

Abstract
Type 1 Diabetes (T1D) is characterized by the immune mediated destruction of β cells. Clinical studies have focused on drug therapies to modulate autoimmunity, yet none of these interventions has resulted in durable preservation of β-cell function. These findings raise the possibility that initiating or propagating events outside of the immune system should be considered in future efforts to prevent or reverse T1D. An emerging concept suggests that defects inherent to the β cell may trigger autoimmunity. A study by Engin et al. in type 1 diabetic NOD mice suggests that excessive β-cell endoplasmic reticulum stress arising from environmental insults results in abnormal protein synthesis, folding, and/or processing. Administration of the chemical protein folding chaperone TUDCA resulted in recovery of β-cell endoplasmic reticulum function and a diminished incidence of diabetes in NOD mice. We propose here that these data and others support a model whereby an inadequate or defective β-cell endoplasmic reticulum response results in the release of β-cell antigens and neoantigens that initiate autoimmunity. Pharmacologic therapies that either mitigate these early β-cell stressors or enhance the ability of β cells to cope with such stressors may prove to be effective in the prevention or treatment of T1D.
Keywords: ATF6, ER stress, autoimmunity, sXBP1, type 1 diabetes, unfolded protein response, β cell
Type 1 diabetes (T1D) arises when loss of islet β-cell function or mass results in the absolute deficiency of insulin. T1D is thought to occur from a breakdown in immune tolerance, resulting in the infiltration into the islet of auto-reactive T cells that target β cells.1 The immunobiology of T1D has been studied most extensively in the non-obese diabetic (NOD) mouse, which exhibits early and gradual infiltration of immune cell types into the pancreas (“insulitis”) with accompanying loss of β-cell mass.2 In the setting of a dysregulated immune system, it has been proposed that externalization of β-cell antigens activates autoreactive T cells and give rise to invasive insulitis.3
Although insulitis of the extent observed in NOD mice has not been as uniformly documented in human autopsy studies, a similar failure of immune tolerance is thought to occur in humans.4 Several immune modulatory drugs with well characterized responses in other autoimmune diseases have been attempted in clinical trials of new-onset T1D subjects. Whereas administration of some of these drugs (such as anti-CD3, anti-CD20, CTLA4-Ig) have led to the preservation of β-cell function (as assessed by C-peptide secretion) for periods of months, subsequent declines in β-cell function paralleled those of placebo.5-8 In no case has true remission, as defined by insulin independence, been observed. These outcomes could be explained by the low replicative capacity of human β cells,9,10 coupled with the possibility that interventions were initiated too late in the disease—at a time in which molecular stress pathways in β cells were so aggressively activated that even a respite from autoimmunity could not prevent β-cell decline. If true, what remains unanswered is the cause and nature of these intrinsic β cell stress pathways, and whether therapies that target these pathways might substantially alter outcomes. It should be noted that these outcomes in humans differ substantially from those seen in NOD mice, in which a number of interventions have been shown to prevent or even lead to remission of T1D.2 Collectively, these considerations have sparked a new perspective in the field, in which greater emphasis has been placed on elucidating the molecular mechanisms within β cells that might initiate or perpetuate autoimmunity and eventual β cell demise.
By all accounts, β cells are a vulnerable cell type with little reserve. Notably, β cell mass is relatively small—less than 1 g in most humans,11 and possibly even lower in individuals susceptible to T1D.12,13 To compound the low mass reserve, β cells have a low proliferative rate. In humans, β cell replication falls exponentially in the immediate postnatal period, to virtually zero by adulthood.9,10 In addition to limited mass and proliferation, β cells also have limited functional reserve. As professional producers and secretors of insulin, β cells rely heavily upon the endoplasmic reticulum (ER) to ensure that proteins are produced robustly and folded efficiently. As such, even minor perturbations in calcium homeostasis, oxidative stress, and peripheral insulin demand can impose stresses that can cause the β-cell ER to decompensate (“ER stress”) and fail to efficiently produce, fold, and process relevant proteins.14 Unfolded or improperly processed proteins that exit the β cell have the potential to trigger autoimmunity.15 Dysfunction of the β cell—as judged by an impaired insulin secretory response to glucose—appears to precede the development of frank T1D in both mice and humans.16-19 Only recently have studies correlated the decline in β-cell function with the appearance of ER stress,16,20 thus raising the question of whether ER stress is a cause or consequence of β-cell dysfunction in T1D.
