

Abstract
Key points
Throughout life, fear learning is indispensable for survival and neural plasticity in the lateral amygdala underlies this learning and storage of fear memories.
During development, properties of fear learning continue to change into adulthood, but currently little is known about changes in amygdala circuits that enable these behavioural transitions.
In recordings from neurons in lateral amygdala brain slices from infant up to adult mice, we show that spontaneous and evoked excitatory and inhibitory synaptic transmissions mature into adolescence.
At this time, increased inhibitory activity and signalling has the ability to restrict the function of excitation by presynaptic modulation, and may thus enable precise stimulus associations to limit fear generalization from adolescence onward.
Our results provide a basis for addressing plasticity mechanisms that underlie altered fear behaviour in young animals.
Abstract
Convergent evidence suggests that plasticity in the lateral amygdala (LA) participates in acquisition and storage of fear memory. Sensory inputs from thalamic and cortical areas activate principal neurons and local GABAergic interneurons, which provide feed‐forward inhibition that tightly controls LA activity and plasticity via pre‐ and postsynaptic GABAA and GABAB receptors. GABAergic control is also critical during fear expression, generalization and extinction in adult animals. During rodent development, properties of fear and extinction learning continue to change into early adulthood. Currently, few studies have assessed physiological changes in amygdala circuits that may enable these behavioural transitions. To obtain first insights, we investigated changes in spontaneous and sensory input‐evoked inhibition onto LA principal neurons and then focused on GABAB receptor‐mediated modulation of excitatory sensory inputs in infant, juvenile, adolescent and young adult mice. We found that spontaneous and sensory‐evoked inhibition increased during development. Physiological changes were accompanied by changes in dendritic morphology. While GABAB heteroreceptors were functionally expressed on sensory afferents already early in development, they could only be physiologically recruited by sensory‐evoked GABA release to mediate heterosynaptic inhibition from adolescence onward. Furthermore, we found GABAB‐mediated tonic inhibition of sensory inputs by ambient GABA that also emerged in adolescence. The observed increase in GABAergic drive may be a substrate for providing modulatory GABA. Our data suggest that GABAB‐mediated tonic and evoked presynaptic inhibition can suppress sensory input‐driven excitation in the LA to enable precise stimulus associations and limit generalization of conditioned fear from adolescence onward.
Abbreviations
- BA
basal nucleus of the amygdala
- BLA
basolateral complex of the amygdala
- CS
conditioned stimulus
- LA
lateral nucleus of the amygdala
- LTP
long‐term potentiation
- PND
postnatal day
- PPD
paired‐pulse depression
- PPR
paired‐pulse ratio
- sEPSC
spontaneous excitatory postsynaptic current
- sIPSC
spontaneous inhibitory postsynaptic current
- US
unconditioned stimulus
Introduction
The amygdala is a key brain structure that controls emotional behaviours, including appetitive behaviour as well as fear and anxiety (LeDoux, 2000). The lateral nucleus of the amygdala (LA) plays a crucial role in the acquisition and storage of fear memory (Maren, 2001; Pape & Pare, 2010). LA principal neurons receive convergent excitatory input from thalamic and cortical sensory brain areas that relay information about sensory stimuli (Romanski et al. 1993; Sah et al. 2003), and long‐term potentiation (LTP) at these inputs is an important mechanism that underlies the association of conditioned (CS) and unconditioned (US) stimuli during Pavlovian fear conditioning (McKernan & Shinnick‐Gallagher, 1997; Rogan et al. 1997; Maren, 2005). Furthermore, the activity of LA principal neurons is under tight control of local GABAergic interneurons (Ehrlich et al. 2009). These can provide sensory input‐driven feed‐forward inhibition which gates synaptic plasticity and modulates fear learning via GABAA and GABAB receptor‐dependent mechanisms (Watanabe et al. 1995; Li et al. 1996; Shumyatsky et al. 2002; Bissiere et al. 2003; Shaban et al. 2006; Tully et al. 2007; Pan et al. 2009). Additional evidence for the importance of GABAergic control comes from findings suggesting that activity of the GABA synthesizing enzyme GAD65, GABA availability, and extracellular GABA levels in the amygdala are modulated by, and influence the expression, generalization, and extinction of, fear in adult animals (Stork et al. 2002; Heldt & Ressler, 2007; Bergado‐Acosta et al. 2008; Sangha et al. 2009; Lange et al. 2014).
To date, most studies have focused on acquired fear behaviour and its underlying amygdala circuits and plasticity mechanisms in adult animals. Only a few studies have addressed developmental changes. Fear learning in rodents, i.e. aversive conditioning to odours, emerges at the infant to juvenile transition at postnatal day (PND) 10–12 (Sullivan et al. 2000; Raineki et al. 2009). Auditory fear conditioning can be observed in animals older than PND 16 (Rudy, 1993; Gogolla et al. 2009; Kim & Richardson, 2010 b) and depends on the activation of NMDA receptors (Langton et al. 2007). However, in younger animals fear conditioning is enhanced and fear is more generalized to unconditioned stimuli compared to adults (Hefner & Holmes, 2007; Ito et al. 2009). Fear extinction is also developmentally regulated with different underlying mechanisms and circuits. In juveniles (PND 16–17) spontaneous recovery, renewal and reinstatement of fear responses are absent after extinction training (Yap & Richardson, 2007; Gogolla et al. 2009; Kim & Richardson, 2010 b). Interestingly, in contrast to adolescents (>PND 24) and adults, extinction acquisition is NMDA‐receptor independent, expression is not influenced by modulators of GABAA receptor activity, and extinction learning and expression depend on the amygdala but not the ventromedial prefrontal cortex (Kim & Richardson, 2007; Kim et al. 2009; Kim & Richardson, 2010 a). These behavioural findings suggest that the supporting neural circuitry in the basolateral amygdala (BLA) and its function change during postnatal development, but these processes are largely unexplored.
At the anatomical level, BLA cytoarchitecture matures between birth and the end of the third postnatal week with marked changes in size, number and density of cells, dendritic morphology, number of synaptic contacts, and changes in expression of calcium binding proteins (Berdel et al. 1997; Morys et al. 1998; Berdel & Morys, 2000; Davila et al. 2008; Ryan et al. 2015). Additionally, GABAergic fibre densities increase between the second and third postnatal week (Brummelte et al. 2007). On a similar timeline, connectivity of the BLA with afferent brain areas is set up. Sensory thalamic areas already project to LA early after birth (PND 7), while caudal cortical areas send weak projections to the BLA at that time but reach mature projection patterns within the second postnatal week (Bouwmeester et al. 2002). Two recent physiological studies have started to outline functional changes within the amygdala. For example, principal neurons in rat basal nucleus of the amygdala (BA) undergo marked changes in passive membrane properties, spiking behaviour and membrane potential oscillations within the first postnatal month (Ehrlich et al. 2012). Also, locally evoked and spontaneous GABAA receptor‐mediated synaptic transmission within the BA matures between PND 7 and 28, exhibiting faster kinetics and an increased IPSC size in older animals, and changes in short‐term plasticity (Ehrlich et al. 2013). However, nothing is known about the developmental changes in sensory‐evoked feed‐forward inhibition and its ability to regulate thalamic and cortical inputs in the LA during development.
Apart from its postsynaptic action, GABAB receptor activity has been shown to modulate presynaptic vesicle release at excitatory synapses by interacting with voltage gated calcium channels, or by directly modulating vesicle priming. On inhibitory synapses, GABAB effectors are potassium channels and voltage gated calcium channels (Chalifoux & Carter, 2011). In adult lateral amygdala, GABAB receptors are located presynaptically on excitatory sensory afferents to principal neurons and interneurons (Pan et al. 2009) as well as on inhibitory inputs to principal neurons where they act as autoreceptors (Szinyei et al. 2000). GABAB receptors on excitatory sensory terminals onto LA principal neurons mediate short‐term heterosynaptic inhibition and gate different forms of LTP (Shaban et al. 2006; Pan et al. 2009; Lange et al. 2014). The importance of GABAB receptors in adult fear and extinction learning has been demonstrated by showing that GABAB(1a)‐knock‐out mice show generalized fear (Shaban et al. 2006) and the application of the GABAB receptor agonist baclofen results in reduced extinction learning (Heaney et al. 2012). While this suggests that presynaptic GABAB receptors play an important role in shaping LA sensory input activity in adult animals, the developmental regulation of this has not been studied.
Here we investigated changes in dendritic morphology of LA principal neurons and their spontaneous and sensory input‐evoked inhibition and then focused on GABAB receptor‐mediated modulation of synaptic transmission during critical developmental stages between infancy and young adulthood. Our data suggest that GABAB receptor‐mediated tonic and evoked presynaptic inhibition can serve to limit sensory input‐driven excitation in the LA to enable precise stimulus associations and to limit generalization of conditioned fear from adolescence onward.
Methods
Ethical approval
All procedures were performed in accordance with the EU directive on use of animals in research and approved by the local animal care and use committee (Regierungspraesidium Tuebingen, state of Baden‐Wuerttemberg, Germany).
Animals
For all experiments, C57BL/6J mice (breeders purchased from Harlan, Netherlands) were used. Breedings were performed in‐house by timed mating, and litter size was reduced to six pups per mother to control for similar levels of maternal care. Pups were housed with their mothers prior to weaning on PND 21. Only male mice of the following four age groups were used for recordings: PND 8–10 (infant), PND 15–18 (juvenile), PND 25–28 (adolescent), and PND 42–56 (young adult). All mice were housed in a 12 h light–dark cycle with food and water available ad libitum. Post‐weaning animals were housed in groups of two to five with their siblings. In this study, 332 cells from 145 mice were recorded (infant: 63/24, juvenile: 98/49, adolescent 83/37, adult 88/35).
Slice preparation
Animals were deeply anaesthetized with isoflurane (Isofluran CP, CP Pharma, Germany, 3% in Oxygen), decapitated and their brains rapidly removed and chilled in ice‐cold artificial cerebrospinal fluid (ASCF) supplemented with 8.7 mm MgSO4. Coronal acute brain slices were cut with a vibrating‐blade microtome (HM650V, Thermo Scientific, Dreieich, Germany) equipped with a sapphire blade (Delaware Diamond Knives, Wilmington, DE, USA) in ice‐cold artificial cerebrospinal fluid (ASCF) supplemented with 8.7 mm MgSO4 at 320 μm thickness. Slices were recovered at 37°C for 45 min in normal ACSF and stored at room temperature in a custom‐built interface chamber until recording. The ACSF was composed of (in mm): 124 NaCl, 1.25 NaH2PO4, 1.3 MgSO4, 2.7 KCl, 26 NaHCO3, 2 CaCl2, 18 d‐glucose and 4 l‐ascorbic acid, and was oxygenated with 95% O2–5% CO2.
Patch clamp recordings
Slices containing the lateral amygdala were transferred to a submersion chamber mounted on an upright microscope (BX51WI, Olympus Germany, Hamburg, Germany) and superfused with ACSF (1–2 ml min−1) at 30°C. Whole‐cell patch‐clamp recordings were performed using pipettes pulled from borosilicate glass capillaries (GB150F‐8P, Science Products, Hofheim, Germany) with resistances of 3–5 MΩ filled with intracellular solution containing (in mm): 135 caesium methylsulphonate, 6 CsCl, 4 Mg‐ATP, 0.4 Na‐GTP, 10 Na2‐phosphocreatine, 10 Hepes, 0.6 EGTA, (osmolarity 290–295 mosmol l−1, pH 7.2–7.3). Data were acquired using a Multiclamp 700B amplifier, Digidata 1440 AD‐board, and Clampex software (all from Molecular Devices, Sunnyvale, CA, USA). Signals were filtered at 2 kHz and digitized at 5 kHz. Electrical stimulation of thalamic and cortical afferent fibres onto LA principal neurons was applied by bipolar tungsten microelectrodes (Science Products, Hofheim, Germany) placed in the internal (medial of LA) and external (dorsal of LA) capsule, respectively with a pulse length of 200 μs. Evoked or spontaneous excitatory and inhibitory postsynaptic currents (EPSCs, IPSCs) were isolated in voltage‐clamp mode at holding potentials of −65 and 0 mV, respectively. In our hands, these were close to the experimentally determined reversal potentials for IPSCs and EPSCs and thus allowed for sequential recordings of EPSCs and IPSCs in the same cells in the absence of blockers. The GABAB receptor agonist baclofen (2 μm), the GABAB receptor antagonists CGP55845A and CGP52432 (both 10 μm) and the GABA reuptake inhibitor SKF89976A (30 μm) were applied after establishment of stable baseline recordings and were allowed to penetrate the slice for at least 8 min. To avoid GABAergic effects on GABAA receptors, the non‐competitive Cl−‐channel blocker picrotoxin (100 μm) was added to the ACSF at the beginning of experiments including SKF89976A application. Series resistance was monitored throughout all experiments and data were excluded if it changed >20%. Between group comparisons for all cells recorded for Figs 1 and 3 showed no difference in access resistance between age groups (data in MΩ: infant (inf): 21.75 ± 1.21 (n = 30); juvenile (juv): 21.53675 ± 1.10 (n = 40); adolescent (adol): 23.80 ± 1.12 (n = 42); adult: 21.01 ± 0.90 (n = 37); one‐way ANOVA: F(3,146) = 0.787, P = 0.503).
Figure 1. Postnatal maturation of spontaneous excitatory and inhibitory drive onto LA principal neurons .
