

Summary
Nuclear receptor subfamily 2, group F, member 6 (NR2F6) is an orphan member of the nuclear receptor superfamily. Here, we show that genetic ablation of Nr2f6 significantly improves survival in the murine transgenic TRAMP prostate cancer model. Furthermore, Nr2f6−/− mice spontaneously reject implanted tumors and develop host-protective immunological memory against tumor rechallenge. This is paralleled by increased frequencies of both CD4+ and CD8+ T cells and higher expression levels of interleukin 2 and interferon γ at the tumor site. Mechanistically, CD4+ and CD8+ T cell-intrinsic NR2F6 acts as a direct repressor of the NFAT/AP-1 complex on both the interleukin 2 and the interferon γ cytokine promoters, attenuating their transcriptional thresholds. Adoptive transfer of Nr2f6-deficient T cells into tumor-bearing immunocompetent mice is sufficient to delay tumor outgrowth. Altogether, this defines NR2F6 as an intracellular immune checkpoint in effector T cells, governing the amplitude of anti-cancer immunity.
Graphical Abstract
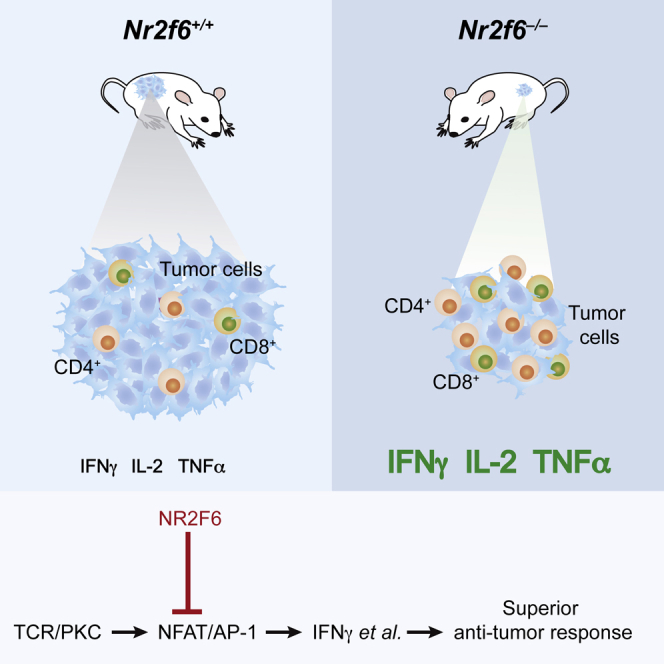
Highlights
-
•
Genetic ablation of Nr2f6 induces immune-mediated cancer surveillance
-
•
Nr2f6−/− mice display an immune contexture favoring anti-tumor responses
-
•
NR2F6 represses transcription of key cytokines in CD4+ and CD8+ effector T cells
-
•
NR2F6 controls the amplitude of tumor immunity
Immune checkpoint blockade has emerged as an effective cancer immunotherapy strategy. Here, Hermann-Kleiter et al. demonstrate a role for the nuclear orphan receptor NRF26 in tumor immune surveillance, effectively identifying an intracellular immune checkpoint in effector T cells that governs the amplitude of anti-cancer immunity.
Introduction
Adaptive immunity together with innate effector immune cells is established to efficiently control malignant cells. The importance of T cells for tumor cell elimination is underscored by the observation that tumor infiltration by T cells represents a valuable prognostic marker in different human cancer types (Hunder et al., 2008; Hodi et al., 2010; reviewed in Motz and Coukos, 2013). Residual T cell reactivity against malignant cells is limited by various components of immune evasion, such as local accumulation of immunosuppressive cell types within the tumor microenvironment. Together this favors tumor escape from the immune system instead of immune-mediated cancer cell elimination (for review, see Zitvogel et al., 2006; Zou, 2005; Motz and Coukos, 2013). Consistently, interleukin 2 (IL-2) and interferon γ (IFN-γ) are both additional key prognostic indicators mirroring a protective anti-tumor immune response in humans. High IL-2 levels thereby favor CD8+ T cell effector functions (Pipkin et al., 2010). High IFN-γ directly modulates both cancer biology (e.g., by inducing tumor cell senescence) (Schwartzentruber et al., 2011; Bedognetti et al., 2013; Braumüller et al., 2013; Galon et al., 2013) and induces an immune contexture favoring continuous tumor cell elimination, a concept coined “cancer immune surveillance” (Shankaran et al., 2001).
Manipulating the immune system to harness anti-tumor immune responses for the treatment of cancer patients has been a major goal for many decades. Promising novel therapeutic advances blocking immune system inhibitory pathways, such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1)/PD-1 ligand (PD-L1), are referred to as “immune checkpoints” (i.e., inhibitory signaling intermediates that control the duration and amplitude of physiological immune responses) are successful entering into clinics (Hodi et al., 2010; Brahmer et al., 2010; Robert et al., 2011; Hamid et al., 2013; Topalian et al., 2012; Powles et al., 2014; Tumeh et al., 2014; Gubin et al., 2014). In addition, adoptive T cell transfer therapy or vaccination approaches are now also providing more encouraging results, especially when combined with cytokines or the above mentioned immune checkpoint-antagonizing antibodies (van den Eertwegh et al., 2012; Kalos and June, 2013; Restifo et al., 2012; Kantoff et al., 2010; Bodor et al., 2012; Motz and Coukos, 2013; Schwartzentruber et al., 2011). Even though these approaches are exciting, there is an unmet medical need, as still only a limited number of patients response to and even less patients are potentially cured by these approaches. Thus, there is a high scientific interest to explore novel cancer immunotherapeutic approaches with the ultimate goal to further strengthen the patient’s immune system.
Notably, mechanistic processes that support immune-mediated tissue destruction appear to be strikingly analogous in autoimmunity and cancer. We previously demonstrated that the nuclear receptor subfamily 2, group F, member 6 (NR2F6; also called COUP-TFIII or Ear2) represents an important gatekeeper of antigen receptor-induced cytokine response thresholds of pro-inflammatory CD4+ Th17 lymphocytes (Hermann-Kleiter et al., 2008). In these autoimmunity-promoting Th17 cells, NR2F6 directly antagonizes the binding of the transcription factors NFAT and, particularly, retinoic acid receptor-related orphan receptor-γ-t (RORc) to the Il17 cytokine locus (Hermann-Kleiter et al., 2012), thereby reducing central nervous system inflammation. Here, we employed various types of transplantable and spontaneous tumor models to define the role of NR2F6 in tumor immunology. Using these model systems, we provide strong experimental evidence that genetic deletion of Nr2f6 is both necessary and sufficient to induce host-protective immune rejection of cancer. Nr2f6 deficiency leads to augmented intratumoral effector CD4+ and CD8+ T cell infiltration and strongly enhances local production of IL-2, IFN-γ, and tumor necrosis factor-α (TNF-α), thereby forming an immune environment that allows strong anti-tumor T cell responses in tumor-bearing mice.