In a recent study, Engin, et al.21 engaged 2 mouse models of autoimmune diabetes and a chemical chaperone of protein folding to address if ER stress is causative of β-cell dysfunction and subsequent T1D. Both models, the NOD mouse and the RIP-LCMV-GP mouse (rat insulin promoter-lymphocytic choriomeningitis virus–glycoprotein, in which diabetes is induced by viral infection), exhibited invasive insulitis and β-cell destruction. The authors first studied the 3 major arms of the unfolded protein response (UPR) cascade—PERK (protein kinase R-like endoplasmic reticulum kinase), ATF6 (activating transcription factor 6), and IRE1α (inositol requiring 1α)—as they became activated in an attempt to resolve ER stress. These arms collectively result in global inhibition of mRNA translation initiation, increased production of protein folding chaperones, and ER biogenesis. Specifically, PERK phosphorylates the translation initiation factor eIF2α, resulting in inhibition of CAP-dependent mRNA translation. ATF6, upon cleavage by S1P and S2P proteases, is translocated to the nucleus to activate gene transcription. IRE1α is an endoribonuclease that splices Xbp1 mRNA to produce sXBP1 protein. Both ATF6 and sXBP1 enhance the transcription of genes encoding ER chaperones and other UPR intermediates (for reviews, see refs. 22 and 23). By pancreas tissue immunofluorescence, Engin, et al. showed that both NOD and RIP-LCMV-GP mice exhibit age-dependent loss of ATF6 and sXBP1 in the weeks preceding the development of T1D, a finding suggestive of a failure of the “proresolution” functions of the UPR. To correlate these findings to human T1D, the authors then studied tissue sections from human subjects. Interestingly, when compared with controls, T1D subjects exhibited reduced ATF6 and sXBP1 staining intensity in residual β cells, a finding more striking in females with diabetes for 8–20 y duration (for unclear reasons).
To address more directly whether failure of the proresolution function of the UPR contributes to β-cell dysfunction and frank T1D, the authors next administered the chemical protein folding chaperone taurine-conjugated ursodeoxycholic acid (TUDCA).24 In both mouse models, TUDCA treatment reduced β cell death, restored insulin secretion, and reduced substantially the incidence of T1D. The molecular mechanism linking TUDCA to its effect appeared to be via stimulation of ATF6 and sXBP1 expressions in β cells. Consistent with this possibility, TUDCA had no effect in preventing T1D in a mouse model of RIP-LCMV-GP lacking ATF6 protein in β cells. Taken together, these data support the notion that ER stress is an important contributor to β-cell dysfunction and eventual T1D in mice, and that β cells in these mouse strains appear to have defective adaptive UPRs.
Whereas prior studies suggested a role for ER stress in the pathogenesis of T1D,16,20 the study by Engin, et al.21 is the first to provide a direct link between ER stress, β-cell dysfunction, and T1D development. Nevertheless, important questions still remain. First, does this study rule out a role for immune cells in triggering β-cell destruction? Because TUDCA was administered systemically, some uncertainty still exists as to whether the drug might have affected immune cells directly. In this regard, immune cells undergo rapid protein production and ER expansion upon antigenic stimulation,25 and therefore it remains possible that systemic TUDCA administration has combined and synergistic effects on both β cells and immune cells. Although the ineffectiveness of TUDCA in the β-cell-specific ATF6-deficient mouse model seemingly discounts this possibility, it is unclear whether the RIP-LCMV-GP mouse model can be used interchangeably with the NOD model, as the viral etiology of T1D remains controversial.26 Therefore, it would seem important to study NOD mice in which ATF6 is either specifically deleted or overexpressed in islet β cells to directly test a role for protein folding in diabetes pathogenesis. Nonetheless, from a β-cell perspective, it is tempting to speculate that the reduced insulitis in TUDCA-treated animals arose from reduced β-cell auto-antigen exposure.
