

Abstract
Key points
The synaptic vesicle associated proteins synapsin I and synapsin II have important functions in synaptic short‐term plasticity.
We investigated their functions in cortical facilitatory feedback to neurons in dorsal lateral geniculate nucleus (dLGN), feedback that has important functions in state‐dependent regulation of thalamic transmission of visual input to cortex.
We compared results from normal wild‐type (WT) mice and synapsin knockout (KO) mice in several types of synaptic plasticity, and found clear differences between the responses of neurons in the synapsin I KO and the WT, but no significant differences between the synapsin II KO and the WT.
These results are in contrast to the important role of synapsin II previously demonstrated in similar types of synaptic plasticity in other brain regions, indicating that the synapsins can have different roles in similar types of STP in different parts of the brain.
Abstract
The synaptic vesicle associated proteins synapsin I (SynI) and synapsin II (SynII) have important functions in several types of synaptic short‐term plasticity in the brain, but their separate functions in different types of synapses are not well known. We investigated possible distinct functions of the two synapsins in synaptic short‐term plasticity at corticothalamic synapses on relay neurons in the dorsal lateral geniculate nucleus. These synapses provide excitatory feedback from visual cortex to the relay cells, feedback that can facilitate transmission of signals from retina to cortex. We compared results from normal wild‐type (WT), SynI knockout (KO) and SynII KO mice, in three types of synaptic plasticity mainly linked to presynaptic mechanism. In SynI KO mice, paired‐pulse stimulation elicited increased facilitation at short interpulse intervals compared to the WT. Pulse‐train stimulation elicited weaker facilitation than in the WT, and also post‐tetanic potentiation was weaker in SynI KO than in the WT. Between SynII KO and the WT we found no significant differences. Thus, SynI has important functions in these types of synaptic plasticity at corticothalamic synapses. Interestingly, our data are in contrast to the important role of SynII previously shown for sustained synaptic transmission during intense stimulation in excitatory synapses in other parts of the brain, and our results suggest that SynI and SynII may have different roles in similar types of STP in different parts of the brain.
Abbreviations
- DKO
double knock‐out
- dLGN
dorsal lateral geniculate nucleus
- EPSC
excitatory post‐synaptic current
- EPSCctr
control EPSC
- KO
knockout
- PPF
paired‐pulse facilitation
- PTP
post‐tetanic potentiation
- RRP
readily releasable pool
- STP
short‐term plasticity
- SV
synaptic vesicle
- SynI
synapsin I
- SynII
synapsin II
- TC
thalamo‐cortical
- WT
wild‐type
Introduction
Synaptic short‐term plasticity (STP) is closely linked to activity‐dependent trafficking of synaptic vesicles (SVs) in nerve terminals (Cesca et al. 2010). SVs are clustered in pools, and functional pools include a readily releasable pool (RRP), a recycling pool, and a resting pool (Denker & Rizzoli, 2010; Alabi & Tsien, 2012). Synapsins, which comprise a family of SV‐ and actin‐associated phosphoproteins, have important roles in the trafficking of SVs in presynaptic terminals (De Camilli et al. 1990; Greengard et al. 1993; Hilfiker et al. 1999; Hosaka & Südhof, 1999; Jovanovic et al. 2001; Chi et al. 2003), and inactivation of synapsins attenuates clustering and trafficking of SVs between pools in the terminals (e.g. Li et al. 1995; Rosahl et al. 1995; Takei et al. 1995; Gabriel et al. 2011; reviewed in Cesca et al. 2010; Bykhovskaia, 2011).
Synapsins in vertebrates are encoded by three genes (Südhof et al. 1989; Hosaka & Südhof, 1998; Kao et al. 1998; Ferreira et al. 2000). Phosphorylation of synapsins, in particular synapsin I (SynI) and II (SynII), depends on synaptic activity, and these proteins have important functions during synaptic transmission (Jovanovic et al. 2001; Chi et al. 2003), including functions related to STP (Cesca et al. 2010; Bykhovskaia 2011). SynI is biochemically more complex and has more phosphorylation sites than SynII (Cesca et al. 2010). Moreover, the activity of the synapsins is regulated through multiple phosphorylation pathways (Cesca et al. 2010; Bykhovsakaia, 2011) that seem to partly differ for SynI and SynII (Verstegen et al. 2014). Thus, the two proteins may influence vesicle trafficking in different manners (Cesca et al. 2010; Bykhovskaia 2011). Little is known about the specific roles of SynI and SynII in synaptic plasticity in different brain regions.
We previously studied the involvement of synapsins in synaptic transmission in the visual system with the main focus on the dorsal lateral geniculate nucleus (dLGN). The principal neurons in dLGN, thalamocortical (TC) neurons, receive primary excitatory input from retinal afferents ending on their proximal dendrites, and massive excitatory feedback from visual cortex ending on their distal dendrites (Wilson et al. 1984). Cortical feedback can provide strong facilitation of thalamic neurons (Andersen et al. 1972), facilitation that has been linked to mechanisms of attention (Singer, 1977). In the visual system a train of stimulus pulses in corticothalamic afferents can facilitate the response of TC neurons to retinal input. The facilitation increases during the train, and is strengthened by increasing pulse frequency (Lindström & Wrόbel, 1990; Turner & Salt, 1998; von Krosigk et al. 1999; Granseth, 2004; Kielland et al. 2006). Thus, the corticothalamic feedback may function as a positive gain‐regulator of thalamocortical transmission (Ahlsen et al. 1985; Lindström & Wrόbel, 1990). Presynaptic mechanisms presumably play a major role in such facilitation (Zucker & Regehr, 2002).
Despite the widespread distribution of synapsins in the brain (Walaas et al. 1988; Südhof et al. 1989), these proteins have distinct cellular distributions in the visual pathway. Synapsins are absent in terminals of all principal neurons from retinal photoreceptors to the layer 4 neurons in visual cortex (Mandell et al. 1990; Kielland et al. 2006; Owe et al. 2013), but are expressed in different subtypes in neurons with modulator functions at each level of the pathway. Specifically, in dLGN the retinal afferent to TC neurons lacks both SynI and SynII, local inhibitory interneurons express only SynI, and the corticothalamic neurons (excitatory feedback from cortical lamina 6 neurons) express both SynI and SynII (Kielland et al. 2006). The function of synapsins in the synaptic facilitation at corticothalamic synapses, studied by comparing response characteristics in Syn I/II double knockout (DKO) mice and wild‐type (WT) mice, showed increased paired‐pulse facilitation (PPF), decreased facilitation during train stimulation, and decreased post‐tetanic potentiation (PTP) in the DKO compared to the WT (Kielland et al. 2006).