Results
Loss of NR2F6 Prolongs Survival of TRAMP Mice, an Autochthonous Model of Prostate Cancer
We employed the murine transgenic adenocarcinoma of the mouse prostate (TRAMP) model, in which prostate-specific expression of SV40 large T antigen results in prostate cancer (Greenberg et al., 1995), to evaluate the role of NR2F6 in cancer immunity. Male TRAMP mice with different Nr2f6 genotypes (Nr2f6+/+TRAMP and Nr2f6−/−TRAMP) were analyzed at week 22 and 28, as well as when terminally ill. While all Nr2f6+/+TRAMP mice had to be sacrificed due to high tumor burden latest at week 40, 50% of Nr2f6−/−TRAMP mice remained alive at this time point (Figure 1A). The majority of the primary tumor masses were composed of atypical epithelial hyperplasia (AH); however, 26% of Nr2f6+/+TRAMP mice developed aggressive neuroendocrine (NE) tumors, as identified through histomorphological analysis (Figures 1B and 1C). In contrast to Nr2f6+/+TRAMP mice and despite a similar prostate tumor incidence, we never observed any NE differentiation in tumors from Nr2f6−/−TRAMP mice. Although tumor growth was significantly delayed in Nr2f6−/−TRAMP mice, all animals had to be sacrificed at week 55. Endpoint analyses indicated an enhanced number of intratumoral CD45+, CD4+, and CD8+ immune cells in Nr2f6−/−TRAMP mice (Figure 1D).
Figure 1.

Loss of NR2F6 Prolongs Survival in the Autochthonous Prostate Cancer Model TRAMP Due to Enhanced Numbers of Cytokine-Secreting Infiltrating T Cells
(A) Endpoint analysis of male Nr2f6+/+TRAMP (n = 14) and Nr2f6−/−TRAMP (n = 10) mice (A) showed a significant survival benefit in Nr2f6−/−TRAMP mice depicted by a Kaplan-Meier curve and statistics analyzed by the log-rank test (p = 0.0018).
(B) Weight of TRAMP urogenital (UG) tract at endpoint was not significantly different between Nr2f6+/+TRAMP and Nr2f6−/−TRAMP mice. Aggressive neuroendocrine (NE) tumors in wild-type mice shown in triangles were absent in the Nr2f6-deficient cohort.
(C) One representative endpoint TRAMP prostate tumor with seminal vesicle metastasis derived from Nr2f6+/+TRAMP (circle, black) and Nr2f6−/−TRAMP (square, green) mice is shown as well as one Nr2f6+/+TRAMP NE tumor (triangle, black).
(D) Phenotypic characterization of CD45+ TILs in Nr2f6+/+TRAMP (black) or Nr2f6−/−TRAMP (green) of prostate tumors taken at the endpoint of the experiment: higher cell percentages of total infiltrating immune cells expressing CD45+ (p = 0.02), CD45+CD4+ (p = 0.009), or CD45+CD8+ (p = 0.03) are detected in Nr2f6−/−TRAMP despite comparable tumor sizes.
(E) Gross examination of urogenital tracts at week 28 of Nr2f6+/+TRAMP and Nr2f6−/−TRAMP mice.
(F and G) (F) Significantly decreased UG tracts relative to body weight (p < 0.0001) and (G) prostate weight (p < 0.0001) were observed in Nr2f6−/−TRAMP mice (n = 21) when compared to Nr2f6+/+TRAMP mice (n = 27). Four prostate tumors within both cohorts were excluded from statistical analysis at this time point as classified as outliers due to being either too small (<0.09 g) or too large (>0.5 g).
(H) Tumor single-cell suspensions isolated from Nr2f6+/+TRAMP or Nr2f6−/−TRAMP mice were analyzed by flow cytometry. The percentage of positively stained cells among total viable cells after gating on forward and side scatter is shown. Representative dot plots of prostate tumor-infiltrating CD45+ single-positive and CD45+CD8+, or CD45+CD4+ double-positive T cells isolated from Nr2f6+/+TRAMP or Nr2f6−/−TRAMP mice are depicted. Numbers adjacent to outlined areas indicate the percentage of positive cells relative to parental gate.
(I) Percentage of TILs at week 28 of Nr2f6+/+TRAMP (n = 11) and Nr2f6−/−TRAMP (n = 7) mice revealed enhanced numbers of CD45+ (p = 0.006), CD45+CD8+ (p = 0.002) and CD45+CD4+ (p = 0.001) cells.
(J) Equal numbers of prostate tumor-infiltrating Treg cells (CD4+CD25+Foxp3+) in Nr2f6+/+TRAMP (n = 11) and Nr2f6−/−TRAMP (n = 7) prostate tumors.
(K) Percentages of total cytokine producing TILs within Nr2f6+/+TRAMP (n = 11) and Nr2f6−/−TRAMP (n = 7) prostate tumors: significantly enhanced numbers of CD8+IL-2+ (p = 0.049), CD8+IFN-γ+ (p = 0.001), CD4+IL-2+ (p = 0.002), and CD4+IFN-γ+ (p = 0.02) cells were found within Nr2f6−/−TRAMP prostate tumors.
(L) Representative cryosection of Nr2f6+/+TRAMP or Nr2f6−/−TRAMP prostate tumors immune-fluorescence staining for the T cell markers CD4 and CD8 in green and Hoechst nuclear stain in blue, respectively. Scale bar, 50 μm.
(M) Graphical representation of increased CD4+ (p = 0.01) and CD8+ (p = 0.03) T cell tumor infiltration in Nr2f6−/− mice, averaged from three fields per mouse and four mice per genotype. Error bars represent SEM and asterisk (∗) indicates statistically significant differences between Nr2f6+/+TRAMP or Nr2f6−/−TRAMP tumors, as calculated using Student’s t test.
In order to define the mechanism underlying the observed survival benefit due to delayed tumor outgrowth in Nr2f6−/−TRAMP mice, we next analyzed in detail prostate tumors at pre-defined time points. At week 28, tumor burden (quantified by calculating urogenital tract weight without the bladder and prostate relative to the total body weight) was significantly reduced in Nr2f6−/−TRAMP compared to Nr2f6+/+TRAMP mice (Figures 1E–1G); notably, no differences in overall body weight could be detected at any time point (Figures S1A–S1C). At week 28, we observed significantly more prostate tumor-infiltrating CD4+ and CD8+ T cells in Nr2f6−/−TRAMP mice than in Nr2f6+/+TRAMP mice (Figures 1H and 1I), whereas comparable numbers of CD4+CD25+Foxp3+ tumor-infiltrating regulatory T cells could be detected in the two genotypes (Figure 1J). Tumor-infiltrating immune cells contained increased frequencies of CD8+IL-2+, CD8+IFN-γ+, CD4+IL-2+, and CD4+IFN-γ+ T lymphocytes in Nr2f6−/−TRAMP mice versus Nr2f6+/+TRAMP mice (Figure 1K). Analysis by immunohistochemistry in tumors with comparable sizes confirmed significantly higher numbers of intratumoral CD4+ and CD8+ T cells in Nr2f6−/−TRAMP mice (Figures 1L and 1M). To address the role of NR2F6 in T cells within the tumor draining lymph node (dLN), we evaluated T cell frequency, activation status, and cytokine production at week 28. Again, the proportions of T cells that were CD45+, CD4+, CD8+, CD44+, or IFN-γ+ were significantly elevated (Figures S1D–S1G). Even though we detected a trend toward higher intratumoral numbers of CD4+RORc+ and γδTCR+ cells in Nr2f6−/−TRAMP mice versus Nr2f6+/+TRAMP mice, the differences between the two genotypes did not reach statistical significance (Figure S1H). Similarly, we also observed a tendency toward larger proportions of CD4+IL-17+, CD8+Tbet+, DX5+, and γδTCR+ cells that also did not reach statistical significance (Figure S1I). The same readouts were used to analyze the immune cell composition of prostate tumors and dLNs at earlier time points (i.e., at week 22). Again, reduced urogenital tract (UG) sizes in Nr2f6−/−TRAMP mice were paralleled by increased numbers of CD45+ immune cells infiltrating prostate tumors, and elevated numbers of CD4+IL-2+ T cells in the dLNs of Nr2f6−/−TRAMP mice (Figures S1J–S1M).