Second, if not autoimmunity, what are the factors that initiate β-cell ER stress in the setting of T1D, and third, are these findings relevant to human Type 1 diabetes? Considerable work has been performed to address triggers of ER stress, which has been shown to be induced by hyperglycemia and resultant oxidative stress, saturated free fatty acids, pro-inflammatory cytokines, proinsulin mutations, and double-stranded RNA, among others.14 Using a bioassay with peripheral blood mononuclear cells in vitro, Wang, et al.27 noted that serum of pre- and recent-onset T1D individuals induce a strong transcriptional signature of innate immunity. Similar findings were noted in the biobreeding rat model of T1D.28-30 These findings suggest that systemic inflammation is present in individuals destined to develop T1D, and that sustained inflammation could lead to ER stress in β cells. The source of this systemic inflammation remains vague, but environmental factors such as viruses or other infections, components of the gut microbiome, prenatal factors, insulin resistance, and diet remain possible candidates.31-33 Observational studies such as TEDDY (The Environmental Determinants of Diabetes in the Young) are focusing on correlation of T1D with such factors.
The observation by Engin, et al.21 that human β cells exhibit similar expression profiles to the mouse models, is reassuring, but by no means definitive that these findings can be extended to human disease. A number of monogenic disorders have been described that lead to β-cell ER stress and death, resulting in early onset of diabetes without apparent autoimmunity. For example, mutations in the WFS1 gene lead to Wolfram syndrome, which is characterized by childhood-onset diabetes, hypoinsulinemia, diabetes insipidus, optic atrophy, and deafness.34,35 Mutations in the gene encoding PERK results in Wolcott-Rallison syndrome, which includes neonatal or early onset diabetes, skeletal dysplasia, and growth retardation.36 At present, genome-wide association studies have not uncovered polymorphisms or T1D risk alleles associated with the UPR. However, studies are beginning to identify UPR genes associated with a number of neurodegenerative disorders including progressive supranuclear palsy and Alzheimer disease.37 Interestingly, while GWAS studies have primarily identified polymorphisms in genes thought to solely impact immune function, over 60% of T1D candidate genes are also expressed in human islets exposed to proinflammatory cytokines.38 Similar to prevailing theories regarding the development of type 2 diabetes, the idea of T1D emerging in the context of a β cell that is inherently susceptible to UPR activation and/or ER stress is intriguing and one that bears further testing. In support of this notion, Donath and colleagues identified a family with T1D stemming from a point mutation in the histone deacetylase SIRT-1 that resulted in increased β-cell nitric oxide synthesis and cytokine production in the context of inflammation.39
Taken together, the study of Engin, et al.21 and an emerging body of work from others suggests a model of T1D pathogenesis that shifts the focus from immunobiology to β-cell biology. The accompanying figure (Fig. 1) depicts a process in which systemic inflammation arising from the environment leads to inflammatory signaling in islet β cells. When inflammation is sustained or in the context of a susceptible islet β cell, there is activation of the UPR and triggering of ER stress that can result in either β-cell death or dysfunction, with the release of β-cell antigens and endogenous “neoantigens” (i.e., in this case an antigenic determinant emanating from misfolded or misprocessed β-cell proteins), which secondarily induce autoimmunity. As noted previously, many of the autoantigens described in T1D, including proinsulin, ZnT8, chromogranin, GAD65, and IA-2, are channeled through the ER.15,40,41 In this hypothetical model, several factors can increase the susceptibility to β-cell dysfunction, such as reductions in starting β-cell mass and genetic factors that may diminish and/or enhance β cell to ER stress-responsiveness.
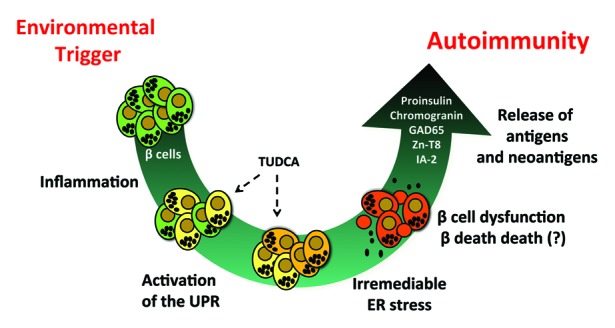
Figure 1. The β cell-centric model of T1D pathogenesis. The model proposes that sustained systemic inflammation arising from environmental factors (e.g., viral infections, gut microbiome, diet) leads to development of ER stress in β cells. Irremediable ER stress, as a result of failed compensatory responses by ATF6 and sXBP1, leads to the dysfunction and death of β cells. The subsequent release of β-cell antigens and endogenous neoantigens triggers secondary autoimmunity. A vicious cycle then ensues, leading to destruction of β-cell mass and development of T1D. The figure shows the potential stages in pathogenesis where intervention with TUDCA might allow for ER stress remediation.