Here we studied the separate involvement of SynI and SynII in these types of STP at the corticothalamic synapse. The results for the SynI KO were similar to the results obtained in the Syn I/II DKO (Kielland et al. 2006) whereas the results from the SynII KO was surprisingly similar to the results for the WT, indicating that SynI contrary to SynII has the major function in these types of STP at this synapse. This differs from results from previous studies of other types of excitatory synapses, where SynII has been shown to have the major role during repetitive transmission (Rosahl et al. 1995, Samigullin et al. 2004, Gitler et al. 2008), and our results suggest that SynI and SynII may have different roles in similar types of STP in different parts of the brain.
Methods
Ethical approval
All procedures were performed in accordance with the European Directive 2010/63/EU and the Norwegian Animal Welfare Act.
Slice preparation
Slices were obtained from wild‐type (WT) C57BL/6 mice, and from SynI KO and SynII KO mice fully backcrossed in the C57 background (Etholm et al. 2012) (Fig. 1). The animals were bred in our local animal facility at the University of Oslo. Animals (27–37 days old, males and females) were anaesthetized with sevofluran (Abbott, Solna, Sweden) and decapitated after reaching a surgical plane of anaesthesia verified by absence of the toe‐pinch reflex. The brain was quickly removed and chilled to 2–4°C in oxygenated (5% CO2–95% O2) solution that contained (mm): 75 glycerol, 87 NaCl, 25 NaHCO3, 2.5 KCl, 0.5 CaCl2, 1.25 NaH2PO4, 7 MgCl2 and 16 d‐glucose. Semi‐parasagittal brain slices (300 μm thick) comprising dLGN were made as described previously (Turner & Salt, 1998) using a vibroslicer (HR‐2, Sigmann Electronik, Hüffenhardt, Germany). The slices were incubated in oxygenated artificial cerebrospinal fluid (ACSF) containing (mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgCl2 and 10 d‐glucose (T = 32°C, osmolarity, 305–308 mosmol l−1).
Figure 1. Animal genotypes .
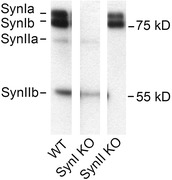
Immunoblotting analysis of dLGN samples from the WT, SynI KO and SynII KO mice, performed with a primary antibody recognizing Syns Ia, Ib, IIa and IIb. Three extracts from the same blot.
Stimulation and recordings
After at least 30 min incubation, the slices were moved to a recording chamber perfused with ACSF at the rate of 5 ml min−1 (T = 34°C). TC neurons in the dLGN were visualized with an upright microscope (Axioscope, Zeiss, Oberkochen, Germany) equipped with DIC optics and an infrared camera (C2400, Hamamatsu, Joko‐cho, Japan). Corticogeniculate fibres were electrically stimulated through a bipolar electrode (WPI, Sarasota, FL, USA; distance between the tips, ∼80 μm). The stimulation electrode was placed rostral to dLGN (Turner & Salt, 1998) clearly outside the nucleus. Excitatory postsynaptic currents (EPSCs) in TC neurons were elicited with short, bipolar current pulses (200 μs, 5–200 μA) using a digital stimulator (DS‐8000, WPI) and a stimulus isolator (HG203/100, HI‐MED, Berkshire, UK). In order to activate a high number of afferents to the recorded neuron, and to obtain consistent responses, we adjusted the stimulus intensity to a level that elicited rather strong single EPSCs (75–100% maximum EPSC). The EPSCs were recorded using a patch‐clamp amplifier (EPC 9, HEKA Elektronik, Lambrecht, Germany) in whole‐cell configuration. Neurons were voltage clamped at −60 mV. The pipette solution contained (mm): 135 caesium gluconate, 5 CsCl, 0.5 EGTA, 10 Hepes, 0.16 CaCl2, 4 Mg‐ATP, 0.4 Na‐GTP and 14 phosphocreatine; pH adjusted to 7.3 with CsOH, osmolarity, 298 mosmol l−1. Micropipettes (3.5–5.5 MΩ) were made of borosilicate glass (WPI), and pulled with a vertical puller (PB‐7, Narishige, Tokyo, Japan). During the recordings, GABAA receptors were blocked with picrotoxin (50 μm), and GABAB receptors with CGP54626 (10 μm). N‐Methyl‐d‐aspartic acid (NMDA) receptors were blocked with (RS)‐3‐(2‐carboxypiperazin‐4‐yl)propyl‐1‐phosphonic acid ((RS)‐CPP; 15 μm). All chemicals were obtained from Tocris Bioscience (Bristol, UK).
Data analyses
Recordings were filtered at 3 kHz and sampled at 10 kHz with Pulse software (Heka Elektronik). The offline data analyses were made with IgorPro 6.34 (WaveMetrics, Lake Oswego, OR, USA), Origin 9.1 (OriginLab Corp., Northampton, MA, USA) and Prism 6.05 (GraphPad Software, La Jolla, CA, USA) software. The amplitude of the EPSCs was measured as the difference between its peak and the baseline current, or the residual decaying current of the preceding EPSC. PPF at a given time interval was calculated as EPSC2/EPSC1. In experiments with train stimulation we applied a series of stimuli at 0.1 Hz before each stimulus train. The average amplitude of the responses to these stimuli was used as estimate of the response to single pulse stimulation (control EPSCs). The EPSCs recorded during the trains were normalized to the average of these control EPSCs (EPSCn/EPSCctr). Functions were fitted to the train data with the method of least squares. Straight lines were fitted to the normalized mean responses of the WT in three separate time segments to estimate the rate of change of facilitation in these periods. Exponential functions were fitted to the normalized mean response values during the whole train using the formula:
where r 0 is the r(t) value at infinite time, w 1 to w 3 are weighting coefficients, and τ1 to τ3 are time constants for the respective exponentials. The contribution of a given exponential to the triple exponential function, r(t), was evaluated by its relative weighting coefficient, i.e. the ratio of the absolute value of a given weighting coefficient to the sum of the absolute values of all three weighting coefficients.
Unless otherwise stated, statistical significance of differences was evaluated by Student's two‐tailed, unpaired t test; P < 0.05 was considered significant. Normality of the data distributions was verified with the Shapiro–Wilk normality test. The experimental data are presented as means ± SEM.
Results
We made whole‐cell patch‐clamp recordings from TC neurons in dLGN in acute brain slices prepared from WT, SynI KO and SynII KO mice (P27–P37). We compared the responses of the three genotypes to paired‐pulse stimulation, pulse‐train stimulation, and tetanic stimulation (see e.g. Zucker & Regehr, 2002).
