NW, Rahman M, Wahl P, Amaral AP, Hamano T, Master SR, Nessel L, Chai B, Xie D, Kallem RR, Chen J, Lash JP, Kusek JW, Budoff MJ, Giachelli CM, Wolf M, Chronic Renal Insufficiency Cohort Study Investigators : Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int 83: 1159–1168, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM: Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87: E10–E17, 2000 [DOI] [PubMed] [Google Scholar]
- 73.Adeney KL, Siscovick DS, Ix JH, Seliger SL, Shlipak MG, Jenny NS, Kestenbaum BR: Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 20: 381–387, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB: Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342: 1478–1483, 2000 [DOI] [PubMed] [Google Scholar]
- 75.Guérin AP, London GM, Marchais SJ, Metivier F: Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant 15: 1014–1021, 2000 [DOI] [PubMed] [Google Scholar]
- 76.Scialla JJ, Wolf M: Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat Rev Nephrol 10: 268–278, 2014 [DOI] [PubMed] [Google Scholar]
- 77.Hu MC, Shi M, Zhang J, Quiñones H, Griffith C, Kuro-o M, Moe OW: Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22: 124–136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, Shelton J, Amaral AP, Faul C, Taniguchi M, Wolf M, Brand M, Takahashi M, Kuro-O M, Hill JA, Moe OW: Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol 26: 1290–1302, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gutiérrez OM: Sodium- and phosphorus-based food additives: Persistent but surmountable hurdles in the management of nutrition in chronic kidney disease. Adv Chronic Kidney Dis 20: 150–156, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gutiérrez OM, Anderson C, Isakova T, Scialla J, Negrea L, Anderson AH, Bellovich K, Chen J, Robinson N, Ojo A, Lash J, Feldman HI, Wolf M, CRIC Study Group : Low socioeconomic status associates with higher serum phosphate irrespective of race. J Am Soc Nephrol 21: 1953–1960, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moe SM, Zidehsarai MP, Chambers MA, Jackman LA, Radcliffe JS, Trevino LL, Donahue SE, Asplin JR: Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 6: 257–264, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Isakova T, Gutiérrez OM, Smith K, Epstein M, Keating LK, Jüppner H, Wolf M: Pilot study of dietary phosphorus restriction and phosphorus binders to target fibroblast growth factor 23 in patients with chronic kidney disease. Nephrol Dial Transplant 26: 584–591, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Isakova T, Barchi-Chung A, Enfield G, Smith K, Vargas G, Houston J, Xie H, Wahl P, Schiavenato E, Dosch A, Gutiérrez OM, Diego J, Lenz O, Contreras G, Mendez A, Weiner RB, Wolf M: Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin J Am Soc Nephrol 8: 1009–1018, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Faul C: Fibroblast growth factor 23 and the heart. Curr Opin Nephrol Hypertens 21: 369–375, 2012 [DOI] [PubMed] [Google Scholar]