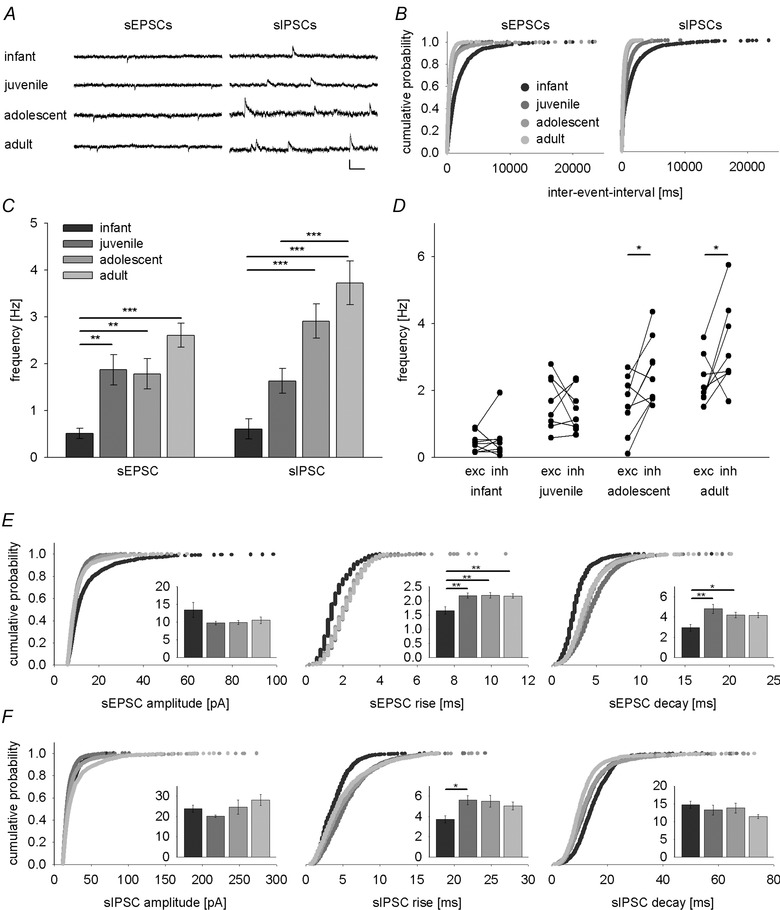
A, example traces of spontaneous EPSCs (sEPSCs, at −65 mV) and IPSCs (sIPSCs, at 0 mV) from representative LA principal neurons in infant, juvenile, adolescent and adult mice illustrating a developmental increase in event frequencies. Scale bar: 20 pA, 100 ms. B, cumulative distribution of inter‐event intervals of sEPSCs and sIPSCs across age groups. C, mean data (±SEM) depicting postnatal maturation of sEPSC and sIPSC frequencies. A major step in sEPSC frequency occurs between infant and juvenile groups while sIPSC frequency develops in a more linear fashion. D, cell‐by‐cell comparisons of sEPSC (exc) and sIPSC (inh) frequency across postnatal development. sIPSC frequency exceeds sEPSC frequency in adolescent and adult mice (paired t test: t(8) = −2.941, P = 0.019; and t(8) = −2.381, P = 0.044, respectively). E and F, cumulative distributions and mean data (±SEM, inset graphs) of amplitude, rise and decay for sEPSCs (E) and sIPSCs (F). Legend for age groups as in panels B and C. Number of recorded cells, mean data, and P values (one‐way ANOVA with Bonferroni correction) are shown in Table 1. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3. Development of sensory‐evoked inhibition onto LA principal neurons .
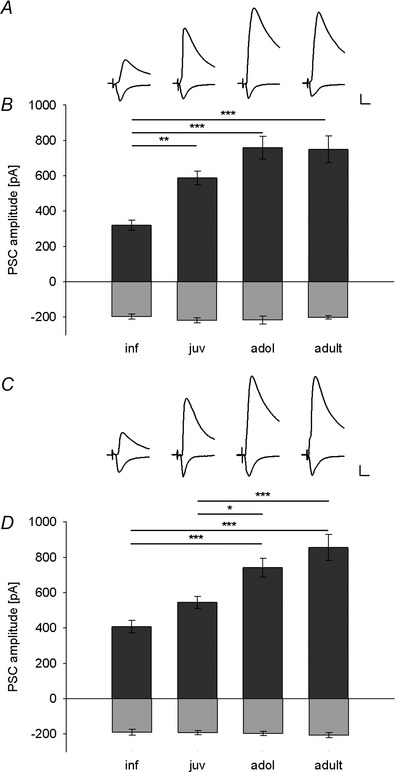
A and C, example traces of EPSCs (at −65 mV) and IPSCs (at 0 mV) in LA principal neurons evoked by thalamic (A) and cortical (C) fibre stimulation in infant, juvenile, adolescent and adult mice, illustrating the developmental increase in sensory‐evoked IPSC amplitude. Scale bar: 100 pA, 10 ms. B and D, mean data (±SEM) of peak EPSC (light grey bars) and IPSC amplitudes (dark grey bars) in LA principal neurons evoked by thalamic (B) and cortical (D) fibre stimulation in different age groups. B, thalamic EPSC and IPSC amplitudes (pA) (number of recorded cells): inf: −196.4 ± 13.6 and 320.6 ± 28.3 (22); juv: −217.3 ± 14.2 and 587.1 ± 39.9 (26); adol: −225.9 ± 20.7 and 759.4 ± 64.5 (23); adult: −200.9 ± 8.5 and 750.6 ± 75.6 (24). Significant differences in IPSC amplitudes (one‐way ANOVA with Bonferroni correction): inf vs. juv: P = 0.006; adol: P < 0.001; and adult: P < 0.001. D, cortical EPSC and IPSC amplitudes (pA) (number of recorded cells): inf: −190.6 ± 16.3 and 408.4 ± 35.5 (21); juv: −192.5 ± 11.9 and 545.4 ± 34.5 (26); adol: −197.6 ± 11.7 and 741.3 ± 53 (24); adult: −206.8 ± 15.0 and 856.5 ± 73.1 (24). Significant differences in IPSC amplitudes (one‐way ANOVA with Bonferroni correction): inf vs. adol and adult: P < 0.001; juv vs. adol: P = 0.044; juv vs. adult: P < 0.001). *P < 0.05, **P < 0.01, ***P < 0.001.
Spontaneous EPSC and IPSC were recorded in gap‐free mode for 5 min. Evoked EPSCs and IPSCs were recorded by alternately stimulating cortical and thalamic input pathways at an inter‐sweep interval of 10 s. To gain information about presynaptic release probabilities and GABAB receptor‐mediated modulation of postsynaptic currents, paired‐pulse stimulation of presynaptic thalamic and cortical fibres was applied with inter‐stimulus intervals of 50 ms (for EPSCs) and 300 ms (for IPSCs), respectively (Szinyei et al. 2000; Zucker & Regehr, 2002). Heterosynaptic inhibition was induced by applying priming stimulation to one sensory input pathway (200 Hz for 50 ms) and assessed by subsequent (100 ms delay) paired‐pulse stimulation of the other sensory input pathway (Pan et al. 2009).
Electrophysiology data analysis
Recordings of spontaneous EPSCs and IPSCs were analysed using MiniAnalysis software (Synaptosoft, Decatur, GA, USA). In brief, excitatory and inhibitory events were first analysed using automatic file analysis with defined parameters. Subsequently, results were crosschecked for false positive and false negative events. The middle 200 events in the 5 min recording period were used for amplitude and frequency analysis. In some cases in the infant age group, the whole file was analysed since less than 200 events were recorded. Total charge transfer was calculated as the area sum of all events. All other data were analysed using the NeuroMatic program suite (developed at University College London, UK; www.neuromatic.thinkrandom.com) and custom‐written macros using the peak detection routines with averaging over a 1 ms time window in Igor Pro (Wavemetrics, Lake Oswego, OR, USA). A minimum of 10 sweeps per pathway and condition were averaged for amplitude and paired‐pulse ratio (PPR) analyses. PPRs were computed by normalizing amplitudes of the second PSC to the first PSC. For Fig. 4, EPSCs and IPSCs were recorded at increasing stimulus intensities and EPSCs larger than 20 pA were sorted into 50 pA bins before plotting them against stimulus intensity or corresponding I/E ratios.
Figure 4. Input–output curves and inhibition/excitation ratios across different EPSC amplitudes .
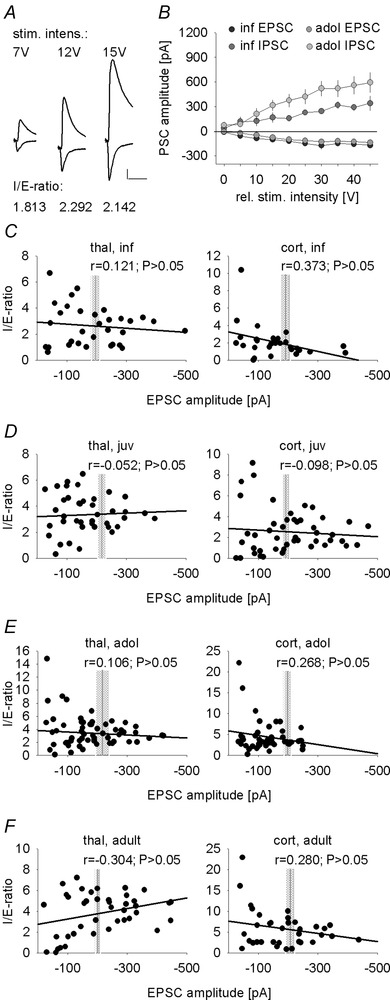
A, example traces of thalamic fibre evoked EPSCs (at −65 mV) and IPSCs (at 0 mV) in a representative LA principal at different stimulation intensities, demonstrating similar I/E ratios for different stimulation intensities. Scale bar: 100 pA, 20 ms. B, mean evoked EPSC and IPSC amplitudes upon thalamic input stimulation across different stimulus intensities in infant and adolescent mice (infant: n = 8 cells; adolescent: n = 11 cells). C–F, scatter plots of I/E ratios across EPSC amplitudes evoked by either thalamic (left) or cortical (right) presynaptic fibre stimulation in infant (A, n = 8 cells), juvenile (B, n = 10), adolescent (C, n = 13) and adult (D, n = 7) mice. Vertical grey lines and shaded areas represent the means ± SEM of EPSC amplitudes obtained from Fig. 3 for comparison.
Cell fills, staining and imaging
LA principal neurons were filled with Biocytin (5 mg ml−1 in intracellular solution) and slices were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 16–24 h at 4°C. Subsequently, slices were permeabilized in 0.5% Triton X‐100 in PBS, and biocytin‐filled cells were revealed using DyLight488‐conjugated Steptavidin (1:750, Dianova, Hamburg, Germany). To achieve sufficient penetration, slices were incubated in 0.5% Triton for 48 h at 4°C. For overview images of filled cells, sections were imaged using a laser scanning microscope (LSM 710, Carl Zeiss, Germany) equipped with a 40 × 1.3 NA objective (4 × 4 tiles, 2 μm optical sections, 1 μm step size, 100 μm total z‐stack thickness). For imaging of dendrites and spines, we used a 63 × 1.4 NA objective, 0.7 μm optical sections and a 0.33 μm step size. To prevent bias due to different imaging depth, special care was taken to achieve similar z‐positions of imaged dendrites (one‐way ANOVA: F(3,45) = 1.893; P = 0.144). Images of filled cells were imported, drawn and quantified in 3D in Neurolucida software (MBF Bioscience, Williston, VT, USA) and collapsed into 2D pictures for display. Analysis of dendritic spines was performed on stack images using Imaris software (Bitplane, Zurich, Switzerland). Specifically, we counted spines on 12 to 13 randomly selected dendritic areas (from 4 cells per age group) located approximately 100 μm from the soma.
Chemicals and drugs
All chemicals were reagent grade and obtained from Roth (Karlsruhe, Germany), Merck (Darmstadt, Germany), or Sigma‐Aldrich (Taufkirchen, Germany). Baclofen and picrotoxin were obtained from Sigma‐Aldrich (Germany). The GABAB receptor antagonists CGP55845A and CGP52432, and the GABA reuptake inhibitor SKF89976A were obtained from Tocris Bioscience (Bristol, UK). Stock solutions of all drugs were stored at −20°C and were diluted 1:1000 in ACSF to final concentration before application.
Statistics
Statistical analysis was performed using SPSS statistics software (IBM, Ehningen, Germany). Interaction of two effects was analysed with one‐tailed bivariate Pearson correlation tests. For simple comparisons between age groups, one‐way ANOVAs were performed followed by Bonferroni post hoc testing. For statistical analysis of within group factors across different age groups, mixed‐model two‐way ANOVAs were used followed by Bonferroni‐corrected Student's paired or unpaired t test as appropriate. All values are shown as the mean ± standard error of the mean (SEM). Differences were considered significant at P < 0.05.
Results
Postnatal maturation of spontaneous excitation and inhibition in LA principal neurons
First, we investigated spontaneous excitatory and inhibitory drive onto LA principal neurons, which arises from action potential‐dependent and ‐independent release of glutamate and GABA from all excitatory and inhibitory synaptic terminals impinging onto a cell. To this end, we performed whole‐cell patch‐clamp recordings of spontaneous EPSCs (sEPSCs, at −65 mV) and IPSCs (sIPSCs, at 0 mV, Fig. 1 A). While there was no significant change in sEPSC or sIPSC amplitude across development (Table 1, Fig. 1 E and F; one‐way ANOVA: F(3,31) = 1.864, P = 0.156; and F(3,31) = 1.665, P = 0.195, respectively), event frequencies were developmentally regulated for both sEPSCs and sIPSCs (Table 1; Fig. 1 A–C; one‐way ANOVA, sEPSCs: F(3,31) = 11.303, P < 0.001; sIPSCs: F(3,31) = 16.087, P < 0.001). For sEPSC frequency, a major increase occurred at the infant to juvenile transition (Bonferroni corrected post hoc comparison P = 0.006), whereas sIPSC frequency increased in a more linear fashion into adulthood (statistically significant post hoc comparisons of sIPSC frequencies: infant vs. adolescent or adult, P < 0.001; juvenile vs. adult, P = 0.001). Similar results were obtained by analysing total charge transfer (Table 1, one‐way ANOVA, sEPSCs: F(3,31) = 7.06, P = 0.001; sIPSCs: F(3,31) = 9.211, P < 0.001). Additionally, we found changes in sEPSC and sIPSC kinetics between infant and juvenile age groups (Table 1 and Fig. 1 E and F), which may be in part related to electrotonic filtering.