Increased paired‐pulse facilitation in the SynI KO mice
Two successive electrical pulses of equal strength to fibres in the optic radiation can elicit PPF in post‐synaptic TC neurons such that the response to the second pulse is stronger than that to the first (Lindström & Wrόbel, 1990; Turner & Salt, 1998; von Krosigk et al. 1999; Granseth et al. 2002; Kielland et al. 2006). We compared PPF between the three genotypes in experiments where the interpulse interval was randomly varied in the range between 10 and 1000 ms. The paired‐pulse effect at a given time interval was calculated as EPSC2/EPSC1. For each neuron a PPF curves was determined based on the mean responses to a seven to ten stimuli presentation at each time interval. The averages of these curves from all recorded neurons of a given genotype are shown in Fig. 2.
Figure 2. Paired‐pulse facilitation .
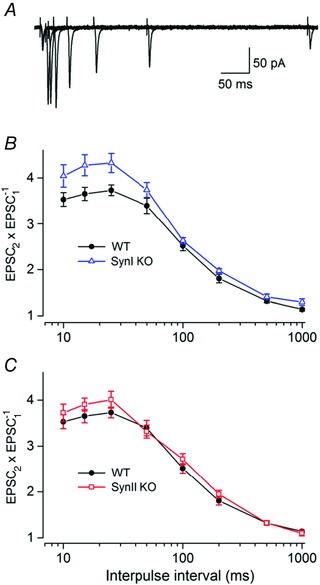
A, overlapping traces from a representative TC neuron from a wild‐type (WT) mouse to stimulation at different interpulse intervals. B, comparisons of PPF of neurons from SynI KO (n = 17) and WT mice (n = 16). Averaged PP ratios (EPSC2/EPSC1) at different interpulse intervals (mean ± SEM). The PPF in SynI KO was significantly stronger than in the WT at the intervals of 15 and 25 ms (P < 0.05). C, comparisons of PPF of neurons from SynII KO (n = 17) and WT mice (n = 16). The data for the WT are the same in B and C. The amplitude of the EPSCs to single pulse stimulation: −52 ± 7 pA (WT), −47 ± 5 pA (SynI KO), −50 ± 6 pA (SynII KO).
The results demonstrated PPF in all three genotypes (Fig. 2). In the WT (number of neurons, n = 16), there was an initial increase in the PPF from 10 ms (3.53 ± 0.15) to 25 ms (3.73 ± 0.11; Fig. 2 B and C). For intervals longer than 25 ms the facilitation gradually decreased, and was almost gone at the 1000 ms interval (1.14 ± 0.03). These characteristics are consistent with previous findings (Granseth et al. 2002; Kielland et al. 2006). In the SynI KO (n = 17; Fig. 2 B), the PPF was significantly stronger than in the WT at the intervals of 15 and 25 ms (P < 0.05). The maximum difference between the WT and the SynI KO was observed at 15 ms (WT: 3.64 ± 0.14; SynI KO: 4.27 ± 0.22; P = 0.02). For the SynII KO (n = 17; Fig. 2 C), we found no statistically significant differences of facilitation compared to the WT (P > 0.05), although there was a tendency toward stronger facilitation in the SynII KO at short intervals (10–25 ms). The data for the WT are the same in Fig. 2 B and C.
Reduced facilitation during train stimulation in SynI KO mice
We studied response differences between TC neurons in the WT and the Syn KOs to stimulation of cortical afferents with pulse trains at 10 Hz and 20 Hz. Before each train we applied a series of stimuli at 0.1 Hz to determine the response amplitude to single pulse stimulation (control EPSCs). The EPSCs recorded during the trains were normalized to the average of the control EPSCs (EPSCn/EPSCctr). In each slice we recorded only one run of a train (10 or 20 Hz) for a single neuron to minimize risks for sequence effects of the synaptic stimulation.
Ten hertz trains
The EPSC amplitudes during a 30 s train showed facilitation in all three genotypes. In the WT neurons (n = 11) we could distinguish three phases of change of the normalized response amplitudes (Fig. 3A and B). The initial phase occurred during the first second of the train (first 10 pulses) when there was a pronounced increase of response. The slope of a line fitted to the response in this interval (Fig. 3 B) indicated an increase at a rate of 2.69 ± 0.48 s−1. The second phase was characterized by slower and almost linear increase to the maximum response that was 5.00 ± 0.24 times stronger than the EPSCctr, and appeared 8.5 s after the start of the train. The slope of a line fitted to the values in this interval indicated a rate of increase of 0.11 ± 0.01 s−1, which is significantly slower than the rate in the first phase (P < 0.0001; extra‐sum‐of‐squares F test). In the third phase there was a slow decrease of the response. Thus, the slope of a line fitted to the values in this interval was negative, and occurred at a rate of −0.038 ± 0.001 s−1.
Figure 3. Facilitation in WT, Syn I KO and Syn II KO mice during 10 Hz pulse‐train stimulation .
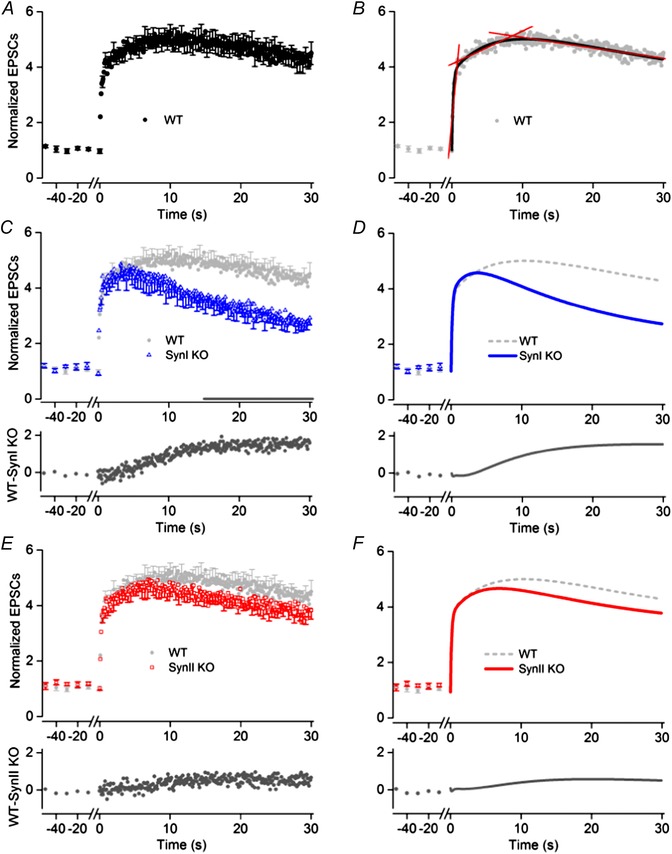
A, C and E, normalized responses of TC neurons to the pulse‐train stimulation (mean and SEM for WT (A; n = 11), SynI KO (C; n = 13) and SynII KO (E; n = 12). Before each train, responses to a series of control stimuli at 0.1 Hz were given (only last five shown). To improve readability, error bars are indicated for only every third data point. The amplitude of the EPSCs to single pulse stimulation: −50 ± 9 pA (WT), −43 ± 6 pA (SynI KO), −47 ± 7 pA (SynII KO). In C and E, the WT response is plotted in light grey to simplify comparisons between the WT and the KO. Below the plots of the data points, the difference between the responses of the WT and the KO are plotted. B, D and F, curves fitted to the data points. B, three different phases of changes in response amplitudes were distinguished: an initial with rapid increase, a second with considerably slower increase to maximum, and a final phase with slow decrease. Straight lines (in red) were fitted to the normalized response in each phase. The slope of the lines indicates the rate of response change in the respective phases (values in text). A three‐phase decay function (curve in black) gave good fit to the data supporting the existence of three phases of the facilitation. D, curves (in blue) obtained by optimal fit of a triple exponential function to the data (cf. Methods) for SynI KO. F, corresponding curve (in red) obtained by optimal fit of the exponential function to the data for SynII KO.