We acknowledge that many aspects of this model remain controversial. Most importantly, several reports suggest a disconnect between β-cell apoptosis and/or death and activation of ER stress. A study by Satoh, et al.42 demonstrated that CHOP is dispensable in the development of type 1 diabetes. There is also disagreement with regard to how cytokines induce β cell death. At least 2 reports suggest that death is independent of ER stress in cell lines43 and the NOD mouse.44 By contrast, a study in INS-1 cells suggests that pro-inflammatory cytokines and classical ER stress inducers like thapsigargin are both capable of inducing death and ER stress, but differ significantly in how they activate specific components of UPR signaling.45
Whereas β-cell death may not be sufficient or necessary for induction of an autoimmune response,46 these studies do not rule out the important possibility that secretion or liberation of of neo-antigens in the context of ER stress leads to the loss of immune tolerance. Based on our understanding of rare disorders such as Wolfram and Wolcott-Rallison syndromes, hyperactivation of ER stress alone is unlikely as well to completely drive this process, as individuals with these disorders lack detectable β-cell autoantibodies. Notwithstanding these controversies, this model should serve as a framework for further refinement using humans and human model systems. Recent application requests from the Juvenile Diabetes Research Foundation (JDRF) and the National Institutes of Health (NIH) increasingly emphasize the need to translate studies from rodent diabetes models to humans and, in this respect, utilization of clinically-approved drugs in rodent models is an important first step in that translation process. As noted by the Engin, et al.,21 TUDCA has been approved in certain cases of liver disease,47,48 and therefore has potential to move into clinical trials of T1D. These strategies will need to be tested in individuals in whom T1D risk is high enough to justify initiation of the drug for prevention studies. Assuming that appropriate biomarkers can be identified that predict—with reasonable certainty—the development of T1D, the opportunity may exist to prevent diabetes using drugs that target β-cell ER function and protein folding. At the very least, this study informs the growing dialog suggesting that intrinsic β-cell stress pathways play important role in T1D progression. This dialog is increasingly important as results from recent large clinical trials in subjects with new onset T1D using drugs that solely impact immune function have met with limited success.7,8,49,50 As such, rational combinations of drugs that address aspects of both T1D immunobiology and β-cell biology are urgently needed.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Research in Dr Mirmira’s lab is supported by NIH grants R01 DK060581and R01 DK083583 and by grants from the George and Frances Ball Foundation, and the Ball Bros. Foundation. Research in Dr Evans-Molina’s lab is supported by NIH grant R01 DK093954, VA MERIT award I01 BX001733, and by grants from the Juvenile Diabetes Research Foundation, the George and Frances Ball Foundation, the Ball Bros. Foundation, and Sigma Beta Sorority. The funders had no role in the preparation of or decision to publish this manuscript.