Taken together, these data show that the numbers of T cells in Nr2f6−/−TRAMP tumor-bearing mice are increased both within the dLNs and at the tumor site at all three time points investigated. The enhanced production rates of IL-2 and IFN-γ may contribute to a local immune-activated microenvironment, which, in turn, significantly contributes to immune-mediated cancer growth control leading to increased survival of Nr2f6−/−TRAMP mice.
Anti-tumor Immune Response in Nr2f6-Deficient Mice
At this point, we were not able to exclude that loss of Nr2f6 function in non-immune cells (for example, in prostate epithelial cells within the autochthonous TRAMP tumor model) may be causally involved in the observed alterations of tumor progression. Therefore, we next used four different highly tumorigenic cancer cell lines (TRAMP-C1, B16-OVA, B16-F10, and EG7) to analyze animal survival, tumor growth, and the tumor/dLN immune microenvironment; of note, all four lines are genetically wild-type for Nr2f6. Similar to the findings from the autochthonous TRAMP model, survival in Nr2f6-deficient mice receiving Nr2f6 wild-type tumor cell lines was significantly enhanced. Figures 2A and 2B demonstrate the delayed growth kinetics of subcutaneously injected TRAMP-C1 and B16-OVA tumors in Nr2f6-deficient mice. TCRVβ repertoire of wild-type and Nr2f6-deficient T lymphocytes prior or post-B16-OVA tumor challenge from naive or tumor dLN was investigated via clonality analysis of the individual Vβ gene families of the CDR3 region. These data indicated unaltered clonal composition of the T cell compartment between wild-type and Nr2f6-deficient T cells in tumor-bearing mice (Figures S2A and S2B). Furthermore, 75% of Nr2f6−/− mice injected with the EG7 lymphoma cell line completely rejected EG7 tumors and remained tumor-free throughout the entire 10-month experimental period; in contrast, all Nr2f6+/+ mice challenged with EG7 cells had to be sacrificed as early as day 21 post-injection because of their high tumor burden (Figures 2C and 2D).
Figure 2.

Nr2f6−/− Mice Reject Transplantable Subcutaneous Tumors
The kinetics of tumor cell growth in 8- to 12-week-old Nr2f6+/+ (black) and Nr2f6−/− (green) mice bearing subcutaneously TRAMP-C1 prostate tumor cells, B16-OVA melanoma, or EG7 lymphoma.
(A) Tumor growth curve of 1 × 106 TRAMP-C1 cells inoculated into Nr2f6+/+ (n = 5) and Nr2f6−/− (n = 5) mice (ANOVA, p < 0.0001).
(B) Kinetics of 1 × 105 (n = 5) B16-OVA cells in Nr2f6+/+ (n = 17) and Nr2f6−/− (n = 17) mice, which were used for subsequent analysis (ANOVA, p = 0.0017).
(C) Significant survival benefit in Nr2f6−/− (n = 8) compared to Nr2f6+/+ (n = 13) mice injected with 1.5 × 105 EG7 tumor cells shown by a Kaplan-Meier curve, statistically analyzed by a log-rank test (p = 0.0001).
(D) Kinetics of EG7 tumor cell growth in Nr2f6+/+ (n = 5) and Nr2f6−/− (n = 5) mice (ANOVA, p < 0.0001). Results shown are derived from at least two independent experiments.
(E) Tumor growth curve in Nr2f6+/+ mice (n = 9) injected s.c. with 5 × 105 B16-OVA cells and administered i.p. with 0.5 mg of an anti-mouse PD-L1 blocking antibody as immune checkpoint inhibitor is comparable to tumor growth curve seen in Nr2f6−/− mice (n = 8) injected with rat immunoglobulin G (IgG) control alone. Summary of three independent experiments is shown, and data are expressed as the mean ± SEM, IgG control in wild-type recipients versus anti-mouse PD-L1 blocking antibody (p = 0.0019) statistically analyzed by two-way ANOVA.
Of note, comparing genetic NR2F6 deletion with blocking interaction of PD-1 with its ligand PD-L1 provides evidence that the observed superior anti-tumor immunity in Nr2f6−/− mice is well comparable to the benefit of PD-L1 blocking therapy in wild-type mice (employing the established protocol with neutralizing Ab10F.9G2; Gros et al., 2014). This suggests that NR2F6 and the PD-1/PD-L1 axis control cancer immunity to a similar extent (Figure 2E).
As next step the B16-OVA subcutaneous (s.c.) tumor model was used for an extensive analysis of tumor-infiltrating immune cells at day 14 after tumor cell injection (Figure 3A), employing a stratified CD45+/CD3+/CD4+ or CD8+ gating strategy (as outlined in Figures S3A and S3B). As in the autochthonous model of prostate cancer, B16-OVA tumors grown in Nr2f6−/− mice exhibited significantly increased numbers of tumor-infiltrating CD45+CD3+CD4+ and CD45+CD3+CD8+ T cells, calculated at the level of total numbers on a weight (i.e., gram) basis when compared to wild-type mice (Figures 3B and 3C). This particular phenotype of Nr2f6−/− mice was independent of the B16-OVA tumor size (Figure S3C). Analysis in tumors with comparable sizes by immunohistochemistry confirmed the markedly enhanced intratumoral CD4+ and CD8+ T cell numbers in Nr2f6−/− mice (Figures S3D and S3E), well reflecting relative levels of tumor-infiltrating lymphocytes (TILs) (i.e., percentage of CD45+, CD45+CD3+, CD45+CD8+, or CD45+CD4+cells; Figure S3F). Pronounced CD8+ and CD4+ T cell effector functions in Nr2f6−/− mice were found to be associated with markedly higher expression of CD25 and PD-1 on both T cell subsets when compared to wild-type animals (Figures 3D and 3E).
Figure 3.

Enhanced Numbers of Tumor-Infiltrating T Cells in Nr2f6-Deficient Mice Bearing B16-OVA Transplantable Melanomas
(A) Gross examination of B16-OVA melanomas from Nr2f6+/+ (top) and Nr2f6−/− (bottom) mice 14 days after subcutaneous injection of 1 × 105 tumor cells.
(B) Tabulation of Nr2f6+/+ (n = 6) and Nr2f6−/− (n = 7) B16-OVA tumor-infiltrating cells on day 14. Significantly increased absolute cell numbers of CD45+ (p = 0.01), CD3+ (p = 0.05), CD8+ (p = 0.007), and CD4+ (p = 0.015) per 0.1 g of tumor tissue were detected in Nr2f6−/− mice.
(C) Dot plots of B16-OVA tumor-infiltrating CD45+ and CD3+ cells derived from Nr2f6+/+ and Nr2f6−/− tumor-bearing mice (for detailed gating strategy, see Figure S3).
(D) Immune cell characterization of Nr2f6+/+ (n = 6) and Nr2f6−/− (n = 7) B16-OVA tumor-infiltrating cells per 0.1 g of tumor tissue, gated on CD45+CD3+ positive cells did reveal increased absolute cell numbers of CD8+CD25+ (p = 0.003); CD8+PD-1+ (p = 0.003); CD4+CD25+ (p = 0.008); CD4+PD-1+ (p = 0.001); and CD4+CD25+Foxp3+ Treg cells (p = 0.007).
(E) Dot plots of B16-OVA tumor-infiltrating CD8+PD-1+ cells derived from Nr2f6+/+ and Nr2f6−/− mice.