The response of neurons from the SynI KO (n = 13; Fig. 3 C) was in the initial phase similar to the response of the WT neurons. The slope of a line fitted to the normalized response values in the time range corresponding to the first phase for the WT indicated an increase rate of 2.63 ± 0.57 s−1, which was not significantly different from the value for the WT (P = 0.94; extra‐sum‐of‐squares F test). However, during the subsequent interval, corresponding to the second phase of the WT response, the response pattern differed markedly from the pattern of the WT neurons. In contrast to the slow and almost linear increase of the EPSC amplitudes in the WT neurons, there was an initial weak and short‐lasting increase to the peak value that was 4.59 ± 0.28 times the control EPSCs appearing already 4.3 s after the start of the train. Subsequently, the degree of facilitation decreased. Overall during this interval, there was a reduction of the response at a rate of −0.014 ± 0.008 s−1. In the interval corresponding to the third response phase of the WT, the response gradually decreased at a rate of −0.068 ± 0.001 s−1. The graph below the plots of the data points in Fig. 3 C shows the differences between the normalized responses of the two genotypes during the three response phases of the WT; no difference in the initial phase, clear increase of differences in the second phase, and a minor increase of differences in the third phase. At the end of the train, the amplitude of the EPSCs in the SynI KO was 2.74 ± 0.24 times larger than the control EPSCs, compared to 4.33 ± 0.23 in the WT (P < 0.0001). For the period from 15 s after start to the end of the train, the differences between the average response amplitudes of the SynI KO and the WT at the various time points were statistically significant (P < 0.05).
The SynII KO neurons (n = 12) had a response pattern that was remarkably similar to the pattern of the WT (Fig. 3 E). There was no significant difference between the average response amplitudes during the train in the two genotypes although the values for the SynII KO tended to be lower than for the WT.
As a relatively simple model to describe the changes of facilitation during the train we selected an exponential decay function, which has previously been used to describe changes of facilitation in corticothalamic synapses in dLGN (e.g. Granseth, 2004). In all three genotypes a triple exponential decay function could adequately describe the changes of facilitation during the train stimulation, whereas application of double exponential functions led to inferior fits in each of the three genotypes, as demonstrated by extra‐sum‐of‐squares F tests. Moreover, fits with a sum of four exponentials did not improve the goodness‐of‐fit, and could not be made without extra constraints.
Curves obtained by optimal fit of a triple exponential function to the data (see Methods) for the WT are shown in Fig. 3 B (curve in black), to the data from the SynI KO in Fig. 3 D, and to the data for the SynII KO in Fig. 3 F. The time constant of the first exponential (τ1), putatively reflecting the initial fast phase of facilitation, was about the same in all three genotypes (WT, 0.18 ± 0.01 s; SynI KO, 0.16 ± 0.01 s; P = 0.37; SynII KO, 0.15 ± 0.01 s; P = 0.22, extra‐sum‐of‐squares F test). The time constant of the second exponential (τ2), putatively reflecting the slowly rising phase of facilitation to the maximum for the WT, was 6.92 ± 0.99 s. The corresponding time constant was 2.40 ± 0.25 s for the SynI KO and 3.88 ± 0.46 s for the SynII KO, and these values differed significantly from the WT (P < 0.05; extra‐sum‐of‐squares F test). The time constants of the third exponential (τ3), putatively reflecting the decay of facilitation, were similar in the WT (22.44 ± 2.78 s), the SynI KO (18.84 ± 0.45 s; P = 0.25) and the SynII KO (20.17 ± 1.30 s; P = 0.49, extra‐sum‐of‐squares F test). Thus, the main differences were related to the second exponential. This result was further emphasized by the reduced value of the relative weight coefficient of the second exponential in the SynI KO (0.21) compared to the WT (0.34) and the SynII KO (0.29). Accordingly, the contribution of the slowly rising exponential to the sum function was less pronounced in the SynI KO genotype.
Twenty hertz trains
Twenty hertz trains (duration, 20 s) elicited faster changes of response and stronger peak facilitation than the 10 Hz trains. In the WT neurons (n = 9) (Fig. 4 A and B) three phases of change could be distinguished, like for the response to the 10 Hz trains, and straight lines were fitted to the normalized response in each phase to estimate the rate of change (Fig. 4 B). During the first phase, limited to the initial ∼0.35 s of the train (first 7 pulses), there was a fast increase of the response. The slope of the line fitted to this interval indicated a rate of change of 9.31 ± 2.48 s−1. In the second phase the increase occurred more slowly up to the maximum of 5.98 ± 0.28 times EPSCctr that occurred after 1.85 s. The slope of the line fitted to the values in this phase indicated a rate of increase of 0.39 ± 0.07 s−1. The third phase was characterized by gradual reduction of the facilitation. The estimated rate of change was −0.48 ± 0.01 s−1 up to ∼12 s, whereafter the reduction gradually became less steep. At the end of the train the average response was 1.26 ± 0.15 times the control EPSCs.
Figure 4. Facilitation in WT, Syn I KO and Syn II KO mice during 20 Hz pulse‐train stimulation .