Glossary
Abbreviations:
- ATF6
activating transcription factor 6
- ER
endoplasmic reticulum
- IRE1α
inositol requiring 1α
- NOD
non-obese diabetic
- PERK
protein kinase R-like endoplasmic reticulum kinase
- RIP-LCMV-GP
rat insulin promoter-lymphocytic choriomeningitis virus–glycoprotein
- sXBP1
spliced X-box binding protein 1
- T1D
type 1 diabetes
- TUDCA
taurine-conjugated ursodeoxycholic acid
- UPR
unfolded protein response
REFERENCES
- 1.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10:501–13. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 2.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 3.Mathis D, Vence L, Benoist C. β-Cell death during progression to diabetes. Nature. 2001;414:792–8. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 4.Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TWH, Atkinson MA, Roep BO, von Herrath MG. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209:51–60. doi: 10.1084/jem.20111187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–8. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 6.Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54:1763–9. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–52. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, et al. Type 1 Diabetes TrialNet Abatacept Study Group Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378:412–9. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97:3197–206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–94. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36:111–7. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fonseca V, Berger LA, Beckett AG, Dandona P. Size of pancreas in diabetes mellitus: a study based on ultrasound. Br Med J (Clin Res Ed) 1985;291:1240–1. doi: 10.1136/bmj.291.6504.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell-Thompson M, Wasserfall C, Montgomery EL, Atkinson MA, Kaddis JS. Pancreas organ weight in individuals with disease-associated autoantibodies at risk for type 1 diabetes. JAMA. 2012;308:2337–9. doi: 10.1001/jama.2012.15008. [DOI] [PubMed] [Google Scholar]
- 14.Evans-Molina C, Hatanaka M, Mirmira RG. Lost in translation: endoplasmic reticulum stress and the decline of β-cell health in diabetes mellitus. Diabetes Obes Metab. 2013;15(Suppl 3):159–69. doi: 10.1111/dom.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arvan P, Pietropaolo M, Ostrov D, Rhodes CJ. Islet autoantigens: structure, function, localization, and regulation. Cold Spring Harb Perspect Med. 2012;2:a007658. doi: 10.1101/cshperspect.a007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, Mirmira RG. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–27. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ize-Ludlow D, Lightfoot YL, Parker M, Xue S, Wasserfall C, Haller MJ, Schatz D, Becker DJ, Atkinson MA, Mathews CE. Progressive erosion of β-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes. 2011;60:2086–91. doi: 10.2337/db11-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sosenko JM, Skyler JS, Beam CA, Krischer JP, Greenbaum CJ, Mahon J, Rafkin LE, Matheson D, Herold KC, Palmer JP, Type 1 Diabetes TrialNet and Diabetes Prevention Trial–Type 1 Study Groups Acceleration of the loss of the first-phase insulin response during the progression to type 1 diabetes in diabetes prevention trial-type 1 participants. Diabetes. 2013;62:4179–83. doi: 10.2337/db13-0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS, DPT-1 Study Group Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes. 2010;59:679–85. doi: 10.2337/db09-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marhfour I, Lopez XM, Lefkaditis D, Salmon I, Allagnat F, Richardson SJ, Morgan NG, Eizirik DL. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia. 2012;55:2417–20. doi: 10.1007/s00125-012-2604-3. [DOI] [PubMed] [Google Scholar]
- 21.Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, Mathis D, Hotamisligil GS. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci Transl Med. 2013;5:ra156. doi: 10.1126/scitranslmed.3006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 24.Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab. 2010;12(Suppl 2):108–15. doi: 10.1111/j.1463-1326.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- 25.Todd DJ, Lee A-H, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–74. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 26.Stene LC, Rewers M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the enterovirus link to type 1 diabetes: critical review of human studies. Clin Exp Immunol. 2012;168:12–23. doi: 10.1111/j.1365-2249.2011.04555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180:1929–37. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- 28.Kaldunski M, Jia S, Geoffrey R, Basken J, Prosser S, Kansra S, Mordes JP, Lernmark A, Wang X, Hessner MJ. Identification of a serum-induced transcriptional signature associated with type 1 diabetes in the BioBreeding rat. Diabetes. 2010;59:2375–85. doi: 10.2337/db10-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YG, Mordes JP, Blankenhorn EP, Kashmiri H, Kaldunski ML, Jia S, Geoffrey R, Wang X, Hessner MJ. Temporal induction of immunoregulatory processes coincides with age-dependent resistance to viral-induced type 1 diabetes. Genes Immun. 