(F) Significantly increased absolute cell numbers of CD11b+Gr-1+ myeloid derived suppressor cells (p = 0.02), and a trend for tumor associated macrophages (p = 0.06) (CD11b+F/80+) could be detected in tumors of Nr2f6−/− (n = 7) mice.
Because immunosuppressive immune cells such as Tregs, tumor-associated macrophages (TAM), and myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment are established to promote T cell dysfunction and reduction of these cells would at least in part explain increased anti-tumor immune responses in Nr2f6−/− mice, we evaluated the extend of tumor infiltration by Treg, MDSC, and TAM (Figures 3D and 3F). In Nr2f6−/− mice, we could not detect any reduction of Treg accumulation but could even observe an increased abundance of these cells in B16-OVA tumor tissues of Nr2f6−/− mice. Nevertheless, the clearly increased numbers of CD4+ and CD8+ effector TILs in Nr2f6−/− mice outweigh this increase of immunosuppressive cell types, as the intratumoral ratios of Teff/Treg did not show a significant difference between mice of both genotypes. The ratio of CD8+ and CD4+ effector T cells to either MDSC or TAM remain even in favor of the effector cell populations in Nr2f6−/− mice.
In tumor-bearing Nr2f6−/− mice, CD4+ and CD8+ tumor-infiltrating T cell subsets expressed significantly increased amounts of IL-2, IFN-γ, and TNF-α (Figures 4A–4C). Among the dLN-derived cells, CD4+ T cells producing IL-2 and IFN-γ as well as CD8+ T cells secreting IFN-γ were significantly more abundant in Nr2f6−/− mice (Figures S4A–S4C) as were activated CD4+ and CD8+ T cells (CD4+CD44hi; CD8+CD44hi) (Figure S4D). Notably, we could not detect any obvious alterations in tumor-infiltrating natural killer (NK) cells (DX5+), macrophages (CD11b+), or DC subsets (CD11c+CD11b+ and CD11c+CD8a+) in Nr2f6−/− compared to wild-type tumor-bearing mice (Figure S4E).
Figure 4.

Nr2f6 Expression Limits Cytokine Secretion of Tumor-Reactive T Cells
(A) Cytokine secretion of Nr2f6+/+ (n = 6) and Nr2f6−/− (n = 7) TILs in tumors larger than 0.022 g revealed significant differences of absolute cell numbers per 0.1 g tumor tissue for the following subsets CD8+IL-2+ (p = 0.01), CD8+IFN-γ+ (p = 0.01), CD8+TNF-α+ (p = 0.002), CD4+IL-2+ (p = 0.002), CD4+IFN-γ+ (p = 0.0006), CD4+TNF-α+ (p = 0.0009) in Nr2f6−/− mice.
(B) qRT-PCR analysis of tumors revealed enhanced Ifng (p = 0.008) as well as Il2 expression (p = 0.052) in Nr2f6−/− mice. Results shown are derived from at least two independent experiments.
(C) Dot plots and histograms of IFN-γ and IL-2 expressing CD8+ and CD4+ tumor-infiltrating cells gated on CD45+CD3+ T cells derived from Nr2f6+/+ and Nr2f6−/− mice. Numbers indicate percentage of positive gated cells. Results shown are derived from at least two independent experiments. Error bars represent SEM; data were analyzed via Student’s t test.
(D) Tumor growth was monitored in Nr2f6−/− mice injected s.c. with 5 × 105 B16-OVA melanoma cells and administered with 0.5 mg of either an anti-mouse IFN-γ (n = 5) or an anti-mouse IL-2 (n = 3) neutralizing antibody or an corresponding IgG1 or IgG2a control every 3 days starting from day 1 of B16-OVA challenge. Comparison with relevant IgG control revealed disrupted tumor protection in Nr2f6−/− mice treated with anti-mouse IFN-γ neutralizing antibody (p = 0.006), but not anti-mouse IL-2 neutralizing antibody (p = 0.4). Summary graphs are the mean ± SEM, statistically analyzed by two-way ANOVA.
Next, the functional importance of increased cytokine secretion by Nr2f6−/− T cells was investigated by blocking IL-2 and IFN-γ function using systemic injection of antagonizing antibodies into tumor-bearing mice in vivo. These experiments identified IFN-γ as key effector cytokine at least in part explaining enhanced anti-tumor immunity of Nr2f6−/− mice, as anti-IFN-γ antibodies abrogated the tumor-protective phenotype of Nr2f6−/− animals (Figure 4D). Increased cytokine production appears to be particularly linked to tumor challenge, as we could not detect an enhanced steady-state cytokine-production potential of CD4+ or CD8+ T cells in tumor-free Nr2f6−/− mice (Figure S5A).
The impact of Nr2f6 deficiency on tumor metastasis was next evaluated by challenging each mouse genotype with intravenously (i.v.) administered B16-F10 cells, which are known to form lung metastases upon i.v. injection. Similar to our previous data, formation of lung metastases was significantly reduced at day 14 and 19 post-injection, as quantified by reduction of the number of tumor foci in the lungs of Nr2f6-deficient mice (Figures 5A and 5B). Thus, deficiency of Nr2f6 in non-cancer cells appears to strongly enhance the anti-metastatic activity of the immune system.
Figure 5.

Reduced Metastasis and Anti-Tumor Memory Depends on NR2F6 in T Cells
(A) Gross examination of representative metastatic tumor lungs at day 14 and day 19 after tumor inoculation of either Nr2f6+/+ mice (n = 10) or Nr2f6−/− mice (n = 9) i.v.-injected B16-F10.
(B) Bar chart depict numbers of lung tumor foci on day 14 (p = 0.002) and day 19 (p = 0.02) after tumor induction in Nr2f6+/+ mice and Nr2f6−/− mice.
(C) Long-lasting anti-tumor memory is demonstrated after injection of EG7 tumor cells: Nr2f6−/− mice (n = 6), which had received 1.5 × 105 EG7 cells subcutaneously at 8–12 weeks of age and rejected the primary tumor, were subsequently re-challenged after 10 months with a 10-fold higher dose of EG7 cells. In contrast to age- and sex-matched naive Nr2f6+/+ mice (n = 6) and naive Nr2f6−/− mice (n = 6) controls, 80% of memory Nr2f6−/− mice were able to reject the 10-fold higher tumor dose.
(D) Kaplan-Meier survival curves of age-matched naive Nr2f6+/+, naive Nr2f6−/−, and memory Nr2f6−/− mice that received 1.5 × 106 EG7 cells subcutaneously (p < 0.001), statistically analyzed by log-rank test.
(E) Enhanced tumor rejection is dependent on Nr2f6−/− T lymphocytes. The kinetics of tumor cell growth in Rag1−/− mice reconstituted with PBS, 1 × 107 CD3+ Nr2f6+/+, or 1 × 107 CD3+ Nr2f6−/− T cells. The average size of Rag1−/− PBS tumors at 17 days was 2,034 mm3 (±156 SEM; n = 5), compared with 1,131 mm3 (±269 SEM; n = 8) in Rag1−/−CD3Nr2f6+/+ and 138 mm3 (±54 SEM; n = 8; p < 0.01) in Rag1−/−CD3Nr2f6−/− mice. One-half of the mice were killed at day 17 after tumor induction in order to analyze tumor dLNs.
(F) Flow cytometry analyses revealed increased CD4+ T cell numbers in the dLNs of tumor-bearing Rag1−/−CD3Nr2f6−/− mice (p = 0.03) when compared to tumor dLNs of Rag1−/−CD3Nr2f6+/+. The data are depicted as bar chart representing CD4+ cells as percentage of total cells. Summary graphs show the mean ± SEM and were statistically analyzed by Student’s t test.