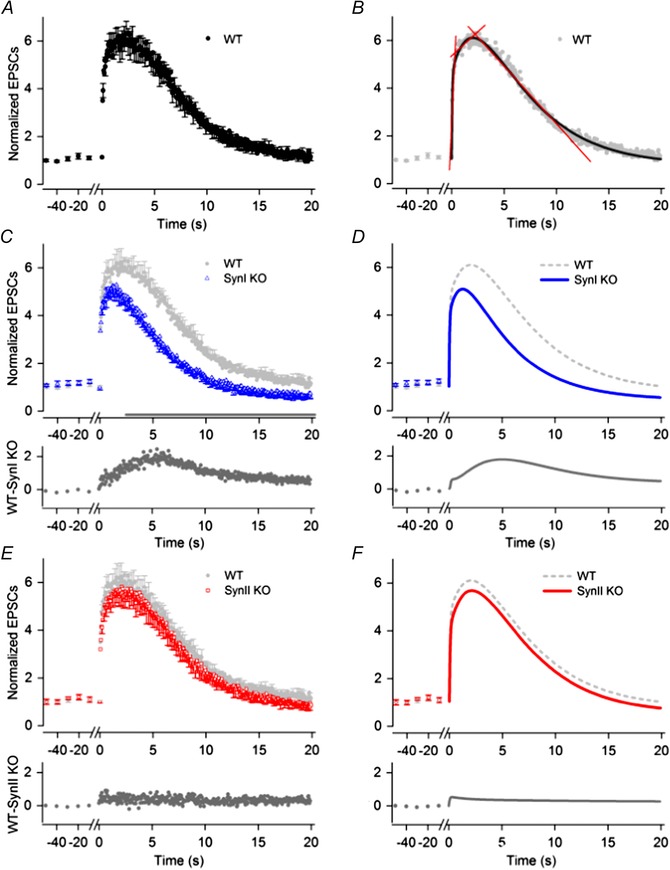
A, C and E, normalized responses of TC neurons to the pulse‐train stimulation (mean and SEM) for WT (A; n = 9), SynI KO (C; n = 10) and SynII KO (E; n = 8). Before each train, responses to a series of control stimuli at 0.1 Hz were given (only last five shown). To improve readability, error bars are indicated for only every third data point. The amplitude of the EPSCs to single pulse stimulation: −49 ± 8 pA (WT), −57 ± 10 pA (SynI KO), −45 ± 9 pA (SynII KO). In C and E, the WT response is plotted in light grey to simplify comparisons between the WT and the KO. Below the plots of the data points, the difference between the responses of the WT and the KO are plotted. B, D and F, curves fitted to the data points. B, three different phases of changes in response amplitudes were distinguished: an initial with rapid increase, a second with considerably slower increase to maximum, and a final phase with slow decrease. Straight lines (in red) were fitted to the normalized response in each phase. The slope of the lines indicates the rate of response change in the respective phases (values in text). A three‐phase decay function (curve in black) gave good fit to the data supporting the existence of three phases of the facilitation. D, curves (in blue) obtained by optimal fit of a triple exponential function to the data for Syn I KO (see Methods). F, corresponding curve (in red) obtained by optimal fit of the exponential function to the data for SynII KO.
In the SynI KO neurons (n = 10), the facilitation initially increased rapidly like in the WT (Fig. 4 C). The estimated rate of change was 7.85 ± 2.22 s−1, which is not significantly different from the value for the WT (P = 0.66; extra‐sum‐of‐squares F test). However, during the interval corresponding to the second phase of the WT, the response deviated markedly from the WT, as for the 10 Hz train. The maximum response was weaker (5.05 ± 0.25 times EPSCctr) and appeared already after ∼700 ms, followed by reduced facilitation. The estimated overall rate of change in this period was −0.16 ± 0.07 s−1. During the period corresponding to the third phase of the WT, the estimated rate of change was −0.46 ± 0.01 s−1 , which is slightly faster than the corresponding rate for the WT (P = 0.015; extra‐sum‐of‐squares F test). At the end of the train, the EPSC amplitude of the SynI KO was smaller than the control responses (0.68 ± 0.12 times EPSCctr) indicating a change from facilitation to depression.
Subtraction of the average response amplitudes of the SynI KO from the WT (Fig. 4 C, lower plot) indicated the trend of the differences between the two genotypes; it increased almost linearly to the maximum at ∼7 s, whereafter it decreased to an almost fixed level after ∼12 s. Comparison of the responses of the two genotypes at the various time points showed that the differences were statistically significant after 3 s from start to the end of train (P < 0.05).
In the SynII KO (n = 8) the response pattern (Fig. 4 E) did not significantly differ from the pattern of the WT (P > 0.05), but the values tended to be slightly lower as illustrated by the difference plot in Fig. 4 E (lower plot). Thus, similar to the responses during the 10 Hz trains, there were no significant difference between the response of the SynII KO and the WT during the 20 Hz stimulation.
As with the 10 Hz trains, the changes of facilitation during the 20 Hz train could be well fitted by a sum of three exponentials (see Methods; Fig. 4 B, D and F). The time constant of the first exponential, τ1, presumably reflecting the initial fast increase of the normalized response, was almost equal for the WT (0.056 ± 0.005 s), the SynI KO (0.043 ± 0.004 s; P = 0.11), and the SynII KO (0.053 ± 0.005 s; P = 0.74). The time constant of the second exponential, τ2, presumably reflecting the slower increase of response, was larger than τ1 in all genotypes. The τ2 was similar in the WT (2.49 ± 0.14 s) and the SynII KO (2.43 s ± 0.11). However, for the SynI KO the τ2 was significantly smaller (1.24 ± 0.05 s) than for the WT (P < 0.0001; extra‐sum‐of‐squares F test). The third exponential, τ3, was remarkably similar in all genotypes (WT, 4.66 ± 0.16 s; SynI KO, 4.57 ± 0.05 s; SynII KO, 4.67 s ± 0.12). Like in the case of the 10 Hz train, there was less contribution of the second exponential to a sum function fitted to the data for the SynI KO; the relative weight coefficient of the slowly rising exponential was smaller in the SynI KO (0.29) than in the WT (0.39) and the SynII KO (0.40).
Reduced PTP in SynI KO mice
We induced PTP with a high‐frequency train (100 pulses at 100 Hz) as in our previous study of SynI/II DKO mice (Kielland et al. 2006). The amplitude of the responses before (control EPSCs) and after (test EPSCs) the tetanic stimulation was monitored with single pulses at 0.1 Hz. The first test pulse was given 10 s after the end of the tetanic stimulation.