Table 1.
Properties of sEPSCs and sIPSCs in LA principal neurons
| Post hoc statistical | |||||
|---|---|---|---|---|---|
| Infant | Juvenile | Adolescent | Adult | comparisons | |
| sEPSC freq. (Hz) | 0.51 ± 0.11 (9)1,2,3 | 1.87 ± 0.32 (8)1 | 1.78 ± 0.32 (9)2 | 2.60 ± 0.26 (9)3 | 1 P = 0.006, 2 P = 0.009, |
| 3 P < 0.001 | |||||
| sEPSC ampl. (pA) | 13.47 ± 2.15 (9) | 9.77 ± 0.52 (8) | 9.93 ± 0.57 (9) | 10.58 ± 0.94 (9) | |
| sEPSC rise (ms) | 1.66 ± 0.03 (9)1,2,3 | 2.19 ± 0.15 (8)1 | 2.2 ± 0.15 (9)2 | 2.18 ± 0.04 (9)3 | 1 P = 0.008, 2 P = 0.005, |
| 3 P = 0.005 | |||||
| sEPSC decay (ms) | 2.95 ± 0.03 (9)1,2 | 4.82 ± 0.15 (8)1 | 4.22 ± 0.15 (9)2 | 4.18 ± 0.04 (9) | 1 P = 0.002, 2 P = 0.049 |
| Total charge sEPSCs (pC) | 6.11 ± 1.64 (9)1,2 | 26.22 ± 5.53 (8) | 23.34 ± 5.13 (9)1 | 34.17 ± 4.87 (9)2 | 1 P = 0.024, 2 P = 0.001 |
| sIPSC freq. (Hz) | 0.61 ± 0.21 (9)1,2 | 1.63 ± 0.26 (8)3 | 2.91 ± 0.37 (9)1 | 3.73 ± 0.47 (9)2,3 | 1 P < 0.001, 2 P < 0.001, |
| 3 P = 0.001 | |||||
| sIPSC ampl. (pA) | 23.85 ± 1.72 (9) | 20.14 ± 0.61 (8) | 24.64 ± 3.50 (9) | 28.07 ± 2.78 (9) | |
| sIPSC rise (ms) | 3.72 ± 0.03 (9)1 | 5.6 ± 0.15 (8)1 | 5.5 ± 0.15 (9) | 5.03 ± 0.04 (9) | 1 P = 0.047 |
| sIPSC decay (ms) | 14.73 ± 0.03 (9) | 13.25 ± 0.15 (8) | 13.79 ± 0.15 (9) | 11.42 ± 0.04 (9) | |
| Total charge sIPSCs (pC) | 61.2 ± 28.3 (9)1,2 | 127.5 ± 27.2 (8)3 | 246.0 ± 42.9 (9)1 | 380.5 ± 70.6 (9)2,3 | 1 P = 0.047, 2 P < 0.001, |
| 3 P = 0.004 |
Values are given as means ± SEM (number of recorded cells). sEPSC, spontaneous excitatory postsynaptic current; sIPSC, spontaneous inhibitory postsynaptic current; freq., frequency; ampl., amplitude. Superscript numbers represent pairs of statistically significant comparisons (Bonferroni corrected).
To directly determine the balance between spontaneous excitatory and inhibitory input in the same cells, we compared inhibitory and excitatory drive in individual cells (Fig. 1 D) and found that event frequencies changed with age (inf, juv, adol, adult) and response type (sEPSC, sIPSC; two‐way ANOVA, age: F(3,31) = 20.266, P < 0.001; response type: F(1,31) = 8.275, P = 0.007; interaction: F(3,31) = 3.620, P = 0.024). While in infants and juveniles, the frequencies of sEPSCs and sIPSCs were similar (post hoc paired t tests: infant: t(8) = −0.0533, P = 0.609; juvenile: t(7) = 0.647, P = 0.538), there was a significant shift towards increased inhibition in adolescent and adult mice (post hoc paired t tests: adolescent: t(8) = −2.941, P = 0.019; adult: t(8) = −2.381, P = 0.044).
Taken together, although both excitation and inhibition increased during development, the balance between spontaneous inhibition and excitation is shifted towards increased inhibition from adolescence onwards. This can be due to increases in probability of transmitter release, increase in the number of active inhibitory synapses, or an increase in spontaneous activity of GABAergic interneurons. Independent of the exact mechanism, our data suggest that LA principal neurons become more tightly controlled by local interneurons.
Development of sensory input‐evoked inhibition in the LA
GABAergic interneurons in the LA can be activated by excitatory sensory inputs to provide feed‐forward inhibition onto principal cells, which gates synaptic plasticity (Ehrlich et al. 2009). We therefore wanted to assess the developmental changes in thalamic and cortical input‐evoked inhibition, and used electrical stimulation of fibres in the internal and external capsule, respectively (Mahanty & Sah, 1999; Weisskopf & LeDoux, 1999). Consistent with previous studies (Weisskopf & LeDoux, 1999; Gambino et al. 2010), evoked synaptic responses were composed of two components, a fast inward current at negative potentials (EPSC, recorded at −65 mV), and a second component with a measured reversal potential of around −60 mV (Fig. 2 C; juvenile: −60.4 ± 2.4 mV, n = 6, and −59.2 ± 2.5 mV, n = 6; adolescent: −58.4 ± 1.4 mV, n = 5, and −58.8 ± 1.3 mV, n = 5, for thalamic and cortical synaptic inputs, respectively). These values were close to the calculated equilibrium potential for chloride (−65 mV), and in agreement with a GABAA receptor‐mediated IPSC. Analysis of synaptic latencies in a subset of cells indicated that IPSC latencies were significantly longer than EPSC latencies in all age groups, and values were consistent with di‐ and monosynaptic activation, respectively (Table 2; Fig. 2 A and B; two‐way ANOVA, thalamic, age: F(3,36) = 0.499, P = 0.685; response type: F(1,36) = 87.709, P < 0.001; cortical, age: F(3,36) = 1.557, P = 0.217; response type: F(1,36) = 100.699, P < 0.001). Furthermore, the glutamate receptor antagonist CNQX (10 μm) dramatically reduced sensory‐evoked IPSCs (Fig. 2 D and E; two‐way ANOVA, thalamic, drug: F(1,28) = 110.423, P < 0.001; cortical, drug: F(3,28) = 90.762, P < 0.001) with equal effect in all age groups (one‐way ANOVA, thalamic: F(3,28) = 0.340, P = 0.796; cortical: F(3,28) = 0.558, P = 0.647). Together, this strongly supports that sensory‐evoked inhibition is mostly driven by glutamatergic inputs that activate feed‐forward interneurons and not by direct activation of an unidentified pool of GABAergic neurons.
Figure 2. Evidence for largely disynaptic inhibition onto LA principal neurons .
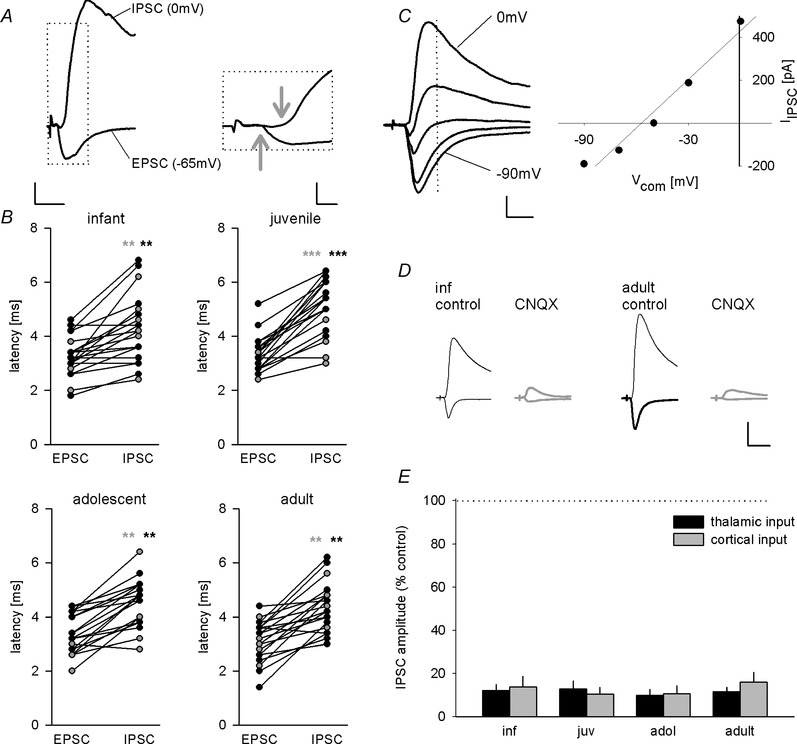
A, example traces of EPSC (at −65 mV) and IPSC (at 0 mV) evoked by cortical presynaptic fibre stimulation illustrating delayed onset of inhibitory current. Arrows indicate onset of excitatory and inhibitory current. Scale bar: left: 100 pA, 10 ms, right: 200 pA, 2 ms. B, scatter plots of latencies in infant, juvenile, adolescent and adult mice. Inhibitory currents show significantly longer latencies compared with excitatory responses (see Table 2; n = 10 for all age groups and input pathways). Black circles, thalamic input; grey circles, cortical input. C, example traces of an EPSC–IPSC sequence evoked by thalamic presynaptic fibre stimulation and corresponding current–voltage plot (holding potentials −90, −70, −50, −30 and 0 mV). Stimulus artefacts are truncated for clarity. Dotted line represents time point of IPSC measurement. Scale bars: 100 pA, 10 ms. The IPSC component had a reversal potential of −59 mV. D, example traces showing the effect of AMPA/kainate receptor antagonist CNQX (10 μm) on IPSCs evoked by thalamic presynaptic fibre stimulation in infant and adult mice. Scale bar: 200 pA, 20 ms. E, mean data (±SEM) of remaining IPSC amplitudes evoked by thalamic and cortical fibre stimulation upon CNQX application in all age groups. Statistical analysis revealed no significant difference across age groups (Table 2; one‐way ANOVA, thalamic input: F(3,28) = 0.340, P = 0.796; cortical input: F(3,28) = 0.558, P = 0.647). **P < 0.01, ***P < 0.001.
Table 2.
Properties of sensory evoked EPSCs and IPSCs
| Post hoc statistical | |||||
|---|---|---|---|---|---|
| Infant | Juvenile | Adolescent | Adult | Comparisons | |
| Latency (ms) | |||||
| Thal. EPSC | 3.0 ± 0.2 (10)1 | 2.8 ± 0.3 (10)2 | 3.1 ± 0.2 (10)3 | 3.2 ± 0.2 (10)4 | 1 P = 0.007; 2 P < 0.001 3 P = 0.001; 4 = 0.001 |
| Thal. IPSC | 4.0 ± 0.4 (10)1 | 4.2 ± 0.6 (10)2 | 4.5 ± 0.3 (10)3 | 4.4 ± 0.2 (10)4 | |
| Cort. EPSC | 3.5 ± 0.3 (10)5 | 3.3 ± 0.4 (10)6 | 3.9 ± 0.2 (10)7 | 3.1 ± 0.3 (10)8 | 5 P = 0.006; 6 P < 0.001 7 P = 0.002; 8 P = 0.001 |
| Cort. IPSC | 4.1 ± 0.4 (10)5 | 5.0 ± 0.6 (10)6 | 4.7 ± 0.2 (10)7 | 4.3 ± 0.4 (10)8 | |
| CNQX effect (% control) | |||||
| Thal. EPSC | 6.1 ± 2.7 (5) | 6.8 ± 0.7 (6) | 6.1 ± 1.6 (3) | 4.7 ± 1 (8) | |
| Thal. IPSC | 14.1 ± 3.5 (8) | 12.9 ± 3.6 (7) | 9.9 ± 2.8 (7) | 11.6 ± 2.1 (10) | |
| Cort. EPSC | 8.8 ± 4 (5) | 6.9 ± 1.9 (6) | 14 ± 9.7 (3) | 7.9 ± 2.8 (8) | |
| Cort. IPSC | 16.3 ± 5 (8) | 10.5 ± 3.1 (7) | 10.6 ± 3.6 (7) | 16.1 ± 4.5 (10) |
Values are given as means ± SEM (number of recorded cells). EPSCs and IPSC were evoked by thalamic (thal.) and cortical (cort.) fibre stimulation. Superscript numbers represent pairs of statistically significant comparisons (paired t test).