(G and H) Adoptive transfer of Nr2f6−/−but not Nr2f6+/+ CD3+ or OT-I cells into B16-OVA tumor-bearing wild-type mice significantly reduces tumor growth. (G) Kinetics of B16-OVA tumor growth after a single therapeutic adoptive transfer of 3 × 106 OT-I donor T cells derived from either Nr2f6−/− or Nr2f6+/+ mice (p = 0.0083) or (H) after single transfer of 1 × 107 CD3+ T cells again derived from either Nr2f6−/− or Nr2f6+/+ mice into wild-type mice on day 7 after tumor induction (p = 0.001). Data are representative of at least two independent experiments (naive recipients n ≥ 7/group, OT-I recipients n = 6/group); graphs show the mean ± SEM statistically analyzed by two-way ANOVA.
To evaluate in detail whether Nr2f6-deficiency also allows induction of immunological memory, we re-challenged Nr2f6−/− mice that had previously rejected EG7 tumor cells (termed “memory Nr2f6−/− mice”) in parallel to 10-month-old sex-matched EG7 tumor antigen-naive Nr2f6+/+ and Nr2f6−/− mice with a 10-fold higher EG7 tumor load than used in Figure 2. As expected, all wild-type mice rapidly developed tumors and had to be killed latest by day 17 post-injection. In line with our previous results, tumor outgrowth in naive Nr2f6−/− mice group was significantly delayed even with this high tumor load. In strict contrast, only one of six memory Nr2f6−/− mice had to be killed because of tumor progression at day 41 post-injection; the other five memory Nr2f6−/− animals even rejected this higher EG7 cell dose. Accordingly, survival of memory Nr2f6−/− mice (80%) was significantly better than the survival of Nr2f6+/+ mice (0%) or the EG7 antigen-naive Nr2f6−/− cohort (20%) (Figures 5C and 5D).
In combination, these results indicate that loss of Nr2f6 in immune cells strongly enhances tumor immune control. This striking survival benefit for tumor-bearing Nr2f6-deficient mice was accompanied by a host-protective induction of immunological memory, which is well known to depend strongly on the T cell compartment.
T Cell Intrinsic Function of NR2F6 in Cancer Immune Surveillance
To define the role of T cells for tumor rejection in Nr2f6−/− mice, we next analyzed EG7 subcutaneous tumor growth in age- and sex-matched Rag1−/− mice reconstituted with T cells derived from either Nr2f6+/+ or Nr2f6−/− mice (designated as Rag1−/−CD3Nr2f6+/+ and Rag1−/−CD3Nr2f6−/−, respectively). Similar to the immunocompetent mice, tumor growth was again strongly reduced in Rag1−/−CD3Nr2f6−/− when compared to Rag1−/−CD3Nr2f6+/+ reconstituted mice (Figures 5E and S5B). Again, T cell numbers in the dLNs were markedly increased (Figures 5F and S5C). Transfer experiments using immunocompetent mice (that also have endogenous immunosuppressive T cells, such as Treg) similarly to the previous experiments demonstrated that adoptive transfer of model tumor antigen-specific T cells isolated from OT-I T cell receptor (TCR) transgenic animals crossed to the Nr2f6−/− background (Figure 5G) and even polyclonal T cells from Nr2f6-deficient animals are sufficient to confer a significant tumor growth delay (Figure 5H). Taken together, these data validate the importance of NR2F6 as T cell-intrinsic suppressor of T cell-mediated tumor growth control in vivo.
NR2F6 Represses Key Cytokine Transcription in Effector CD4+ Th1 and CD8+ T Cells
Next, we investigate the underlying molecular mechanisms mediating enhanced anti-tumor immune reactivity in Nr2f6−/− mice and particularly increased cytokine production in Nr2f6−/− CD4+ T cells in vitro. Intriguingly, the established repressor of the antigen receptor activation threshold NR2F6 in Th17 cells (Hermann-Kleiter et al., 2008) is substantially upregulated upon in vitro CD3/CD28 stimulation in CD4+ T cells (Figure 6A), indicating a dynamic regulation of Nr2f6 expression as a potential negative feedback loop limiting CD4+ T cell activation. When culturing wild-type and Nr2f6-deficient naive CD4+ T cells under Th1 conditions, cytokine expression pattern analyses confirmed enhanced cytokine responses for IL-2, IFN-γ and TNF-α (Figures 6B–6E). Of note, in contrast to its negative regulatory role for effector T cell biology, NR2F6 is not required for CD4+ Treg cell function (Figures S6A–S6F).
Figure 6.

Nr2f6 Suppresses Th1 CD4+ T Cell Activation
(A) In vitro qRT-PCR analysis of Nr2f6 mRNA in wild-type CD4+ T cells during Th1 differentiation activated with anti-CD3 mAb (5 μg) and anti-CD28 mAb (1 μg) at the indicated time points (n = 3).
(B) Bioplex technology was used to demonstrate significantly increased secretion of the pro-inflammatory cytokines IL-2 (p = 0.045), IFN-γ (p = 0.047), and TNF-α (p = 0.046) in the supernatant of in-vitro-activated Nr2f6−/− versus wild-type CD4+ T cells at day 1 and day 2 of differentiation under Th1-polarizing conditions (n = 3).
(C) In vitro qRT-PCR analysis similarly detected enhanced transcript expression levels of Il2 (p = 0.003), Ifng (p = 0.044), Tnfa (p = 0.017), but not Tbx21 (p = 0.17) mRNA in Nr2f6−/− CD4+ Th1 cells compared to Nr2f6+/+ cells upon activation with anti-CD3 (5 μg) and anti-CD28 (1 μg) at the indicated time points (n = 3). Expression was normalized to the housekeeping gene GAPDH and presented as fold induction of unstimulated cells. Summary graphs represent the mean ± SD, data are representative for at least two independent experiments, and statistical differences were evaluated by applying two-way ANOVA.
(D and E) (D) Analysis of IL-2 and IFN-γ producing CD4+ Th1 T Nr2f6+/+ or Nr2f6−/− cells by flow cytometry after 3 days (3d) of Th1 driving conditions and (E) followed by a restimulation with anti-CD3 (5 μg) overnight (d4/re). Numbers within outlined areas indicate the percentage of cytokine-expressing cells, and one out of three representative experiments is shown.
Similar to CD4+ T cells, expression of Nr2f6 mRNA is low in resting CD8+ T cells, whereas its expression level is strongly induced upon CD3/CD28 stimulation in a time-dependent manner both in murine and human CD8+ T cells (Figures 7A and 7B). Reminiscent to the in vivo data generated in the different tumor models, deficiency of the murine Nr2f6 gene is associated with significantly elevated IL-2, IFN-γ, and TNF-α secretion levels in CD8+ T cells after CD3/CD28 stimulation, as shown by quantification of secreted cytokines as well as intracellular staining and fluorescence-activated cell sorting (FACS) (Figures 7C and S7A). Accordingly, qRT-PCR revealed significantly enhanced transcript levels of Il2, Ifng, and Tnfa as well as Il21, Tbx21, Il2ra mRNA when compared to wild-type T cells (Figure 7D). Enhanced cytokine secretion was not attributable to altered survival of Nr2f6−/− CD8+ and CD4+ T cells, respectively (Figures 7E, S6G, S6H, and S7B). Of note, Nfat2 but not Nfat1 mRNA was found to be strongly enhanced in Nr2f6-deficient T cells (Figure 7F). NFAT2 protein levels are known to be markedly induced by constitutively expressed NFAT1 upon T cell activation (Serfling et al., 2006). By directly binding to promoters with NFAT, where it is thought to, i.e., actively suppress NFAT/AP-1-mediated gene transcription, NR2F6 simultaneously antagonizes the well-established amplification of Nfat2 transcription, together maintaining the level of DNA-bound NFAT proteins below what is required for robust transcriptional activation of the Il2 and Ifng promoters.