2013;14:387–400. doi: 10.1038/gene.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levy H, Wang X, Kaldunski M, Jia S, Kramer J, Pavletich SJ, Reske M, Gessel T, Yassai M, Quasney MW, et al. Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes. Genes Immun. 2012;13:593–604. doi: 10.1038/gene.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eringsmark Regnéll S, Lernmark A. The environment and the origins of islet autoimmunity and Type 1 diabetes. Diabet Med. 2013;30:155–60. doi: 10.1111/dme.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, Drew JC, Ilonen J, Knip M, Hyöty H, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knip M, Veijola R, Virtanen SM, Hyöty H, Vaarala O, Akerblom HK. Environmental triggers and determinants of type 1 diabetes. Diabetes. 2005;54(Suppl 2):S125–36. doi: 10.2337/diabetes.54.suppl_2.S125. [DOI] [PubMed] [Google Scholar]
- 34.Strom TM, Hörtnagel K, Hofmann S, Gekeler F, Scharfe C, Rabl W, Gerbitz KD, Meitinger T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet. 1998;7:2021–8. doi: 10.1093/hmg/7.13.2021. [DOI] [PubMed] [Google Scholar]
- 35.Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, Mueckler M, Marshall H, Donis-Keller H, Crock P, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat Genet. 1998;20:143–8. doi: 10.1038/2441. [DOI] [PubMed] [Google Scholar]
- 36.Julier C, Nicolino M. Wolcott-Rallison syndrome. Orphanet J Rare Dis. 2010;5:29. doi: 10.1186/1750-1172-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Lee VM, Trojanowski JQ, Devlin B, Schellenberg GD, PSP Genetics Study Group The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:31. doi: 10.1186/2051-5960-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, Ortis F, Santin I, Colli ML, Barthson J, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012;8:e1002552. doi: 10.1371/journal.pgen.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biason-Lauber A, Böni-Schnetzler M, Hubbard BP, Bouzakri K, Brunner A, Cavelti-Weder C, Keller C, Meyer-Böni M, Meier DT, Brorsson C, et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab. 2013;17:448–55. doi: 10.1016/j.cmet.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Sullivan-Murphy B, Urano F. ER stress as a trigger for β-cell dysfunction and autoimmunity in type 1 diabetes. Diabetes. 2012;61:780–1. doi: 10.2337/db12-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherr J, Sosenko J, Skyler JS, Herold KC. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab. 2008;4:334–43. doi: 10.1038/ncpendmet0832. [DOI] [PubMed] [Google Scholar]
- 42.Satoh T, Abiru N, Kobayashi M, Zhou H, Nakamura K, Kuriya G, Nakamura H, Nagayama Y, Kawasaki E, Yamasaki H, et al. CHOP deletion does not impact the development of diabetes but suppresses the early production of insulin autoantibody in the NOD mouse. Apoptosis. 2011;16:438–48. doi: 10.1007/s10495-011-0576-2. [DOI] [PubMed] [Google Scholar]
- 43.Chambers KT, Unverferth JA, Weber SM, Wek RC, Urano F, Corbett JA. The role of nitric oxide and the unfolded protein response in cytokine-induced beta-cell death. Diabetes. 2008;57:124–32. doi: 10.2337/db07-0944. [DOI] [PubMed] [Google Scholar]
- 44.Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, King C, Biden TJ, Laybutt DR. Cytokine-induced β-cell death is independent of endoplasmic reticulum stress signaling. Diabetes. 2008;57:3034–44. doi: 10.2337/db07-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tonnesen MF, Grunnet LG, Friberg J, Cardozo AK, Billestrup N, Eizirik DL, Størling J, Mandrup-Poulsen T. Inhibition of nuclear factor-kappaB or Bax prevents endoplasmic reticulum stress- but not nitric oxide-mediated apoptosis in INS-1E cells. Endocrinology. 2009;150:4094–103. doi: 10.1210/en.2009-0029. [DOI] [PubMed] [Google Scholar]
- 46.Carrington EM, Kos C, Zhan Y, Krishnamurthy B, Allison J. Reducing or increasing β-cell apoptosis without inflammation does not affect diabetes initiation in neonatal NOD mice. Eur J Immunol. 2011;41:2238–47. doi: 10.1002/eji.201141476. [DOI] [PubMed] [Google Scholar]
- 47.Rubin RA, Kowalski TE, Khandelwal M, Malet PF. Ursodiol for hepatobiliary disorders. Ann Intern Med. 1994;121:207–18. doi: 10.7326/0003-4819-121-3-199408010-00009. [DOI] [PubMed] [Google Scholar]
- 48.Combes B, Carithers RL, Jr., Maddrey WC, Munoz S, Garcia-Tsao G, Bonner GF, Boyer JL, Luketic VA, Shiffman ML, Peters MG, et al. Biliary bile acids in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology. 1999;29:1649–54. doi: 10.1002/hep.510290618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rigby MR, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE, Patel CM, Griffin KJ, Tsalikian E, Gottlieb PA, et al. T1DAL Study Team Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013;1:284–94. doi: 10.1016/S2213-8587(13)70111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Jr., Bode B, Aronoff S, Holland C, Carlin D, et al. Protégé Trial Investigators Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–97. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]