To compare the relative size of sensory‐evoked IPSCs in LA principal cells across development, we used a similar approach as Gambino et al. (2010), i.e. thalamic and cortical presynaptic fibre stimulation was adjusted such that the amount of excitation (EPSC amplitude at −65 mV) was held constant in both pathways in all age groups (Fig. 3 B and D; one‐way ANOVA: F(3,91) = 0.850, P = 0.470 and F(3,91) = 0.278, P = 0.841, respectively). Under these conditions, we found that the amplitude of the sensory input‐evoked IPSCs increased with age in both thalamic and cortical pathways (one‐way ANOVA: F(3,91) = 12.943, P < 0.001 and F(3,91) = 14.275, P < 0.001, respectively). For the thalamic input pathway, a major increase occurred between the infant and juvenile age groups (Fig. 3 A and B; Bonferroni corrected post hoc comparison: P = 0.006), while in the cortical input pathway, inhibition increased significantly between juvenile and adolescent age groups (Fig. 3 C and D; significant Bonferroni corrected post hoc comparisons: inf vs. adol: P < 0.001; inf vs. adult: P < 0.001; juv vs. adol: P = 0.044; juv vs. adult: P < 0.001). We found no difference in IPSC amplitude between the two input pathways (one‐way ANOVA: F(1,90) = 1.144, P = 0.288), suggesting that in our hands, there is no asymmetry in recruitment of inhibition as previously described (Shin et al. 2006). To rule out that our data are biased by stimulation intensity, we compared input–output curves of IPSCs and EPSCs and found a concomitant increase in both a constant inhibition/excitation ratio (I/E ratio) and a larger contribution of inhibition in older animals (Fig. 4 A and B). If EPSCs were shunted by differential recruitment of inhibition with higher stimulation intensities, the I/E ratio should not stay constant, but increase disproportionally. To address this for all age groups and inputs, we performed correlation analysis between EPSC size (obtained by different stimulation strengths) and the resulting I/E ratio (Fig. 4 C–F). As we detected no correlation, we conclude that this does not affect comparison of I/E ratios under our experimental conditions.
In summary, our data strongly suggest that sensory input‐evoked inhibition onto LA principal neurons increases in an input‐specific manner during development. Thalamic inhibition matures earlier than cortical and may already be required for tuning fear learning‐related sensory plasticity already in juveniles. This is different from the spontaneous excitatory and inhibitory drive, which more likely represents local connectivity amongst LA neurons, both excitatory and inhibitory.
Maturation of LA principal neuron dendritic morphology and spine density
To gain insights into morphological changes associated with functional increases in synaptic drive, we used cell fills and reconstructions to investigate maturation of LA principal neuron dendritic morphology. In keeping with recent results by others in the rat BLA (Ryan et al. 2015) we found a moderate increase in total dendrite length and area of dendritic field with no apparent changes in number of primary dendrites, branch points and endings (Fig. 5 A–D and data not shown; one‐way ANOVA, total dendritic length: F(3,16) = 2.372, P = 0.109; area of dendritic field: F(3,16) = 1.555, P = 0.239). Because most glutamaterigc synapses are located on spines, we then assessed dendritic spine density in an area of the dendritic tree previously shown to also receive thalamic and cortical sensory inputs (Humeau et al. 2005). We show that spine density increased several‐fold between infancy and adolescence (Fig. 5 E–G; one‐way ANOVA: F(3,45) = 36.401, P < 0.001), suggesting a developmental increase in the number of excitatory synapses that impinge onto individual LA neurons.
Figure 5. Postnatal changes in dendritic morphology and spine density in LA principal neurons .
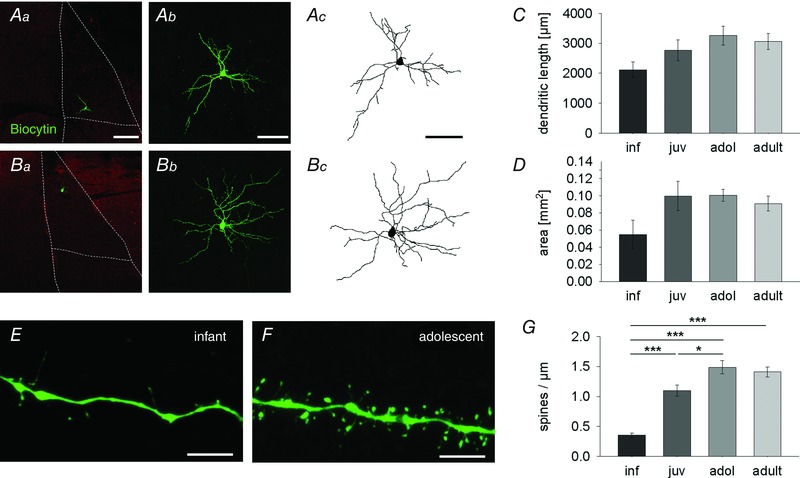
A–D, postnatal maturation of dendritic morphology. Aa and Ba, location of representative principal neurons within the LA (dotted lines) in an infant (Aa) and adolescent (Ba) mouse. Sections are counterstained with vesicular GABA transporter antibody (red). Scale bar: 200 μm. Ab and Bb, maximum intensity projection confocal images of neurons shown in Aa and Ba. Scale bar: 100 μm. Ac and Bc, Neurolucida drawing of cells from Ab and Bb. Scale bar: 100 μm. C, mean data (±SEM) of total dendritic length (μm) in all age groups (number of filled cells): inf: 2116.8 ± 261.9 (4); juv: 2764.4 ± 352.5 (5); adol: 3258.5 ± 312.9 (5); adult: 3059.6 ± 265.8 (6)). D, mean data (±SEM) of area of dendritic field (mm2): inf: 0.055 ± 0.017 (4); juv: 0.1 ± 0.017 (5); adol: 0.101 ± 0.007 (5); adult: 0.091 ± 0.009 (6)). E–G, postnatal changes in spine density. E and F, confocal images (maximum intensity projection) from infant (E) and adolescent (F) dendrites illustrating an increase in spine density. Scale bars: 5 μm. G, mean data (±SEM) of spine density per μm (number of imaged dendrites): inf: 0.35 ± 0.01 (12); juv: 1.10 ± 0.01 (12); adol: 1.49 ± 0.02 (13); adult: 1.41 ± 0.02 (12); significant differences (one‐way ANOVA with Bonferroni correction): inf vs. juv, adol and adult: P < 0.001; juv vs. adol: P = 0.013). *P < 0.05, ***P < 0.001.
Taken together, our anatomical data show a moderate increase in dendritic length and area of dendritic field. More pronounced was a change in dendritic spine density, which might contribute to the observed increase in excitatory drive onto LA principal neurons.
GABAB receptor modulation of sensory‐evoked inhibition is differentially regulated
In the LA, GABA released from GABAergic interneurons can regulate its own release onto LA principal cells via GABAB autoreceptors (Yamada et al. 1999; Szinyei et al. 2000). At the same time, sensory input‐evoked GABA release has been shown to be ineffective in modulating excitatory inputs onto local interneurons (Szinyei et al. 2000; Pan et al. 2009). Here, we asked at which age sensory‐evoked GABAA‐mediated inhibition becomes regulated by GABAB mechanisms. We used paired‐pulse stimulation with a 300 ms inter‐stimulus interval, the shortest interval previously shown to elicit GABAB modulation (Szinyei et al. 2000). Under control conditions, we observed a similar amount of paired‐pulse depression (PPD) of sensory‐evoked IPSCs in LA principal neurons in all age groups (Fig. 6 A–C; one‐way ANOVA, thalamic: F(3,81) = 1.375, P = 0.256; cortical: F(3,81) = 1.997, P = 0.121). To address the mechanism of PPD, we applied the GABAB receptor antagonist CGP55845A (10 μm), which showed a significant effect on PPD in both input pathways (Fig. 6 B and C; thalamic, age: F(3,80) = 0.853, P = 0.469; drug: F(1,80) = 22.66, P < 0.001; cortical, age: F(3,81) = 0.971, P = 0.411; drug: F(1,81) = 14.596, P < 0.001). As previously reported, CGP55845A significantly increased the paired‐pulse‐ratio (PPR) in young adult mice of thalamic and cortical input evoked IPSCs (Fig. 6 B and C). Interestingly, while in the thalamic pathway CGP55845A also significantly increased the PPR of IPSCs in infant and juvenile animals, cortical modulation was GABAB receptor‐dependent only in adults (Fig. 6 A–C).
Figure 6. Maturation of GABAB‐modulation of sensory‐evoked inhibition in LA principal neurons .
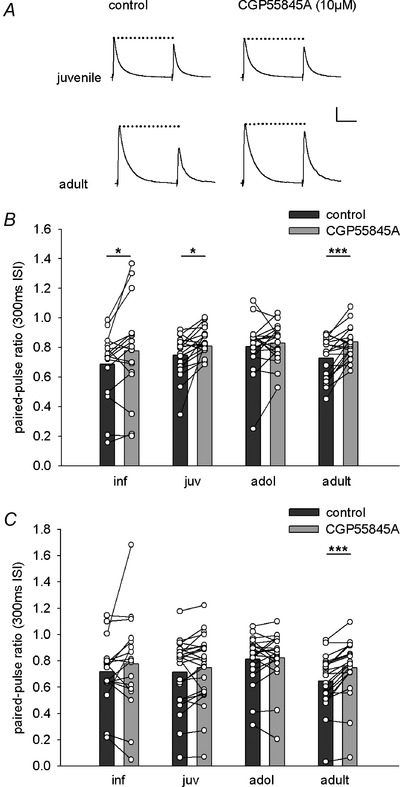
A, representative IPSCs evoked by cortical fibre stimulation in juvenile and adult mice. Paired pulses were applied at an inter‐stimulus interval of 300 ms. Traces show IPSCs under control condition (left) and after application of the GABAB receptor antagonist CGP55845A (right). CGP55845A reduces paired‐pulse depression in adult mice. Scale bar: 100 pA, 100 ms. B and C, single cell and mean data showing the effect of CGP55845A application on paired‐pulse ratios of IPSCs evoked by thalamic (B) or cortical (C) fibre stimulation in the different age groups. Paired‐pulse ratios under control condition (dark grey bars) and after CGP55845A application (light grey bars). B, thalamic input. Inf: 0.69 ± 0.06 vs. 0.78 ± 0.08 (n = 17), P = 0.049; juv: 0.75 ± 0.03 vs. 0.81 ± 0.02 (25), P = 0.035; adol: 0.81 ± 0.04 vs. 0.83 ± 0.03 (19), P = 0.443; adult: 0.73 ± 0.03 vs. 0.84 ± 0.02 (23), P < 0.001). C, cortical input. Inf: 0.72 ± 0.06 vs. 0.78 ± 0.09 (n = 17), P = 0.271; juv: 0.71 ± 0.05 vs. 0.75 ± 0.05 (25), P = 0.073; adol: 0.81 ± 0.04 vs. 0.82 ± 0.04 (20), P = 0.653; adult: 0.65 ± 0.04 vs. 0.75 ± 0.04 (23), P < 0.001). P values are from paired t tests. *P < 0.05; ***P < 0.001.
In summary, PPD of GABAA receptor‐mediated sensory‐evoked inhibition was prominent in all age groups, but its control via GABAB receptors is differentially regulated during development.
Exogenous GABAB receptor activation induces presynaptic depression of excitatory sensory inputs in all age groups
Assuming that increases in inhibition measured in LA principal neurons results from increased GABA release from recruited GABAergic neurons, we wanted to investigate its consequences for heterosynaptic modulation of excitatory inputs. A prerequisite enabling heterosynaptic modulation is the functional expression of GABAB receptors on sensory afferents. In a first step, we investigated the presence of GABAB receptors on sensory inputs onto LA principal neurons by application of the GABAB receptor agonist baclofen (2 μm) and subsequent co‐application of the antagonist CGP55845A (10 μm). Indeed, baclofen and co‐application of CGP55845A significantly affected EPSC amplitudes in both input pathways with no effect of age (Fig. 7 A–D; two‐way ANOVA, thalamic, age: F(3,33) = 0.237, P = 0.870; drug: F(2,66) = 97.087, P < 0.001; age × drug: F(6,66) = 0.574, P = 0.750; cortical, age: F(3,32) = 0.580, P = 0.632; drug: F(2,64) = 45.352, P < 0.001; age × drug: F(6,64) = 0.832, P = 0.550). Supporting a presynaptic mechanism, baclofen and co‐application of CGP55845A also altered the PPR at an inter‐stimulus interval of 50 ms (Fig. 7 A, B, E and F; two‐way ANOVA, thalamic, age: F(3,33) = 0.790, P = 0.508; drug: F(2,66) = 35.901, P < 0.001; age × drug: F(6,66) = 0.782, P = 0.587; cortical, age: F(3,32) = 1.050, P = 0.384; drug: F(2,64) = 57.311, P < 0.001; age × drug: F(6,64) = 2.128, P = 0.062). Post hoc comparisons of baclofen effects in individual age groups revealed a significant decrease in EPSC amplitude and concomitant increase in PPR in both inputs and for all individual age groups (Table 3; Fig. 7 C–F). Importantly, all changes in EPSC amplitude and PPR were reversed by subsequent co‐application of the GABAB receptor antagonist CGP55845A (Table 3; Fig. 7 C–F).
Figure 7. GABAB receptors are present at excitatory sensory inputs onto LA principal neurons throughout development .