Figure 7.

Nr2f6 Suppresses CD8+ T Cell Activation
(A and B) Nr2f6 expression is induced in a TCR-dependent manner in both (A) mice and (B) human CD8+ T cells activated with anti-CD3 (5 μg) and anti-CD28 (1 μg) in vitro (n = 3).
(C) Bioplex technology was used to analyze the secretion of IL-2 (p = 0.0002), IFN-γ (p = 0.03), and TNF-α (p = 0.016) in the supernatant of in vitro activated murine CD8+ T cells at day 1 and day 2 (n = 3).
(D) qRT-PCR analysis of in-vitro-stimulated Nr2f6+/+ and Nr2f6−/− CD8+ T cells at the indicated time points revealed enhanced Il2 (p = 0.0003), Ifng (p = 0.02), Tnfa (p = 0.002), Il21 (p = 0.002), Tbx21 (p = 0.02), and Il2ra (p = 0.0006) expression levels (n = 3). Expression was normalized to the housekeeping gene GAPDH and presented as fold induction versus unstimulated controls. Summary graphs represent the mean ± SD, data are representative of at least two independent experiments, and statistics were calculated using two-way ANOVA.
(E) Using FACS, CD4+ T cells were sorted from Nr2f6+/+ and Nr2f6−/− mice and stimulated in vitro with plate-bound anti-CD3 (5 μg/ml) plus soluble anti-CD28 (1 μg/ml). Viability was assessed by Annexin V/Propidiumiodid (PI) staining in a flow cytometer after 48 hr.
(F) qRT-PCR analysis of in-vitro-stimulated Nr2f6+/+ and Nr2f6−/− CD8+ T cells revealed enhanced Nfat2 (p = 0.015) expression levels (n = 3).
(G) Nr2f6+/+ and Nr2f6−/− CD8+ T cells were activated with anti-CD3 (0.5 or 5 μg) and anti-CD28 (1 μg) for 20 hr in vitro, nuclear extracts were isolated, and electromobility assays were performed for AP-1 and NFAT2. AP-1 and NFAT2 DNA binding is induced in an anti-CD3 mAb concentration-dependent manner and was strongly enhanced in Nr2f6−/− CD8+ T cells. Supershift analysis was performed by using antibodies against c-fos and NFAT2 as indicated. Loading was controlled by immunoblotting of lamin B using the corresponding nuclear extracts (Figure S7C). One representative experiment out of three is shown.
(H) Nr2f6 binding to the Il2 promoter was evaluated by ChIP. Therefore, either Nr2f6+/+ or Nr2f6−/− CD8+ resting or activated for 20 hr with anti-CD3 (5 μg) and anti-CD28 (1 μg) activated cells were used with anti-NR2F6 or IgG control, and cytokine promoter sequence was quantified by real-time PCR. The data are presented as percentage of input samples before immunoprecipitation.
Mechanistically, we have previously established that NR2F6 is able to directly suppress DNA binding of the activation-dependent transcription factor NFAT at promoter regions within the Il17 locus in Th17-polarized CD4+ T cells (Hermann-Kleiter et al., 2012). Employing band-shift assays in CD8+ T cells, we complement our recent findings by demonstrating augmented NFAT/AP-1 binding to their bona fide consensus motif defined within the minimal Il2 promoter region in Nr2f6−/− T cells (Figures 7G and S7C). Using chromatin immunoprecipitation (ChIP), we further show that NR2F6 directly binds to the Il2 promoter in resting wild-type CD8+ T cells; its DNA-binding capability is reduced in a dose-dependent manner upon CD3/CD28 cross-linking (Figures 7H and S7D). Complementary NFAT2 binding at the Il2 minimal promoter locus, as demonstrated by ChIP analysis, increases in a stimulation-dependent manner that is strongly enhanced in the Nr2f6−/− T cells (Figure S7D). Transcription factor binding analysis (Matys et al., 2006) revealed NFAT – NR2F6 DNA binding sites also at the defined distal regulatory region (−1.4 kb) (Ono et al., 2007) as well as at the minimal Ifng promoter. Consistently, ChIP analyses with NR2F6 and NFAT2 revealed binding of NR2F6 to these regions in resting cells and enhanced NFAT binding capability in activated Nr2f6-deficient T cells (Figures S7E and S7F).
Taken together, the data indicate that T cell-intrinsic NR2F6 directly antagonizes the DNA binding capabilities of the NFAT/AP-1 complex on the Il2 and Ifng promoters, thereby inhibiting the antigen receptor-mediated amplitude of cytokine transcription of these established NFAT/AP-1-dependent target genes. As a consequence, enhanced IL-2 and IFN-γ secretion in Nr2f6−/− mice favors T cell-mediated cancer cell elimination, which may, at least in part, explain the improved anti-tumor immunity.
Discussion
Uncovering mechanisms that govern immune control of cancer is of high clinical relevance to further develop improved immune-oncological therapies (Chen and Mellman, 2013; Galon et al., 2013; Motz and Coukos, 2013; Zitvogel et al., 2013). In the present study, we describe that NR2F6 plays a crucial role for effector T cell-dependent anti-tumor immunity. First, we defined the enhanced survival of Nr2f6-deficient tumor-bearing mice and their superior immune cell composition using the prostate tumor model TRAMP. Second, we validated these findings by using various transplantable tumor models showing the survival benefit and the induction of a protective anti-tumor immune response including long-lasting immunological memory in Nr2f6-deficient mice. Third, we highlight that the observed anti-tumor effects depend on NR2F6 function in T cells, as adoptive transfer of both OT-I TCR-transgenic and even polyclonal Nr2f6-deficient T cells into tumor-bearing immunocompetent wild-type mice is sufficient to delay tumor growth.
Albeit one cannot exclude the possibility that other immune or non-immune cells are at least to some extent involved, consistent results generated in all of our experimental approaches validate the negative regulatory role of NR2F6 during T cell activation in cancer. Thus, NR2F6 acts as a bona fide intracellular immune checkpoint in T cells. This statement is further supported by our finding that NR2F6 deletion is equally effective than blocking PD-1/PD-L1 interaction as an established immune-checkpoint mechanism.
We observed significantly elevated numbers of tumor-infiltrating CD4+ and CD8+ T cells, as well as increased IL-2, IFN-γ, and TNF-α secretion rates in ex vivo analysis as well as at the tumor site in Nr2f6−/− tumor-bearing mice. Deficiency of Nr2f6 thus leads to T cells with an a priori lowered TCR activation threshold, finally resulting in enhanced cytokine secretion of IL-2 as an established key cytokine potently favoring tumor rejection (reviewed by Liao et al., 2013). Along this line, we also observed increased production of IFN-γ by effector T cells. Importantly, IFN-γ has been shown to play a pivotal role in tumor-protective immune responses (Weiss et al., 2011): it is critically required for the “productive” immune surveillance of spontaneous malignancies and chemical carcinogen methylcholanthrene (MCA)-induced sarcomas, immune memory formation, and especially senescence of cancer cells (Braumüller et al., 2013).