In all three genotypes, the tetanic stimulation induced PTP (Fig. 5 A and B). The first test EPSC elicited maximum potentiation, whereafter the amplitudes of the test EPSCs gradually decreased to the level of the control EPSCs. In the WT (n = 11), the post‐tetanic responses reached control levels after ∼300 s (Fig. 5 A and B). In contrast, in the SynI KO (n = 11; Fig. 5 A) the potentiation was weaker than in the WT, and the potentiation reached the baseline level after ∼150 s. The maximum potentiation for the SynI KO, 2.18 ± 0.12, was significantly different from the maximum potentiation in the WT (3.14 ± 0.23, P = 0.001), and the response curve for the SynI KO was consistently below the curve for the WT. However, the differences between the pairs of SynI KO and WT values were not statistically significant beyond the first test pulse. In the SynII KO (n = 10) there was no statistically significant difference with respect to degree of PTP compared to the WT at any test pulse interval; the maximum potentiation for the SynII KO was 2.69 ± 0.12 (P = 0.11).
Figure 5. Post‐tetanic potentiation .
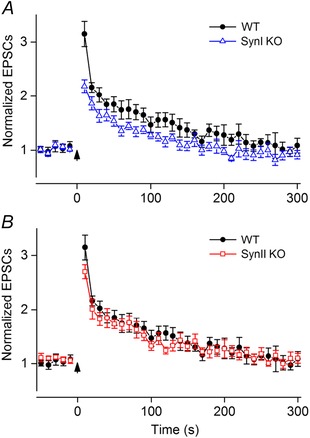
A, comparisons between neurons from SynI KO (n = 11) and WT mice (n = 11) (means ± SEM). The maximum PTP (responses to the first test stimuli) in SynI KO was smaller than in WT (P = 0.001). B, comparisons between neurons from SynII KO (n = 10) and WT mice (n = 11). No differences between SynII KO and WT were observed (P > 0.05). A tetanic pulse train, 100 pulses at 100 Hz, started at t = 0 (see arrow). Before the pulse train, responses to a series of control pulses at 0.1 Hz were recorded (only last five shown). Ten seconds after the train, responses to test pulses at 0.1 Hz were recorded. Notice that the data for the WT are the same in A and B. The amplitude of the EPSCs to single pulse stimulation: −60 ± 8 pA (WT), −58 ± 8 pA (SynI KO), −47 ± 5 pA (SynII KO).
Discussion
We studied functions of SynI and SynII in synaptic short‐term plasticity by comparing response patterns of WT mice and SynI and SynII KO mice in three paradigms of synaptic STP at corticothalamic synapses on TC neurons in dLGN. By PPF, we found stronger facilitation at short interpulse intervals in the SynI KO than in the WT, but no significant differences between the WT and the SynII KO. During train stimulation we distinguished three phases of facilitation in the WT: an initial phase with fast increase, a second phase with moderate increase, and a third phase with decreasing facilitation. In the KOs the response in the initial phase was similar to the response of the WT, and in the SynII KO the response pattern was similar to the WT also in the next two phases. However, in the SynI KO the response pattern in the interval corresponding to the second phase of the WT deviated markedly from the pattern of the WT and the SynII KO. Instead of the steadily increasing facilitation seen in the WT and the SynII KO, there was a weak and short‐lasting increase followed by gradually reduced facilitation. In the third phase there was a similar gradual decrease of facilitation in all three genotypes. With respect to PTP, the effect was smaller in the SynI KO than in the WT, but no significant differences occurred between the SynII KO and the WT. Thus, the major new result was that there are clear differences between the SynI KO and the WT in the types of STP we studied, but no clear differences between SynII KO and the WT, indicating that SynI but not SynII have important roles in the corticogeniculate facilitation.
Despite the general finding that the response pattern in the SynII KO was similar to the response in the WT, there were indications of minor systematic differences. Thus, during train stimulation the response in the SynII KO in the second phase tended to be slightly weaker than in the WT (see Figs 3 E and 4 E). Moreover, during the third phase the response values in the SynII KO were consistently below the response values in the WT. Also during PTP the responses tended to be lower in the SynII KO than in the WT. These differences could reflect a reduced level of SynI, since inactivation of SynII appears to be accompanied by a minor decrease in the expression of SynI (Rosahl et al. 1995). However, it could also indicate a minor implication of SynII during the train stimulation.
Another indication for a minor implication of SynII is suggested by comparing the PTP effects in the SynI KO and the SynII KO with the PTP effects in our previous study of the Syn I/II DKO (Kielland et al. 2006). The maximum PTP effect was almost the same in the DKO and the SynI KO (2.1 vs. 2.18 times control EPSCs), but the DKO responses appeared to decline faster and reach the control baseline earlier than the SynI KO responses in our present study (1.5 vs. 2.5 min). Thus, deficiency of both the SynI and SynII isoforms appears to have a stronger effect on PTP than the lack of SynI alone. The role of SynI and SynII in PTP is still not quite clear, but it could be that the presence of both isoforms is necessary for full expression of PTP. Interesting this ‘sign of cooperation’ is also noticeable in earlier work by Rosahl et al. (1995) when the response by PTP for the WT is compared with the response for the Syn DKO, SynI KO and SynII KO.
The increased PPF at short stimulus intervals in the SynI KO compared to the WT corresponds to previous results from excitatory synapses in hippocampus (Rosahl et al. 1993, 1995), and from autapses on neocortical neurons (Chiappalone et al. 2009). PPF is a short‐lived process that occurs within milliseconds and is assumed to be related to the latest stages of exocytosis of SVs (Zucker & Regehr, 2002). Thus, this difference between the results for the SynI KOs and the WT suggests that SynI has a specific role during transmitter release, consistent with the proposed involvement in functions of the RRP (Rosahl et al. 1993, 1995; Ryan et al. 1996; Hilfiker et al. 1999, 2005, Chiappalone et al. 2009, Hvalby et al. 2006, Cesca et al. 2010; Bykhovskaia, 2011).
The change of response during train stimulation in the WT and the SynII KO from the initial fast to the secondary slower increase of facilitation indicates a shift from an initial phase with fast increase of transmitter release to a phase with slower increase of release, presumably to a phase where mobilization of vesicles from the recycling pool becomes important after partial depletion of the RRP (Alabi & Tsien, 2012). This mobilization clearly involves SynI in the corticothalamic synapses since the secondary increase of facilitation was almost lacking in the SynI KO. This is in agreement with a general contribution of synapsins in clustering of synaptic vesicles and in their activity‐dependent dissociation from vesicles and actin that is important for increase of the number of vesicles available for release (Cesca et al. 2010). Moreover, the disrupted balance between the depletion of the RRP and its replenishment from the recycling pool, which presumably occurred already during the secondary slow increase of facilitation, has consequences for the response to the subsequent part of the train. This might have contributed to the reduced facilitation during the third phase in SynI KO.