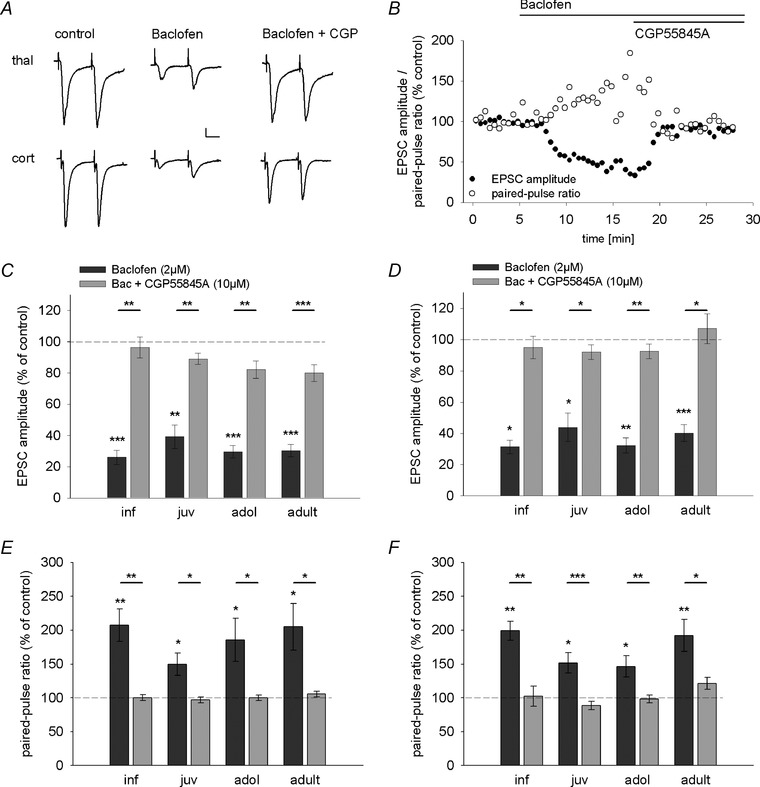
A, example EPSC traces depicting the effect of baclofen and baclofen + CGP55845A application on EPSC amplitudes and paired‐pulse ratios (50 ms inter‐stimulus interval) in thalamic (top) and cortical (bottom) inputs in an adult mouse. Scale bar: 20 pA, 20 ms. B, time course of the effect of baclofen and CGP55845A application showing a reduction of EPSC amplitude and concurrent increase in paired‐pulse ratio in a representative LA principal neuron in an adult mouse. Bars indicate time of baclofen and CGP55845A application. C and D, mean drug effect (as a percentage of control condition ± SEM) showing the effect of baclofen (dark grey bars) and CGP55845A (light grey bars) application on EPSC amplitudes evoked by thalamic (C) and cortical (D) fibre stimulation across development. Mean EPSC amplitudes, number of recorded cells, and post hoc statistics are shown in Table 3. E and F, mean drug effect (as a percentage of control condition ± SEM) showing the effect of baclofen (dark grey bars) and CGP55845A (light grey bars) application on paired‐pulse ratios (50 ms inter‐stimulus interval) evoked by thalamic (E) and cortical (F) fibre stimulation across development. Mean paired‐pulse ratios, number of recorded cells and post hoc statistics are shown in Table 3. *P < 0.05, **P < 0.01, ***P < 0.001.
Table 3.
EPSC amplitudes and paired‐pulse ratios after baclofen and baclofen+CGP55845A application
| Post hoc statistical | |||||
|---|---|---|---|---|---|
| Infant | Juvenile | Adolescent | Adult | comparisons | |
| EPSC amplitude (pA) | |||||
| Thalamic, control | −154.3 ± 16.6 (8)1 | −169.9 ± 29.3 (10)3 | −171.6 ± 23.5 (9)5 | −152.1 ± 22.9 (10)7 | 1 P < 0.001, 2 P = 0.004 |
| Thalamic, Bac | −39.3 ± 6.9 (8)1,2 | −64.1 ± 19.2 (10)3,4 | −51.4 ± 9.4 (9)5,6 | −50.1 ± 12.4 (10)7,8 | 3 P = 0.004, 4 P = 0.007 |
| Thalamic, Bac. + CGP | −150.2 ± 22.8 (8)2 | −148.4 ± 25.3 (10)4 | −143.8 ± 26.1 (9)6 | −120.3 ± 18.4 (10)8 | 5 P < 0.001, 6 P = 0.004 |
| 7 P < 0.001, 8 P < 0.001 | |||||
| Cortical, control | −99.0 ± 23.7 (7)1 | −143.9 ± 33.1 (10)3 | −162.4 ± 27.4 (9)5 | −119.2 ± 25.8 (10)7 | 1 P = 0.016, 2 P = 0.013 |
| Cortical, Bac | −27.4 ± 5.8 (7)1,2 | −50.0 ± 16.5 (10)3,4 | −53.2 ± 12.2 (9)5,6 | −56.8 ± 19.6 (10)7,8 | 3 P = 0.013, 4 P = 0.037 |
| Cortical, Bac. + CGP | −92.9 ± 20.8 (7)2 | −125.1 ± 27.9 (10)4 | −145.0 ± 24.0 (9)6 | −141.0 ± 44.2 (10)8 | 5 P = 0.002, 6 P = 0.001 |
| 7 P < 0.001, 8 P = 0.023 | |||||
| PPR | |||||
| Thalamic, control | 0.76 ± 0.03 (8)1 | 0.94 ± 0.08 (10)3 | 0.90 ± 0.05 (9)5 | 0.89 ± 0.05 (10)7 | 1 P = 0.004, 2 P = 0.003 |
| Thalamic, Bac | 1.55 ± 0.15 (8)1,2 | 1.41 ± 0.18 (10)3,4 | 1.59 ± 0.23 (9)5,6 | 1.91 ± 0.37 (10)7,8 | 3 P = 0.019, P = 0.013 |
| Thalamic, Bac. + CGP | 0.76 ± 0.04 (8)2 | 0.90 ± 0.08 (10)4 | 0.90 ± 0.06 (9)6 | 0.95 ± 0.08 (10)8 | 5 P = 0.044, 6 P = 0.032 |
| 7 P = 0.032, 8 P = 0.048 | |||||
| Cortical, control | 1.00 ± 0.19 (7)1 | 1.02 ± 0.07 (10)3 | 0.93 ± 0.09 (9)5 | 0.91 ± 0.06 (10)7 | 1 P = 0.001, 2 P = 0.001 |
| Cortical, Bac | 1.85 ± 0.26 (7)1,2 | 1.49 ± 0.11 (10)3,4 | 1.28 ± 0.09 (9)5,6 | 1.73 ± 0.22 (10)7,8 | 3 P = 0.010, 4 P < 0.001 |
| Cortical, Bac. + CGP | 1.10 ± 0.27 (7)2 | 0.87 ± 0.04 (10)4 | 0.90 ± 0.08 (9)6 | 1.10 ± 0.10 (10)8 | 5 P = 0.030, 6 P = 0.002 |
| 7 P = 0.006, 8 P = 0.026 |
Values are given as means ± SEM (number of recorded cells). EPSCs were evoked by thalamic and cortical fibre stimulation. Paired‐pulse ratios were determined at 50 ms inter‐stimulus interval. PPR, paired‐pulse ratio; Bac., baclofen; CGP, CGP55845A. Superscript numbers represent pairs of statistically significant comparisons (Bonferroni‐corrected paired t tests).
In summary, our data show that GABAB receptors are functionally expressed on thalamic and cortical afferents onto LA principal neurons throughout development, and their activation depresses glutamate release at these terminals. This raises the question at which developmental stages and under which conditions endogenous release of GABA can activate this signalling pathway.
GABAB receptors mediate heterosynaptic inhibition of sensory inputs in older animals
Modulation of sensory afferents by GABA released from a synaptically recruited interneuronal network is achieved by priming stimulation of one sensory input pathway while assessing changes in the other pathway (Shaban et al. 2006; Pan et al. 2009; Lange et al. 2014). GABAB receptors play a critical role in mediating heterosynaptic inhibition between thalamic and cortical input pathways, limiting the amount of glutamatergic transmission onto LA principal neurons and gating presynaptic LTP in the cortical input (Shaban et al. 2006; Pan et al. 2009). To investigate the development of heterosynaptic inhibition onto the cortical pathway, we adopted a protocol from Pan et al. (2009) and applied presynaptic fibre stimulation at 200 Hz for 50 ms (priming stimulation) in the thalamic input pathway (Fig. 8 A). When comparing cortical EPSCs before and after priming stimulation, we found a significant change in EPSC amplitude with no effect of age (two‐way ANOVA, EPSC amplitude, age: F(3,63) = 0.67, P = 0.99; stimulation: F(1,63) = 107.794, P < 0.001), and post hoc comparisons in individual age groups showed a significant reduction of EPSC amplitude in all age groups (Fig. 8 B and C). In addition, priming stimulation significantly altered the PPR, which was also affected by age group (two‐way ANOVA, PPR, age: F(3,63) = 4.63, P = 0.005; stimulation: F(1,63) = 8.083, P = 0.006). Post hoc analysis revealed a significant increase in PPR in adolescent mice and a good correlation of EPSC amplitude decrease with an increase in PPR in adults (r(20) = −0.639, P = 0.001), while changes in PPR were not detectable in younger age groups (Table 4). This suggests that heterosynaptic inhibition was mediated by a presynaptic mechanism in adolescent and adult mice. To probe if this depended on activation of presynaptic GABAB receptors, we compared the effect of priming stimulation under control condition and in the presence of the GABAB receptor antagonist CGP55845A (Fig. 8 D and E). Here, we found a significant drug effect on EPSC amplitude and PPR after priming stimulation with no effect of age (two‐way ANOVA, EPSC amplitude, age: F(3,63) = 0.844, P = 0.475; drug: F(1,63) = 4.755, P = 0.033; PPR, age: F(3,62) = 1.512, P = 0.220; drug: F(1,62) = 17.989, P < 0.001). Post hoc analysis revealed that blockade of GABAB receptors reduced the effect of priming stimulation only in adolescent and adult mice (Table 4; Fig. 8 D and E). Again, consistent with a presynaptic effect, the increase in EPSC amplitude was accompanied by a significant reduction in PPR (Table 4; adol: r(15) = −0.508, P = 0.027; adult: r(21) = −0.549, P = 0.005). Thus, thalamic priming stimulation initiates GABAB receptor‐dependent presynaptic depression of the cortical input from adolescence onward.
Figure 8. Developmental regulation of heterosynaptic inhibition of cortical inputs onto LA principal neurons .
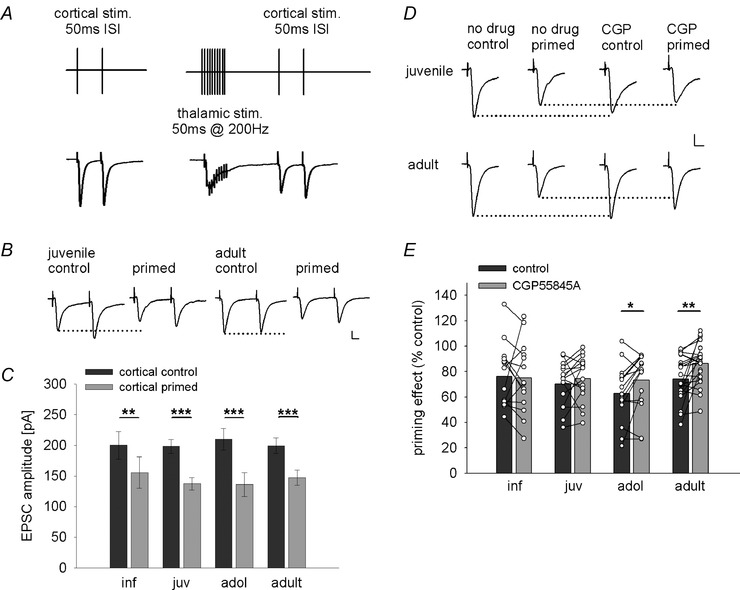
A, scheme depicting experimental design and stimulation protocol, and example traces of heterosynaptic inhibition of cortical EPSCs induced by priming burst stimulation applied to the thalamic pathway. B, example traces of EPSCs from juvenile and adult mice evoked by paired‐pulse stimulation (50 ms inter‐stimulus interval) under control condition and after thalamic burst simulation (primed) illustrating heterosynaptic depression in both age groups. Scale bar: 50 pA, 10 ms. C, mean data of cortical EPSC amplitudes under control condition (cortical control, dark grey bars) and after thalamic burst stimulation (cortical primed, light grey bars) in different age groups. Mean EPSC amplitudes, number of recorded cells, and post hoc statistics are shown in Table 4. D, example traces of EPSCs from juvenile and adult mice under control condition and after thalamic burst stimulation (primed) in the absence and presence of the GABAB receptor antagonist CGP55845A illustrating the differential block of heterosynaptic priming in adult vs. juvenile mice. Scale bar: 50 pA, 10 ms. E, single cell and mean data showing the effect (as a percentage of control) of thalamic burst stimulation on cortical EPSC amplitudes under control condition (dark grey bars) and after application of the GABAB antagonist CGP55845A (light grey bars). Mean EPSC amplitudes, paired‐pulse ratios, number of recorded cells, and post hoc statistics are shown in Table 4. *P < 0.05, **P < 0.01, ***P < 0.001.
Table 4.