Mechanistically, our data suggest that NR2F6 acts as a transcriptional suppressor of NFAT/AP-1-mediated signaling in T cells and its deletion results in enhanced sensitivity to CD3/CD28 activation. Similar to the action of other NRs, NR2F6 appears to directly bind to sites that are otherwise occupied by NFAT/AP-1 to prevent transcriptional activation. This supports a model in which the balance of pre-bound NR2F6 versus TCR-triggered NFAT/AP-1 DNA-binding-capabilities directly dictates the outcome of, e.g., IFN-γ cytokine production. As NFAT/AP-1 transcription factors are known to bind to the Il2, Ifng, and Tnfa promoters, it is therefore likely that NR2F6 mediates its suppressive effects in tumor-reactive effector CD4+ Th1 and CD8+ T cells via this cell-intrinsic mechanism. Importantly, however, CD3/CD28-induced activation apparently leads to an induced displacement of NR2F6 from the given cytokine promoter, thereby promoting unopposed DNA binding of NFAT/AP-1-complexes at the critical cytokine gene loci.
The immunological microenvironment observed in Nr2f6-deficient tumor-bearing mice assemble a positive immune contexture potentially supporting cancer cell rejection. This hypothesis is strengthened by correlative clinical data showing that an increased rate of tumor-infiltrating T cells is an important predictor of clinical outcome in cancer patients (reviewed in Galon et al., 2013; Fridman et al., 2012). Moreover, increased expression levels of PD-1 within the TIL compartment in Nr2f6−/− mice fit to the recent observation that PD-1-expressing CD8+ T cells are the tumor-reactive ones (Gros et al., 2014). Intriguingly, Nr2f6 is substantially upregulated ex vivo in CD3/CD28-stimulated CD4+ and CD8+ T cell cultures, respectively, indicating a dynamic regulation of Nr2f6 expression as a negative feedback loop. Nr2f6 gene ablation therefore results in immune response outcomes that, albeit by a different mode of action, appears reminiscent to the blockade of immune tolerance signaling induced by CTLA-4 or PD-1/PD-L1 (Robert et al., 2011; Hodi et al., 2010; Hamid et al., 2013; Brahmer et al., 2010; Chen and Mellman, 2013; Fridman et al., 2012; Powles et al., 2014; Tumeh et al., 2014).
Interestingly, a correlation of NR2F6 expression and human cancer has been observed: a compendium of data sets indicates that Nr2f6 expression is predominately upregulated in human ovarian cancer, colon cancer, and lymphoma (King et al., 2011; Li et al., 2011; Eckerle et al., 2009; Ichim et al., 2011). Genome-wide association studies and expression analysis have linked NR2F6 to breast cancer, when compared with the expression pattern of the other 46 human nuclear receptors (Antoniou et al., 2010; Muscat et al., 2013). Although it is impossible from these published data sets to determine whether the origin of NR2F6 expression is the malignant or the immune cell, in one breast cancer study tumor-infiltrating T cells were isolated and shown to have upregulated Nr2f6 expression compared to peripheral T cells (Gu-Trantien et al., 2013).
Because of the established T cell dysfunction as a consequence of cancer-mediated immunosuppression, our data strongly suggest that T cell-based therapies could significantly benefit from modulation of this NR2F6 inhibitory signaling pathway. Importantly, the ligand binding domain (LBD) on NR2F6 is evolutionarily highly conserved and appears to be essential for NR2F6 transcriptional repressor activity (Hermann-Kleiter et al., 2012). This potential drugability of its LBD for a “small molecule checkpoint blockade drug” may provide a rational mechanistic basis envisioning targeted manipulation of NR2F6 in T cells as a promising intracellular checkpoint-targeting strategy to improve the efficacy and broaden the applicability of cancer immunotherapy regimens, finally enabling prolonged patient survival.
In sum, orphan nuclear receptor NR2F6 appears to be an intracellular immune checkpoint, directly repressing transcription of cytokine genes in T cells relevant for cancer cell rejection, such as IL-2, IFN-γ, and TNF-α. Thus, in the presence of NR2F6 effector T cell activation is limited within the tumor microenvironment. The exact molecular pathway by which NR2F6 impairs the transcriptional amplitude of NFAT/AP-1 gene transactivation including all target genes of NR2F6 demands further investigations.
Experimental Procedures
Mice
Nr2f6-deficient mice (Warnecke et al., 2005) backcrossed on C57BL/6 background were used. Wild-type C57BL/6 TRAMP (Tg[TRAMP]8247Ng) mice were crossed into Nr2f6-deficient mice to generate Nr2f6−/−TRAMP and Nr2f6+/+TRAMP mice. All animal experiments have been performed in accordance with national and European guidelines and have been reviewed and authorized by the committee on animal experiments (BMWFW-66.0ll/0128-WF/V/3b/2014).
Tumor Induction and Adoptive Cell Transfer
1 × 105 B16-OVA, 3 × 105 B16-F10 cells, 1.5 × 105 EG7 cells, or 1 × 106 TRAMP-C1 cells (purchased from the ATCC, CRL-2730) were injected subcutaneously (s.c.) into the left flank of 8- to 12-week-old mice. Tumor memory experiments were performed via s.c. injection of 1.5 × 106 EG7 cells into 1-year-old Nr2f6−/− mice, which rejected primary tumor challenge. Tumor growth was monitored three times per week by measuring tumor length and width. Tumor volume was calculated according to the following equation: 1/2(length × width2). For survival analysis, mice with tumors greater than the length limit of 15 mm were sacrificed and counted as dead.
Preparation of Tumor-Infiltrating Cells
Mononuclear infiltrating cells were isolated from both subcutaneous and autochthonous tumors at the indicated time points. Briefly, tumor tissues from sacrificed mice were prepared by mechanical disruption followed by digestion for 45 min with collagenase D (2.5 mg/ml; Roche, 11088858001) and DNase I (1 mg/ml; Roche, 11284932001) at 37°C. Digested tissues were incubated 5 min at 37°C with EDTA (0.5 M) to prevent DC/T cell aggregates and mashed through filters.
Statistics
Data were analyzed using Prism 5.03 software (GraphPad). Experiments were repeated at least two or three times. Data are represented as indicated (either mean ± SEM or ± SD) for all figure panels in which error bars are shown. The p values were assessed using two-tailed unpaired Student’s t test, log-rank test, or ANOVA. A p value of <0.05 was considered statistically significant: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Author Contributions
All authors have contributed substantially to the work. S.W., K.S., S. Klepsch, S.T., S. Kaminski, A.V., T.G., and C.P.-O. performed key experiments and/or analyses of the data. N.H.-K. outlined and performed the overall experimental design of tumor and memory experiments, in vitro experiments, ChIP analysis, and qRT-PCR, prepared figures, and wrote the initial paper draft; V.K. performed flow cytometry, tumor, and adoptive transfer experiments. G.B. and D.W. provided the study conception, and G.B. provided the overall supervision of the research at all stages.
Acknowledgments
This work was supported by grants from the FFG Austrian Research Promotion Agency (BRIDGE-842388-CBL-AIM) and FWF Austrian Science Fund (W1101-B18, P23537-B13, P25044-B21, “Hertha-Firnberg” T264-B13, and “Lise-Meitner” M1636-B23), the Austrian Cancer Society, Tyrol as well as the Deutsche Forschungsgemeinschaft (DFG WO1877/1-1). We are grateful to Michaela Kind and Nina Posch (both from our institute in Innsbruck) for technical assistance.
Published: September 17, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.08.035.
Contributor Information
Natascha Hermann-Kleiter, Email: natascha.kleiter@i-med.ac.at.
Gottfried Baier, Email: gottfried.baier@i-med.ac.at.