Synapsins have been reported to be important in establishing PTP where high frequency stimulation of presynaptic terminals elicits a reversible enhancement of transmitter release through Ca2+ influx activating multiple signalling pathways (reviewed in Cesca et al. 2010). In our results from corticothalamic synapses, the maximum PTP was reduced by ∼30% in the SynI KO mice compared to the WT, and the PTP effect disappeared earlier than in the WT. No significant difference was found between SynII KO and the WT. This is in agreement with recent findings from hippocampal cultures where autapses lacking SynI showed weakened PTP, presumably due to reduced capability to increase the size of the RRP in response to the high frequency stimulation, a capability assumed to be normally provided by SynI (Valente et al. 2012).
The coticothalamic feedback can regulate the transmission of retinal signals to cortex through TC neurons in distinctly different manners. This is related to the property of TC neurons to operate in two state‐dependent functional modes depending on the prevailing membrane potential (e.g. Llinas & Steriade, 2006). At hyperpolarized membrane potentials, typical during slow‐wave sleep, slow oscillations of the membrane potential involving T‐type Ca2+ conductances are generated, and this can elicit an intrinsic rhythmic burst of action potentials that largely blocks the signal transmission from retina to cortex (burst mode). At depolarized membrane potentials, which are typical during wakefulness, this rhythmicity is inactivated. Thus, depolarization induced by the cortical feedback may have two alternative state‐dependent effects on the thalamocortical transmission: in burst mode, to induce a switch to tonic mode (e.g. McCormick & von Krosigk, 1992), and in tonic mode, to regulate, depending on the degree of depolarization, the number of retinal signals that elicit action potentials in TC neurons such that they are transmitted to cortex (e.g. Augustinaite & Heggelund, 2007). In this connection it is interesting that the corticothalamic facilitation during pulse trains can be remarkably strong with a maximum in the WT of ∼5 times the control response during the 10 Hz trains, and ∼6 times during the 20 Hz trains (cf. also e.g. Granseth, 2004). In our experiments the responses in the TC neurons were mediated by AMPA receptors; the NMDA receptors were pharmacologically blocked to get as precise information as possible about changes in transmitter release from presynaptic terminals. Thus, with intact postsynaptic NMDA receptors, the effect of the depolarization induced by the corticothalamic feedback would be considerably stronger (Augustinaite & Heggelund, 2007). Moreover, the cortical feedback can elicit long‐lasting dendritic plateau potentials in TC neurons that may further boost the transmission of retinal signals through dLGN (Augustinaite et al. 2014). The results from the present study clearly demonstrate that SynI can play important roles in the effects of the cortical feedback in thalamus.
Our results are partly in contrast to the results of Rosahl et al. (1995) that showed reduced response during train stimulation and during PTP in hippocampal neurons in brain slices from SynII KO mice. Two methodological differences could be relevant here. First, the temperature during the recordings of Rosahl et al. (1995) was lower than in our experiments (27–29°C vs. 34°C). However, Jensen et al. (2007) studied the response pattern of WT hippocampal neurons during train stimulation at different temperatures and showed that the patterns at 29°C and at 37°C were qualitatively similar to the pattern in the study of Rosahl et al. (1995), despite quantitative differences. Thus, it is unlikely that the discrepancies between the results are due to temperature differences. Second, Rosahl et al. (1995) recorded field potentials whereas we made whole‐cell patch‐clamp recordings. However, Gabriel et al. (2011) could confirm the results of Rosahl et al. (1995) using whole‐cell recordings. Accordingly, we think it is reasonable to conclude that there is a real difference between the types of synapsin involved in these kinds of STP at corticothalamic synapses and at Schaffer‐collateral synapses on CA1 neurons. How these effects are related to mechanisms of vesicle trafficking in the terminals, and whether the synapsin inactivation is affecting the same or different underlying molecular mechanisms in the corticothalamic synapses and hippocampal Schaffer‐collateral to CA1 synapses are not known. However, our results suggest that the SynI KO mouse can be an interesting model for investigations of SynI‐specific phosphorylation sites and pathways.
This selective utilization of different synapsins in similar STP paradigms in different brain areas raises questions concerning the functional redundancy of the synapsins. In a wider perspective the fact that SynI is biochemically more complex and has more phosphorylation sites than SynII (Cesca et al. 2010) may be important here. Thus, SynII is the type of synapsin that seems to have the major role in synaptic STP in hippocampus, which belongs to the phylogenetically older archicortex, whereas SynI has the major role in synaptic STP of the corticothalamic synapses that provide feedback from neocortex (lamina 6 neurons in visual cortex). This may reflect a general phylogenetic adaptation of the synapsin family such that SynI may have evolved from SynII in parallel with the development of the more advanced processing that appeared during the evolution of neocortex.
Additional information
Competing interests
The authors declare no competing interests
Author contributions
P.H. and M.N. conceived and designed the experiments; M.N. collected the data; M.N. and P.H. analysed and interpreted the data, wrote and edited the manuscript, and approved the final version of the manuscript.
Funding
The studies were supported by a grant from the Letten Foundation, and made in laboratory facilities provided by the Institute of Basic Medical Sciences at the University of Oslo.
Acknowledgements
We thank Iren Sefland and Marit Johanne Nielsen for assistance with the genotyping.