Priming effects on EPSC amplitudes and paired‐pulse ratios under control condition and after CGP55845A application
| Post hoc statistical | |||||
|---|---|---|---|---|---|
| Infant | Juvenile | Adolescent | Adult | comparisons | |
| Thal → cort | |||||
| EPSC (pA), control | −200 ± 22.3 (14)1 | −198.3 ± 11.5 (16)2 | −209.9 ± 17.3 (15)3 | −199.3 ± 12.5 (22)4 | 1 P = 0.008; 2 P < 0.001 |
| EPSC (pA), primed | −155.6 ± 25.8 (14)1 | −137.2 ± 9.9 (16)2 | −135.9 ± 19.2 (15)3 | −147.5 ± 12.3 (22)4 | 3 P < 0.001; 4 P < 0.001 |
| PPR, control | 0.79 ± 0.07 (14) | 1.01 ± 0.04 (16) | 0.93 ± 0.05 (15)1 | 1.06 ± 0.04 (22) | 1 P = 0.038 |
| PPR, primed | 0.83 ± 0.07 (14) | 1.04 ± 0.05 (16) | 1.14 ± 0.13 (15)1 | 1.13 ± 0.05 (22) | |
| EPSC primed (%), control | 76.2 ± 6.7 (14) | 70.4 ± 4.3 (16) | 62.8 ± 5.9 (15)1 | 74.1 ± 3.8 (21)2 | 1 P = 0.028; 2 P = 0.007 |
| EPSC primed (%), CGP | 75.2 ± 7.4 (14) | 74.4 ± 4.5 (16) | 73.1 ± 5.8 (15)1 | 86.2 ± 3.6 (21)2 | 3 P = 0.002; 4 P = 0.027 |
| PPR primed (%), control | 108 ± 6.5 (14) | 104 ± 6.2 (16) | 120.6 ± 7.3 (15)3 | 107.7 ± 4.3 (21)4 | |
| PPR primed (%), CGP | 92.1 ± 4.1 (14) | 100.5 ± 3.3 (16) | 104.2 ± 5.1 (15)3 | 99.4 ± 2.2 (21)4 | |
| Cort → thal | |||||
| EPSC (pA), control | −217.1 ± 15.2 (14)1 | −213.6 ± 16.3 (16)2 | −223.3 ± 24.9 (15)3 | −201.7 ± 8.9 (21)4 | 1 P < 0.001; 2 P = 0.002 |
| EPSC (pA), primed | −136 ± 19.2 (14)1 | −176.5 ± 13.1 (16)2 | −172.5 ± 25.7 (15)3 | −143.6 ± 10.9 (21)4 | 3 P < 0.001; 4 P < 0.001 |
| PPR, control | 0.65 ± 0.07 (14) | 0.97 ± 0.05 (16) | 1.01 ± 0.06 (15)1 | 1.06 ± 0.03 (21) | 1 P = 0.013 |
| PPR, primed | 0.62 ± 0.07 (14) | 0.96 ± 0.03 (16) | 0.87 ± 0.06 (15)1 | 1.07 ± 0.05 (21) | |
| EPSC primed (%), control | 60.8 ± 6.3 (14) | 83.9 ± 3.9 (16) | 76 ± 5.2 (15) | 71.7 ± 5.2 (20)1 | 1 P < 0.001 |
| EPSC primed (%), CGP | 63.6 ± 5.9 (14) | 89 ± 4.3 (16) | 81.6 ± 4.8 (15) | 83.2 ± 5.6 (20)1 | 2 P = 0.019 |
| PPR primed (%), control | 106.8 ± 11 (14)2 | 105.5 ± 9.1 (16) | 88.6 ± 5.1 (15) | 100.3 ± 4.3 (20) | |
| PPR primed (%), CGP | 87.1 ± 5.1 (14)2 | 103.7 ± 6.8 (16) | 86.4 ± 4.8 (15) | 95.9 ± 2.9 (20) |
Values are given as mean ± SEM (number of recorded cells). EPSCs were evoked by thalamic and cortical fibre stimulation. Paired‐pulse ratios were determined at 50 ms inter‐stimulus interval. PPR, paired‐pulse ratio; CGP, CGP55845A; thal → cort, thalamic priming cortical input; cort → thal, cortical priming thalamic input. Superscript numbers represent pairs of pairs of statistically significant comparisons (paired t tests).
Subsequently, we investigated the effect of cortical priming on the thalamic pathway (Pan et al. 2009; Lange et al. 2014). We also found a significant effect on EPSC amplitudes with no effect of age (two‐way ANOVA, EPSC amplitude, age: F(3,62) = 0.683, P = 0.566; stimulation: F(1,62) = 102.415, P < 0.001) and post hoc comparisons in individual age groups showed a significant reduction of EPSC amplitudes in all age groups (Table 4). Application of the GABAB receptor antagonist CGP55845A showed a significant drug effect on EPSC amplitudes and PPR (two‐way ANOVA, EPSC amplitude, age: F(3,61) = 3.937, P = 0.012; drug: F(1,61) = 10.265, P = 0.002; PPR, age: F(3,61) = 1.421, P = 0.245; drug: F(1,61) = 8.063, P = 0.006). Post hoc analysis revealed a significant increase of EPSC amplitudes and decrease in PPR, as well as a correlation between these changes only in young adults (Table 4; r(20) = 0.384, P = 0.047).
In summary, we show that heterosynaptic inhibition is a common feature of activity‐dependent crosstalk between sensory inputs at all developmental stages. While the mechanisms in early development remain unclear, presynaptic GABAB receptors participate in mediating heterosynaptic interaction in older animals, when more released GABA is available in the LA. In agreement with an earlier increase in evoked inhibition in the thalamic pathway, the ability of this pathway to prime the cortical input emerges slightly earlier than cortical priming of the thalamic input.
Tonic activation of GABAB receptors
Next, we asked if the observed spontaneous GABAergic drive that becomes more prominent from adolescence onward (Fig. 1) is sufficient to modulate excitatory sensory inputs in the absence of priming. Towards this end, we stimulated sensory inputs at low frequency (0.1 Hz) and assessed the effects of GABAB receptor blockade. Indeed, application of the GABAB antagonist CGP55845A alone resulted in a small but significant increase in the amplitude of sensory‐evoked EPSCs in adolescent and adult animals in both input pathways (Fig. 9 A, B, D and E; two‐way ANOVA, thalamic, age: F(3,98) = 1.91, P = 0.133; drug: F(1,98) = 10.225, P = 0.002; cortical, age: F(3,101) = 2.451, P = 0.068; drug: F(1,101) = 27.957, P < 0.001). The increase in EPSC amplitude correlated well with a concomitant decrease in the PPR (Fig. 9 C and F; thalamic, adol: r(27) = −0.459, P = 0.009; adult: r(27) = −0.613, P < 0.001; cortical, adol: r(29) = −0.393, P = 0.017; adult: r(27) = −0.494, P = 0.004), indicating a tonic presynaptic depression via GABAB receptors. The most parsimonious explanation is that ambient GABA provided by spontaneous GABA release in basolateral amygdala leads to tonic activation of presynaptic GABAB receptors on excitatory sensory terminals to restrict glutamate release. Interestingly, this tonic presynaptic inhibition appeared to be specific to excitatory synapses on LA principal neurons since we did not observe any changes in sensory‐evoked IPSCs in the presence of the GABAB antagonist (thalamic: two‐way ANOVA, age: F(3,80) = 12.94, P < 0.001; drug: F(1,80) = 1.007, P = 0.319; post hoc paired t tests, adol: t(18) = −1.888, P = 0.075; adult: t(22) = −0.536, P = 0.598; cortical, two‐way ANOVA, age: F(3,81) = 18.395, P < 0.001; drug: F(1,81) = 0.966, P = 0.329; post hoc paired t tests, adol: t(19) = −1.572, P = 0.133; adult: t(22) = −0.545, P = 0.591).
Figure 9. Developmental regulation of GABAB‐mediated tonic presynaptic inhibition of sensory inputs .
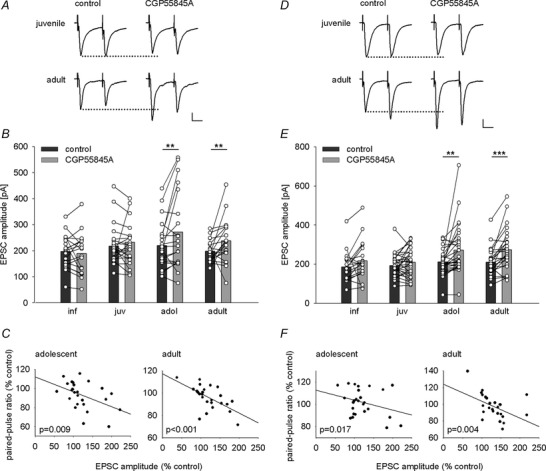
A and D, example traces of EPSCs evoked by thalamic (A) or cortical (D) paired‐pulse stimulation (50 ms inter‐stimulus interval) from juvenile and adult mice under control conditions and after CGP55845A application showing amplitude and paired‐pulse ratio changes in adults. Scale bar: 50 pA, 20 ms. B and E, single cell and mean data illustrating the effect of CGP55845A application on EPSC amplitudes evoked by thalamic (B) or cortical (E) fibre stimulation across development. B, thalamic EPSC amplitudes (pA) under control condition and after CGP55845A application, paired t tests: inf: −196.9 ± 14.7 vs. −189.7 ± 18.1 (19), P = 0.562; juv: −217.8 ± 12.2 vs. −233.6 ± 17.6 (30), P = 0.330; adol: −219.7 ± 15.1 vs. −269.7 ± 25.8 (27), P = 0.008; adult: −198.4 ± 7.2 vs. −238 ± 14.1 (27), P = 0.005). E, cortical EPSC amplitudes (pA) under control condition and after CGP55845A application, paired t tests: inf: −186 ± 17.6 vs. −217.3 ± 23.4 (n = 19), P = 0.096; juv: −193.3 ± 10.3 vs. −210.3 ± 13 (30), P = 0.136; adol: −211.1 ± 11.6 vs. −271.8 ± 23.5 (29), P = 0.006; adult: −210 ± 13.1 vs. −273.8 ± 19.9 (27), P < 0.001). C and F, plots showing correlations between changes in EPSC amplitude and paired‐pulse ratio following application of CGP55845A in thalamic and cortical inputs in adolescent and adult mice. C, thalamic input: adol: n = 26, r = −0.459, P = 0.009; adult: n = 27, r = −0.613, P < 0.001). F, cortical input: adol: n = 29, r = −0.393, P = 0.017; adult: n = 27, r = −0.494, P = 0.004). **P < 0.01, ***P < 0.001.
Increasing ambient GABA levels enables GABAB receptor‐mediated tonic inhibition in juveniles
Although GABAB receptors are expressed and functional on excitatory terminals across all investigated age groups (Fig. 7), we found no evidence for tonic activation of GABAB receptors in infants and juveniles. We hypothesized that this results from lower ambient GABA levels. Therefore, increasing ambient GABA levels experimentally in slices from young animals should enable tonic heterosynaptic inhibition via presynaptic GABAB receptors. This effect should also be observable in adult animals if presynaptic GABAB signalling is not saturated. We tested this by using the GABA reuptake blocker SKF89976A (30 μm) in juvenile and young adult animals. We first confirmed that SKF89976A enhanced extracellular GABA as evidenced by longer decay times of GABAA receptor‐mediated IPSCs (n = 3 juveniles, n = 4 adults, data not shown). To prevent potential adverse effects via GABAA receptor‐mediated inhibition in other parts of the network, we then applied SKF89976A in the presence of picrotoxin (100 μm). Under these conditions, GABA reuptake block decreased EPSC amplitudes evoked by thalamic and cortical presynaptic fibre stimulation in both juveniles and young adults (Fig. 10 A and C; two‐way ANOVA, thalamic, age: F(1,22) = 2.295, P = 0.144; drug: F(1,22) = 30.099, P < 0.001; cortical, age: F(1,21) = 0.115, P = 0.738; drug: F(1,21) = 48.91, P < 0.001). To further show that this decrease was due to activation of GABAB receptors, we pre‐incubated slices for 10–12 min with the GABAB receptor antagonist CGP52432 (10 μm) before the application of SKF89976A. As expected, we found a significant effect of antagonist pre‐treatment (SKF vs. SKF+CGP) in both input pathways (thalamic: F(1,44) = 10.613, P = 0.002; cortical: F(1,42) = 15.775, P < 0.001) with no effect of age group (thalamic: F(1,44) = 0.693, P = 0.410; cortical: F(1,42) = 0.213, P = 0.647). Post hoc analysis revealed that CGP52432 significantly decreased the effect of GABA reuptake inhibition on EPSC amplitude in both juveniles and young adults (Fig. 10 E and F). This indicates that activation of GABAB receptors play a role in mediating the effect by SKF89976A.
Figure 10. Increased ambient GABA levels enable GABAB‐mediated tonic inhibition in juvenile mice .