Supplemental Information
References
- Antoniou A.C., Wang X., Fredericksen Z.S., McGuffog L., Tarrell R., Sinilnikova O.M., Healey S., Morrison J., Kartsonaki C., Lesnick T., EMBRACE. GEMO Study Collaborators. HEBON. kConFab. SWE-BRCA. MOD SQUAD. GENICA A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010;42:885–892. doi: 10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedognetti D., Spivey T.L., Zhao Y., Uccellini L., Tomei S., Dudley M.E., Ascierto M.L., De Giorgi V., Liu Q., Delogu L.G. CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and IL-2. Br. J. Cancer. 2013;109:2412–2423. doi: 10.1038/bjc.2013.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor J., Bopp T., Vaeth M., Klein M., Serfling E., Hünig T., Becker C., Schild H., Schmitt E. Cyclic AMP underpins suppression by regulatory T cells. Eur. J. Immunol. 2012;42:1375–1384. doi: 10.1002/eji.201141578. [DOI] [PubMed] [Google Scholar]
- Brahmer J.R., Drake C.G., Wollner I., Powderly J.D., Picus J., Sharfman W.H., Stankevich E., Pons A., Salay T.M., McMiller T.L. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braumüller H., Wieder T., Brenner E., Aßmann S., Hahn M., Alkhaled M., Schilbach K., Essmann F., Kneilling M., Griessinger C. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- Chen D.S., Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Eckerle S., Brune V., Döring C., Tiacci E., Bohle V., Sundström C., Kodet R., Paulli M., Falini B., Klapper W. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia. 2009;23:2129–2138. doi: 10.1038/leu.2009.161. [DOI] [PubMed] [Google Scholar]
- Fridman W.H., Pagès F., Sautès-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- Galon J., Angell H.K., Bedognetti D., Marincola F.M. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Greenberg N.M., DeMayo F., Finegold M.J., Medina D., Tilley W.D., Aspinall J.O., Cunha G.R., Donjacour A.A., Matusik R.J., Rosen J.M. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A., Robbins P.F., Yao X., Li Y.F., Turcotte S., Tran E., Wunderlich J.R., Mixon A., Farid S., Dudley M.E. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu-Trantien C., Loi S., Garaud S., Equeter C., Libin M., de Wind A., Ravoet M., Le Buanec H., Sibille C., Manfouo-Foutsop G. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Invest. 2013;123:2873–2892. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin M.M., Zhang X., Schuster H., Caron E., Ward J.P., Noguchi T., Ivanova Y., Hundal J., Arthur C.D., Krebber W.J. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O., Robert C., Daud A., Hodi F.S., Hwu W.J., Kefford R., Wolchok J.D., Hersey P., Joseph R.W., Weber J.S. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann-Kleiter N., Gruber T., Lutz-Nicoladoni C., Thuille N., Fresser F., Labi V., Schiefermeier N., Warnecke M., Huber L., Villunger A. The nuclear orphan receptor NR2F6 suppresses lymphocyte activation and T helper 17-dependent autoimmunity. Immunity. 2008;29:205–216. doi: 10.1016/j.immuni.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann-Kleiter N., Meisel M., Fresser F., Thuille N., Müller M., Roth L., Katopodis A., Baier G. Nuclear orphan receptor NR2F6 directly antagonizes NFAT and RORγt binding to the Il17a promoter. J. Autoimmun. 2012;39:428–440. doi: 10.1016/j.jaut.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi F.S., O’Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunder N.N., Wallen H., Cao J., Hendricks D.W., Reilly J.Z., Rodmyre R., Jungbluth A., Gnjatic S., Thompson J.A., Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichim C.V., Atkins H.L., Iscove N.N., Wells R.A. Identification of a role for the nuclear receptor EAR-2 in the maintenance of clonogenic status within the leukemia cell hierarchy. Leukemia. 2011;25:1687–1696. doi: 10.1038/leu.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M., June C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff P.W., Schuetz T.J., Blumenstein B.A., Glode L.M., Bilhartz D.L., Wyand M., Manson K., Panicali D.L., Laus R., Schlom J. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010;28:1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King E.R., Tung C.S., Tsang Y.T.M., Zu Z., Lok G.T.M., Deavers M.T., Malpica A., Wolf J.K., Lu K.H., Birrer M.J. The anterior gradient homolog 3 (AGR3) gene is associated with differentiation and survival in ovarian cancer. Am. J. Surg. Pathol. 2011;35:904–912. doi: 10.1097/PAS.0b013e318212ae22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-B., Jiao S., Sun H., Xue J., Zhao W.-T., Fan L., Wu G.-H., Fang J. The orphan nuclear receptor EAR2 is overexpressed in colorectal cancer and it regulates survivability of colon cancer cells. Cancer Lett. 2011;309:137–144. doi: 10.1016/j.canlet.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Liao W., Lin J.-X., Leonard W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz G.T., Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat G.E.O., Eriksson N.A., Byth K., Loi S., Graham D., Jindal S., Davis M.J., Clyne C., Funder J.W., Simpson E.R. Research resource: nuclear receptors as transcriptome: discriminant and prognostic value in breast cancer. Mol. Endocrinol. 2013;27:350–365. doi: 10.1210/me.2012-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Yaguchi H., Ohkura N., Kitabayashi I., Nagamura Y., Nomura T., Miyachi Y., Tsukada T., Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- Pipkin M.E., Sacks J.A., Cruz-Guilloty F., Lichtenheld M.G., Bevan M.J., Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T., Eder J.P., Fine G.D., Braiteh F.S., Loriot Y., Cruz C., Bellmunt J., Burris H.A., Petrylak D.P., Teng S.L. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- Restifo N.P., Dudley M.E., Rosenberg S.A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C., Thomas L., Bondarenko I., O’Day S., Weber J., Garbe C., Lebbe C., Baurain J.F., Testori A., Grob J.J. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber D.J., Lawson D.H., Richards J.M., Conry R.M., Miller D.M., Treisman J., Gailani F., Riley L., Conlon K., Pockaj B. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011;364:2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serfling E., Chuvpilo S., Liu J., Höfer T., Palmetshofer A. NFATc1 autoregulation: a crucial step for cell-fate determination. Trends Immunol. 2006;27:461–469. doi: 10.1016/j.it.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Shankaran V., Ikeda H., Bruce A.T., White J.M., Swanson P.E., Old L.J., Schreiber R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- Topalian S.L., Drake C.G., Pardoll D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Eertwegh A.J., Versluis J., van den Berg H.P., Santegoets S.J., van Moorselaar R.J., van der Sluis T.M., Gall H.E., Harding T.C., Jooss K., Lowy I. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:509–517. doi: 10.1016/S1470-2045(12)70007-4. [DOI] [PubMed] [Google Scholar]
- Warnecke M., Oster H., Revelli J.P., Alvarez-Bolado G., Eichele G. Abnormal development of the locus coeruleus in Ear2(Nr2f6)-deficient mice impairs the functionality of the forebrain clock and affects nociception. Genes Dev. 2005;19:614–625. doi: 10.1101/gad.317905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G.R., Grosh W.W., Chianese-Bullock K.A., Zhao Y., Liu H., Slingluff C.L., Jr., Marincola F.M., Wang E. Molecular insights on the peripheral and intratumoral effects of systemic high-dose rIL-2 (aldesleukin) administration for the treatment of metastatic melanoma. Clin. Cancer Res. 2011;17:7440–7450. doi: 10.1158/1078-0432.CCR-11-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L., Tesniere A., Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat. Rev. Immunol. 2006;6:715–727. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- Zitvogel L., Galluzzi L., Smyth M.J., Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.