References
- Alabi AA & Tsien RW (2012). Synaptic vesicle pools and dynamics. Cold Spring Harb Persp Biol 4, a013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlsen G, Lindström S & Lo F‐S (1985). Interaction between inhibitory pathways to principal cells in the lateral geniculate nucleus of the cat. Exp Brain Res 58, 134–143. [DOI] [PubMed] [Google Scholar]
- Andersen P, Junge K & Sveen O (1972). Cortico‐fugal facilitation of thalamic transmission. Brain Behav Evol 6, 170–184. [DOI] [PubMed] [Google Scholar]
- Augustinaite S & Heggelund P (2007). Changes in firing pattern of lateral geniculate neurons caused by membrane potential dependent modulation of retinal input through NMDA receptors. J Physiol 582, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinaite S, Kuhn B, Helm PJ & Heggelund P (2014). NMDA spike/plateau potentials in dendrites of thalamocortical neurons. J Neurosci 34, 10892–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykovskaia M (2011). Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev Biol 22, 387–392. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F & Benfenati F (2010). The synapsins: Key actors of synapse function and plasticity. Prog Neurobiol 91, 313–348. [DOI] [PubMed] [Google Scholar]
- Chiappalone M, Casagrande S, Tedesco M, Valtorta F, Balitelli P, Martinoia S & Benfenati F (2009). Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I‐deficient cortical networks. Cereb Cortex 19, 1422–1439. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P & Ryan TA (2003). Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38, 69–78. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Benfenati F, Valtorta F & Greengard P (1990). The synapsins. Annu Rev Cell Biol 6, 433–460. [DOI] [PubMed] [Google Scholar]
- Denker A & Rizzoli SO (2010). Synaptic vesicle pools: an update. Front Synaptic Neurosci 2, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etholm L, Bahonjic E, Walaas SI, Kao H‐T & Heggelund P (2012). Neuroethologically delineated differences in the seizure behavior of Synapsin 1 and Synapsin 2 knock‐out mice. Epilepsy Res 99, 252–259. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Kao HT, Feng J, Rapoport M & Greengard P (2000). Synapsin III: developmental expression, subcellular localization, and role in axon formation. J Neurosci 20, 3736–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel T, Garcia‐Perez E & Mahfooz K (2011). A new kinetic framework for synaptic vesicle trafficking tested in synapsin knock‐outs. J Neurosci 31, 11563–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Cheng Q, Greengard P & Augustine GJ (2008). Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci 28, 10835–10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Ahlstrand E & Lindström S (2002). Paired pulse facilitation of corticogeniculate EPSCs in the dorsal lateral geniculate nucleus of the rat investigated in vitro. J Physiol 544, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B (2004). Dynamic properties of corticogeniculate excitatory transmission in the rat dorsal lateral geniculate nucleus in vitro . J Physiol 556, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ & Benfenati F (1993). Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259, 780–785. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ & Greengard P (1999). Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Benfenati F, Doussau F, Nairn AC, Czernik AJ, Augustine GJ & Greengard P (2005). Structural domains involved in the regulation of transmitter release by synapsins. J Neurosci 25, 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M & Südhof TC (1998). Synapsin III, a novel synapsin with an unusual regulation by Ca2+ . J Biol Chem 273, 13371–13374. [DOI] [PubMed] [Google Scholar]
- Hosaka M & Südhof TC (1999). Homo‐ and heterodimerization of synapsins. J Biol Chem 274, 16747–16753. [DOI] [PubMed] [Google Scholar]
- Hvalby Ø, Jensen V, Kao HT & Walaas SI (2006). Synapsin‐regulated synaptic transmission from readily releasable synaptic vesicles in excitatory hippocampal synapses in mice. J Physiol 571, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen V, Walaas SI, Hilficker S, Ruiz A & Hvalby Ø (2007). A delayed response enhancement during hippocampal presynaptic plasticity in mice. J Physiol 583, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC Jr, Greengard P & Czernik AJ (2001). Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+‐dependent glutamate release in isolated nerve terminals. J Neurosci 21, 7944–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, Benfenati F & Greengard P (1998). A third member of the synapsin gene family. Proc Natl Acad Sci U S A 95, 4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielland A, Erisir A, Walaas SI & Heggelund P (2006). Synapsin utilization differs among functional classes of synapses on thalamocortical cells. J Neurosci 26, 5786–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO & Greengard P (1995). Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I‐deficient mice. Proc Natl Acad Sci USA 92, 9235–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström S & Wrόbel A (1990). Frequency dependent corticofugal excitation of principal cells in the cat's dorsal lateral geniculate nucleus. Exp Brain Res 79, 313–318. [DOI] [PubMed] [Google Scholar]
- Llinas RR & Steriade M (2006). Bursting of thalamic neurons and states of vigilance. J Neurophysiol 95, 3297–3308. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Townes‐Anderson E, Czernik AJ, Cameron R, Greengard P & DeCamilli P (1990). Synapsins in the vertebrate retina‐absence from ribbon synapses and heterogeneous distribution among conventional synapses. Neuron 5, 19–33. [DOI] [PubMed] [Google Scholar]
- McCormick DA & Von Krosigk M (1992). Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci U S A 89, 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owe SG, Erisir A & Heggelund P (2013). Terminals of the major thalamic input to visual cortex. Neuroscience 243, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spitlane D, Herx J, Hammer RE, Malenka RC & Südhof TC (1993). Short‐term synaptic plasticity is altered in mice lacking synapsin I. Cell 75, 661–670. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC & Südhof TC (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Li L, Chin L‐S, Greengard P & Smith SJ (1996). Synaptic vesicle recycling in synapsin I knock‐out mice. J Cell Biol 134, 1219−1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigullin D, Bill CA, Coleman WL & Bykhovskaia M (2004). Regulation of transmitter release by synapsin II in mouse motor terminals. J Physiol 561,149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer W (1977). Control of thalamic transmission by corticofugal and ascending reticular pathways in the visual system. Physiol Rev 57, 386–420. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, et al (1989). Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science 245, 1474–1480. [DOI] [PubMed] [Google Scholar]
- Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T & Hirokawa N (1995). Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol 131, 1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP & Salt TE (1998). Characterization of sensory and corticothalamic excitatory inputs to rat thalamocortical neurones in vitro . J Physiol 510, 829–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente P, Casagrande S, Nieus T, Verstegen AMJ, Valorta F, Benfenati F & Baldelli P (2012). Site‐specific Synapsin I phosphorylation participates in the expression of post‐tetanic potentiation and its enhancement by BDNF. J Neurosci 32, 5868–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstegen AM, Tagliatti E, Lignani G, Marte A, Stolero T, Atias M, Corradi A, Valtorta F, Gitler D, Onofri F, Fassio A & Benfenati F (2014). Phosphorylation of synapsin I by cyclin‐dependent kinase‐5 sets the ratio between the resting and recycling pools of synaptic vesicles at hippocampal synapses. J Neurosci 34, 7266–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Krosigk M, Monckton JE, Reiner PB & McCormick DA (1999). Dynamic properties of corticothalamic excitatory postsynaptic potentials and thalamic reticular inhibitory postsynaptic potentials in thalamocortical neurons of the guinea‐pig dorsal lateral geniculate nucleus. Neuroscience 91, 7–20. [DOI] [PubMed] [Google Scholar]
- Walaas SI, Browning MA & Greengard P (1988). Synapsin Ia, synapsin Ib, protein IIIa and protein IIIb, four related synaptic vesicle‐associated phosphoproteins, share regional and cellular localization in rat brain. J Neurochem 51, 1214–1220. [DOI] [PubMed] [Google Scholar]
- Wilson JR, Friedlander MJ & Sherman SM(1984). Fine structural morphology of identified X‐ and Y‐cells in the cat's lateral geniculate nucleus. Proc R Soc Lond B Biol Sci 221, 411–436. [DOI] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG (2002). Short‐term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]