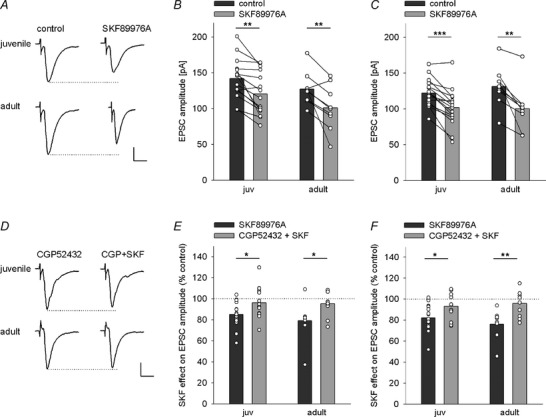
A, example trace of thalamic EPSCs under control condition and after application of the GABA reuptake inhibitor SKF89976A in juvenile and adult mice illustrating a reduction in EPSC amplitude. Scale bar: 50 pA, 10 ms. B and C, single cell and mean data showing the reduction of EPSC amplitudes in thalamic (B) and cortical (C) inputs after application of the GABA reuptake inhibitor SKF89976A. B, thalamic EPSC amplitudes (pA) under control condition and after SKF89976A application, paired t tests: juv: −141.9 ± 7.3 vs. −120.7 ± 7.8 (15), P = 0.001; adult: −127.4 ± 7.7 vs. −101.1 ± 10.4 (9), P = 0.009). C, cortical EPSC amplitudes (pA) under control condition and after SKF89976A application, paired t tests: juv: −122.4 ± 5.2 vs. −101.8 ± 7 (15), P < 0.001; adult: −131.3 ± 10.5 vs. −100.4 ± 12.1 (8), P = 0.005). D, example traces of thalamic EPSCs upon pre‐treatment with the GABAB antagonist CGP52432 and after subsequent application of the GABA reuptake inhibitor SKF89976A in juvenile and adult mice illustrating no effect of GABA reuptake inhibition when GABAB receptors are blocked in both ages. Scale bar: 50 pA, 10 ms. E and F, comparison of single cell and mean data showing the effect of GABA reuptake block on EPSC amplitudes (% of control) under SKF89976A (dark grey bars) and after pre‐treatment with the GABAB antagonist CGP52432 (CGP52432 + SKF, light grey bars) in juvenile and adult mice. E, thalamic EPSC amplitudes (%): juv: 85.1 ± 3.2 (15) vs. 96.3 ± 4 (14); adult: 79.1 ± 6.1 (9) vs. 95.3 ± 3.7 (10); initial EPSC amplitudes under CGP (pA): juv: −151.5 ± 8.1; adult: −150.1 ± 7.2; unpaired t tests SKF alone vs. CGP+SKF condition: juv: t(27) = −2.22, P = 0.035; adult: t(17) = −2.32, P = 0.033. F, cortical EPSC amplitudes (%): juv: 82.3 ± 3.3 (15) vs. 93.5 ± 3.4 (13); adult: 76.2 ± 5.2 (8) vs. 96 ± 3.9 (10); initial amplitudes under CGP (pA): juv: −130.5 ± 8.9; adult: −140.8 ± 8.2; unpaired t tests SKF alone vs. CGP+SKF condition: juv: t(26) = −2.354, P = 0.026; adult: t(16) = −3.119, P = 0.007. *P < 0.05, **P < 0.01, ***P < 0.001.
Taken together, our data strongly suggest that the amount of ambient GABA available in the slice, which is naturally low in young animals but increases during development, is a critical factor in mediating tonic GABAB receptor‐dependent presynaptic inhibition of sensory presynaptic inputs in the LA of mice.
Discussion
Thalamic and cortical inputs convey sensory information relevant in Pavlovian fear conditioning to LA principal neurons. They also target local interneurons that provide feed‐forward inhibition, which has a crucial role in gating synaptic plasticity and modulating fear learning and expression. Here, we have shown that the LA inhibitory system and sensory input modulation by inhibition undergo postnatal maturation from infancy into young adulthood. This includes changes in spontaneous inhibitory and excitatory drive onto principal neurons that are paralleled by anatomical changes, increases in the amount of sensory‐evoked inhibition, as well as age‐ and input‐dependent activation of auto‐ and heterosynaptic inhibition via GABAB receptors. Additionally, we provide evidence that the availability of ambient GABA critically determines whether presynaptic GABAB receptors can be activated to mediate tonic inhibition of excitatory inputs.
Maturation of spontaneous and sensory‐evoked feed‐forward inhibition
In many brain regions, maturation of GABAergic drive includes an increase in GABAergic synapse numbers and innervation of excitatory neurons, changes in release probability and the activity of interneurons (Kilb, 2012). Our finding that sIPSC frequency in LA principal neurons increases into early adulthood is consistent with, and extends previous functional data from, the BLA of younger animals (Vislay et al. 2013). Anatomical data reporting an overall increase in GABAergic fibres and presynaptic markers in the BLA into early adulthood (Brummelte et al. 2007; Avila et al. 2011) suggest that inhibitory synaptogenesis takes place. Thus, we favour the hypothesis, that a developmental increase in GABAergic synapse number as well as spontaneous spiking activity of interneurons (sIPSCs frequency surpassing mIPSC frequency from adolescence onward; authors’ unpublished results) contribute to increases in GABAergic drive onto principal neurons. While others report that sIPSC kinetics become significantly faster during postnatal development in hippocampus, cortex and amygdala (Hollrigel & Soltesz, 1997; Bosman et al. 2002; Ehrlich et al. 2013), our results do not show a developmental decay effect, which is most likely due to technical limitations of our recording conditions.
The increase in sensory‐evoked IPSCs relative to EPSCs in LA principal neurons resembles the development of evoked inhibitory postsynaptic responses in other brain areas (Banks et al. 2002; Morales et al. 2002). Since we did not detect differences in sIPSC amplitude (although this may have been below our detection threshold), this may reflect an increase in efficacy of recruiting feed‐forward interneurons and/or the number of their inhibitory synapses onto principal cells. Although assessing sensory‐evoked inhibition/excitation balance using electrical stimulation of the internal and external capsule has limitations, we believe that recruitment of intra‐amygdala inputs via antidromic activation of projection neurons, or a direct stimulation of interneurons is unlikely. Firstly, all recorded LA neurons showed delayed responses, and interconnectivity of projection neurons within basolateral amygdala appears to be low (2%, Abatis et al. 2014, FENS abstract). Secondly, we have good evidence that given the placement of our stimulation electrodes in the internal capsule and external capsule (dorsal to the tip of the LA), the evoked IPSCs were disynaptic and driven by glutamatergic inputs from stimulated fibre tracts. This could occur via activation of local interneurons within the LA (Ehrlich et al. 2009) or additionally via recruitment of GABAergic lateral or medial intercalated cells (Marowsky et al. 2005; Morozov et al. 2011; Asede et al. 2015). Together, maturation of local and input‐evoked inhibition may provide the tight inhibitory control that maintains low firing rates and high plasticity thresholds in LA principal neurons (Ehrlich et al. 2009). Furthermore, it may provide sufficient GABA in the neuropil for activity‐driven (priming) and tonic heterosynaptic inhibition of sensory inputs, limiting excitation onto excitatory neurons to enable inhibitory dominance later in development.
Postnatal emergence of synaptic modulation via presynaptic GABAB receptors
Our study is the first to delineate the functional development of presynaptic control via GABAB receptors in the amygdala. A general theme is that this modulation emerges during adolescence in the LA. Anatomical and physiological studies in other brain areas suggest that presynaptic GABAB receptors are expressed on glutamatergic and GABAergic terminals at birth and are functional in the first postnatal week (Fukuda et al. 1993; Gaiarsa et al. 1995; Fritschy et al. 1999; Lopez‐Bendito et al. 2002; Lopez‐Bendito et al. 2004; Hassfurth et al. 2010). In contrast to our findings, the only other study investigating postnatal development shows a reduction of EPSC modulation by presynaptic GABAB receptors in the brainstem (Hassfurth et al. 2010).
Sensory evoked IPSCs and control by GABAB receptors
Our results in adult mice are consistent with previous findings in rat BLA principal neurons, where GABAB receptors mediate paired‐pulse depression (PPD) of sensory evoked IPSCs (Szinyei et al. 2000). Extending this, we show that contribution of GABAB receptors to overall PPD of IPSCs is differentially regulated during development. While PPD at an inter‐stimulus interval of 300 ms suggests a metabotropic mechanism, we cannot completely exclude a contribution of basal release probability (Zucker & Regehr, 2002). Indeed, in the presence of GABAB blockers, intra‐BLA inhibitory synapses show pronounced PPD early in development, which was reduced from the third postnatal week (Ehrlich et al. 2013). This is consistent with our results that PPD in older animals is partially controlled by GABAB receptors. Based on previous studies, we can largely exclude a secondary effect via GABAB‐mediated depression of EPSCs onto interneurons (Szinyei et al. 2000; Pan et al. 2009). We cannot completely rule out a contribution of ionic plasticity, however, the reversal potential of IPSCs was largely determined by the chloride concentration in our recording solution. Overall, a use‐dependent depression of sensory‐evoked IPSCs, irrespective of the contributing mechanisms, may contribute to favour high pass filtering of excitatory inputs (Abbott & Regehr, 2004; George et al. 2011), and thus may be implemented by GABAB‐dependent and ‐independent mechanisms throughout development.
Two mechanisms could account for the observed variability in PPR: (1) variability in release probability at the two synapses mediating feed‐forward inhibition (i.e. glutamatergic input onto interneurons and interneuron input onto LA principal neuron); and (2) recruitment of a heterogeneous set of local interneurons and intercalated cells with different release probabilities. Nevertheless, the observed variability of PPR is in line with previous reports on feed‐forward inhibition or unitary IPSCs onto principal neurons in amygdala und hippocampus (Szinyei et al. 2000; Hefft & Jonas, 2005; Woodruff & Sah, 2007). Further work that selectively targets interneuron subtypes in the BLA is needed to decipher their roles in sensory feed‐forward inhibition and the precise mechanism of its modulation.
Heterosynaptic GABAB receptor activation at excitatory inputs
We find that presynaptic GABAB receptors are present on sensory inputs to LA principal neurons and can be activated by exogenous agonists as early as PND 8. However, in keeping with previous reports (Shaban et al. 2006; Pan et al. 2009; Lange et al. 2014), GABAB receptor‐dependent priming was only detected in adolescents and adults. It is likely that the developmental increase in sensory input‐evoked inhibition enhances the amount of released GABA that can spill over to modulate excitatory sensory terminals. Consistent with this, GABAB‐dependent priming critically depended on availability of GABA and local accumulation in the vicinity of the target neuron (Pan et al. 2009; Lange et al. 2014). Early on, priming‐induced depression appears to be mediated by other mechanisms, perhaps via modulatory receptors activated by glutamate or other released factors (Schmid & Fendt, 2006; Muly et al. 2007; Rau et al. 2014). A potential concern is specificity of our electrical stimulation. Because EPSC size in response to co‐stimulation of the two pathways was additive at all ages (data not shown), we can rule out overlapping recruitment of fibres, which may have led to depression due to synaptic fatigue.
GABAB receptor‐mediated tonic inhibition of excitatory inputs
Tonic inhibition resulting from ambient GABA has been mostly studied assessing activation of extrasynaptic GABAA receptors in various brain regions (Farrant & Nusser, 2005; Belelli et al. 2009) including the BLA (Marowsky et al. 2012). Tonic presynaptic control of IPSCs and EPSCs via GABAB receptors was detected in thalamus, hippocampus, cerebellum and medial prefrontal cortex (Emri et al. 1996; Jensen et al. 2003; Mapelli et al. 2009; Wang et al. 2010). Here, we provide the first evidence for a GABAB receptor‐mediated tonic inhibition of sensory inputs onto LA principal neurons. Specifically, blocking GABAB heteroreceptors increased sensory EPSCs with a correlated decrease in PPR in the absence of priming in adolescent and adult animals. Conversely, increasing GABA levels led to GABAB‐dependent depression. Furthermore, the lack of GABAB‐mediated depression in younger mice was overcome by increasing ambient GABA levels. Together, this strongly suggests a tonic activation of presynaptic GABAB receptors. This is likely to be enabled by a developmental increase in spontaneous inhibitory drive, which could provide sufficient ambient GABA levels. In a similar manner, ambient adenosine levels via presynaptic adenosine‐1 receptors can tonically depress EPSCs in the BA (Rau et al. 2014), suggesting that ambient levels of several neurotransmitters limit excitation in different parts of the amygdala.
Mechanisms of differential activation of GABAB heteroreceptors on sensory inputs
Several factors can contribute to the emerging activation of GABAB receptors during development. One is differential expression of GABAB receptors. Although no quantitative expression data are available for developing amygdala, our data suggest that GABAB receptors are functional on presynaptic terminals from infancy onward. Other key factors may be differential location of GABAB receptors on presynaptic terminals, the amount of GABA released by GABAergic synapses in the vicinity, and reuptake by GABA transporters. In adult mice differences in GABAB receptor distance from the active zone determines presynaptic inhibition (Pan et al. 2009). It is feasible that a similar mechanism contributes during development. Furthermore, reduced levels of GABA release resulting in lower GABA concentrations in the neuropil of infant and juvenile mice may limit GABA availability in the vicinity of sensory afferents. Lastly, extracellular GABA concentrations are regulated by GABA transporters, of which GAT‐1 is the most abundant in rodent brain (Radian et al. 1990). GAT‐1 is present in the mouse BLA at birth, and its expression levels are developmentally upregulated paralleling those of GABAergic markers (Avila et al. 2011). Thus, it seems unlikely that a decrease in GABA reuptake efficacy is an important contributing factor.
Conclusions and implications for fear learning and extinction during development
Studies in knock‐out animals have demonstrated that two factors are linked to behavioural expression of fear generalization to non‐reinforced stimuli (Shaban et al. 2006; Bergado‐Acosta et al. 2008; Lange et al. 2014). One is reduced availability of presynaptically released GABA in GAD‐65 knock‐out animals, resulting in impairment of GABA spillover to presynaptic GABAB receptors and a loss of the associative nature of LTP at cortical inputs. The other is expression of presynaptic GABAB receptors, as genetic deletion of the presynaptic GABAB1a subunit unmasks non‐associative LTP at cortico‐LA synapses. Furthermore, increasing thalamic input strength by exogenously increasing CREB levels in thalamic areas projecting to the LA also enhances fear generalization (Han et al. 2008). In young mice, the absence of physiologically activated tonic and evoked heterosynaptic inhibition at sensory inputs onto LA principal neurons could promote excitatory drive and lower LTP thresholds leading to a loss of the associative nature of cortical LTP. This is in agreement with the observation that expression of fear memory and generalization to non‐conditioned stimuli is enhanced in young animals (Hefner & Holmes, 2007; Ito et al. 2009). GABAergic mechanisms and availability of GABA also play a critical role in extinction (Ehrlich et al. 2009; Sangha et al. 2009; Herry et al. 2010). However, in juvenile rodents, extinction is independent of GABAA receptor activation, and a second inhibitory memory trace is not created (Kim & Richardson, 2007, 2010 b). Since in LA (our study) and BA (Ehrlich et al. 2013) the GABAergic system is immature in young mice, it is feasible that extinction mechanisms do not modulate or engage this system, but extinction is rather mediated by other factors or reversal of learning‐associated LTP in the BLA (Kim et al. 2007).
To understand the functional implications of immature inhibitory control at sensory inputs to the BLA, future studies will have to delineate exactly how these shape plasticity mechanisms, which plasticity mechanisms are engaged upon learning, and how these lead to the altered behavioural expression of acquired fear in young animals.
Additional information
Competing interests
The authors declare no competing interests, financial or otherwise.
Author contributions
All experiments were conducted in the lab of I.E. D.B. and I.E. designed the experiments. D.B. conducted and analysed all the experiments. D.B. prepared the figures I.E. and D.B. interpreted the data, wrote and revised the manuscript, and approved of the final version.
Funding
This work was supported by the Werner Reichardt Centre for Integrative Neuroscience (CIN) at the University of Tuebingen, an Excellence Initiative funded by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Excellence Initiative (EXC 307), and by funds from the Charitable Hertie Foundation.
Acknowledgements
We would like to thank Andrea Gall for technical assistance, and Alaa Sharif for performing some of the recordings shown in Fig. 7. We also thank Drs Douglas Asede, Kaspar Vogt and Holger Lerche for comments on an earlier version of the manuscript.
References
- Abatis M, Perin R, Niu R, Markram H & Stoop R. Characterizing connectivity and signal propagation in the lateral amygdala through local networks. 9th FENS Forum of Neuroscience, July 5‐9, Milano, Italy, Abstract 2794.
- Abbott LF & Regehr WG (2004). Synaptic computation. Nature 431, 796–803. [DOI] [PubMed] [Google Scholar]
- Asede D, Bosch D, Luthi A, Ferraguti F & Ehrlich I (2015). Sensory inputs to intercalated cells provide fear‐learning modulated inhibition to the basolateral amygdala. Neuron 86, 541–554. [DOI] [PubMed] [Google Scholar]
- Avila MA, Real MA & Guirado S (2011). Patterns of GABA and GABA transporter‐1 immunoreactivities in the developing and adult mouse brain amygdala. Brain Res 1388, 1–11. [DOI] [PubMed] [Google Scholar]
- Banks MI, Hardie JB & Pearce RA (2002). Development of GABAA receptor‐mediated inhibitory postsynaptic currents in hippocampus. J Neurophysiol 88, 3097–3107. [DOI] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC & Cope DW (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci 29, 12757–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdel B & Morys J (2000). Expression of calbindin‐D28k and parvalbumin during development of rat's basolateral amygdaloid complex. Int J Dev Neurosci 18, 501–513. [DOI] [PubMed] [Google Scholar]
- Berdel B, Morys J & Maciejewska B (1997). Neuronal changes in the basolateral complex during development of the amygdala of the rat. Int J Dev Neurosci 15, 755–765. [DOI] [PubMed] [Google Scholar]
- Bergado‐Acosta JR, Sangha S, Narayanan RT, Obata K, Pape HC & Stork O (2008). Critical role of the 65‐kDa isoform of glutamic acid decarboxylase in consolidation and generalization of Pavlovian fear memory. Learn Mem 15, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissiere S, Humeau Y & Luthi A (2003). Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci 6, 587–592. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Rosahl TW & Brussaard AB (2002). Neonatal development of the rat visual cortex: synaptic function of GABAA receptor α subunits. J Physiol 545, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwmeester H, Smits K & Van Ree JM (2002). Neonatal development of projections to the basolateral amygdala from prefrontal and thalamic structures in rat. J Comp Neurol 450, 241–255. [DOI] [PubMed] [Google Scholar]
- Brummelte S, Witte V & Teuchert‐Noodt G (2007). Postnatal development of GABA and calbindin cells and fibers in the prefrontal cortex and basolateral amygdala of gerbils (Meriones unguiculatus). Int J Dev Neurosci 25, 191–200. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR & Carter AG (2011). GABAB receptor modulation of synaptic function. Curr Opin Neurobiol 21, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila JC, Olmos L, Legaz I, Medina L, Guirado S & Real MA (2008). Dynamic patterns of colocalization of calbindin, parvalbumin and GABA in subpopulations of mouse basolateral amygdalar cells during development. J Chem Neuroanat 35, 67–76. [DOI] [PubMed] [Google Scholar]
- Ehrlich DE, Ryan SJ, Hazra R, Guo JD & Rainnie DG (2013). Postnatal maturation of GABAergic transmission in the rat basolateral amygdala. J Neurophysiol 110, 926–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich DE, Ryan SJ & Rainnie DG (2012). Postnatal development of electrophysiological properties of principal neurons in the rat basolateral amygdala. J Physiol 590, 4819–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C & Luthi A (2009). Amygdala inhibitory circuits and the control of fear memory. Neuron 62, 757–771. [DOI] [PubMed] [Google Scholar]
- Emri Z, Turner JP & Crunelli V (1996). Tonic activation of presynaptic GABAB receptors on thalamic sensory afferents. Neuroscience 72, 689–698. [DOI] [PubMed] [Google Scholar]
- Farrant M & Nusser Z (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci 6, 215–229. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Meskenaite V, Weinmann O, Honer M, Benke D & Mohler H (1999). GABAB‐receptor splice variants GB1a and GB1b in rat brain: developmental regulation, cellular distribution and extrasynaptic localization. Eur J Neurosci 11, 761–768. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Mody I & Prince DA (1993). Differential ontogenesis of presynaptic and postsynaptic GABAB inhibition in rat somatosensory cortex. J Neurophysiol 70, 448–452. [DOI] [PubMed] [Google Scholar]
- Gaiarsa JL, McLean H, Congar P, Leinekugel X, Khazipov R, Tseeb V & Ben‐Ari Y (1995). Postnatal maturation of gamma‐aminobutyric acidA and B‐mediated inhibition in the CA3 hippocampal region of the rat. J Neurobiol 26, 339–349. [DOI] [PubMed] [Google Scholar]
- Gambino F, Khelfaoui M, Poulain B, Bienvenu T, Chelly J & Humeau Y (2010). Synaptic maturation at cortical projections to the lateral amygdala in a mouse model of Rett syndrome. PLoS One 5, e11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AA, Lyons‐Warren AM, Ma X & Carlson BA (2011). A diversity of synaptic filters are created by temporal summation of excitation and inhibition. J Neurosci 31, 14721–14734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Luthi A & Herry C (2009). Perineuronal nets protect fear memories from erasure. Science 325, 1258–1261. [DOI] [PubMed] [Google Scholar]
- Han JH, Yiu AP, Cole CJ, Hsiang HL, Neve RL & Josselyn SA (2008). Increasing CREB in the auditory thalamus enhances memory and generalization of auditory conditioned fear. Learn Mem 15, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassfurth B, Grothe B & Koch U (2010). The mammalian interaural time difference detection circuit is differentially controlled by GABAB receptors during development. J Neurosci 30, 9715–9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaney CF, Bolton MM, Murtishaw AS, Sabbagh JJ, Magcalas CM & Kinney JW (2012). Baclofen administration alters fear extinction and GABAergic protein levels. Neurobiol Learn Mem 98, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S & Jonas P (2005). Asynchronous GABA release generates long‐lasting inhibition at a hippocampal interneuron‐principal neuron synapse. Nat Neurosci 8, 1319–1328. [DOI] [PubMed] [Google Scholar]
- Hefner K & Holmes A (2007). Ontogeny of fear‐, anxiety‐ and depression‐related behavior across adolescence in C57BL/6J mice. Behav Brain Res 176, 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA & Ressler KJ (2007). Training‐induced changes in the expression of GABAA‐associated genes in the amygdala after the acquisition and extinction of Pavlovian fear. Eur J Neurosci 26, 3631–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I & Luthi A (2010). Neuronal circuits of fear extinction. Eur J Neurosci 31, 599–612. [DOI] [PubMed] [Google Scholar]
- Hollrigel GS & Soltesz I (1997). Slow kinetics of miniature IPSCs during early postnatal development in granule cells of the dentate gyrus. J Neurosci 17, 5119–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Herry C, Kemp N, Shaban H, Fourcaudot E, Bissiere S & Luthi A (2005). Dendritic spine heterogeneity determines afferent‐specific Hebbian plasticity in the amygdala. Neuron 45, 119–131. [DOI] [PubMed] [Google Scholar]
- Ito W, Pan BX, Yang C, Thakur S & Morozov A (2009). Enhanced generalization of auditory conditioned fear in juvenile mice. Learn Mem 16, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA & Mody I (2003). GABA transporter‐1 (GAT1)‐deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol 90, 2690–2701. [DOI] [PubMed] [Google Scholar]
- Kilb W (2012). Development of the GABAergic system from birth to adolescence. Neuroscientist 18, 613–630. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, Kim H, Lee C, Sun W, Kim K, Shin KS & Choi S (2007). Amygdala depotentiation and fear extinction. Proc Natl Acad Sci USA 104, 20955–20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Hamlin AS & Richardson R (2009). Fear extinction across development: the involvement of the medial prefrontal cortex as assessed by temporary inactivation and immunohistochemistry. J Neurosci 29, 10802–10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH & Richardson R (2007). A developmental dissociation of context and GABA effects on extinguished fear in rats. Behav Neurosci 121, 131–139. [DOI] [PubMed] [Google Scholar]
- Kim JH & Richardson R (2010. a). Extinction in preweanling rats does not involve NMDA receptors. Neurobiol Learn Mem 94, 176–182. [DOI] [PubMed] [Google Scholar]
- Kim JH & Richardson R (2010. b). New findings on extinction of conditioned fear early in development: theoretical and clinical implications. Biol Psychiatry 67, 297–303. [DOI] [PubMed] [Google Scholar]
- Lange MD, Jungling K, Paulukat L, Vieler M, Gaburro S, Sosulina L, Blaesse P, Sreepathi HK, Ferraguti F & Pape HC (2014). Glutamic acid decarboxylase 65: a link between GABAergic synaptic plasticity in the lateral amygdala and conditioned fear generalization. Neuropsychopharmacology 39, 2211–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langton JM, Kim JH, Nicholas J & Richardson R (2007). The effect of the NMDA receptor antagonist MK‐801 on the acquisition and extinction of learned fear in the developing rat. Learn Mem 14, 665–668. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23, 155–184. [DOI] [PubMed] [Google Scholar]
- Li XF, Armony JL & LeDoux JE (1996). GABAA and GABAB receptors differentially regulate synaptic transmission in the auditory thalamo‐amygdala pathway: an in vivo microiontophoretic study and a model. Synapse 24, 115–124. [DOI] [PubMed] [Google Scholar]
- Lopez‐Bendito G, Shigemoto R, Kulik A, Paulsen O, Fairen A & Lujan R (2002). Expression and distribution of metabotropic GABA receptor subtypes GABABR1 and GABABR2 during rat neocortical development. Eur J Neurosci 15, 1766–1778. [DOI] [PubMed] [Google Scholar]
- Lopez‐Bendito G, Shigemoto R, Kulik A, Vida I, Fairen A & Lujan R (2004). Distribution of metabotropic GABA receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus during prenatal and postnatal development. Hippocampus 14, 836–848. [DOI] [PubMed] [Google Scholar]
- Mahanty NK & Sah P (1999). Excitatory synaptic inputs to pyramidal neurons of the lateral amygdala. Eur J Neurosci 11, 1217–1222. [DOI] [PubMed] [Google Scholar]
- Mapelli L, Rossi P, Nieus T & D'Angelo E (2009). Tonic activation of GABAB receptors reduces release probability at inhibitory connections in the cerebellar glomerulus. J Neurophysiol 101, 3089–3099. [DOI] [PubMed] [Google Scholar]
- Maren S (2001). Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24, 897–931. [DOI] [PubMed] [Google Scholar]
- Maren S (2005). Synaptic mechanisms of associative memory in the amygdala. Neuron 47, 783–786. [DOI] [PubMed] [Google Scholar]
- Marowsky A, Rudolph U, Fritschy JM & Arand M (2012). Tonic inhibition in principal cells of the amygdala: a central role for α3 subunit‐containing GABAA receptors. J Neurosci 32, 8611–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K & Vogt KE (2005). A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron 48, 1025–1037. [DOI] [PubMed] [Google Scholar]
- McKernan MG & Shinnick‐Gallagher P (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611. [DOI] [PubMed] [Google Scholar]
- Morales B, Choi SY & Kirkwood A (2002). Dark rearing alters the development of GABAergic transmission in visual cortex. J Neurosci 22, 8084–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov A, Sukato D & Ito W (2011). Selective suppression of plasticity in amygdala inputs from temporal association cortex by the external capsule. J Neurosci 31, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morys J, Berdel B, Kowianski P & Dziewiatkowski J (1998). The pattern of synaptophysin changes during the maturation of the amygdaloid body and hippocampal hilus in the rat. Folia Neuropathol 36, 15–23. [PubMed] [Google Scholar]
- Muly EC, Mania I, Guo JD & Rainnie DG (2007). Group II metabotropic glutamate receptors in anxiety circuitry: correspondence of physiological response and subcellular distribution. J Comp Neurol 505, 682–700. [DOI] [PubMed] [Google Scholar]
- Pan BX, Dong Y, Ito W, Yanagawa Y, Shigemoto R & Morozov A (2009). Selective gating of glutamatergic inputs to excitatory neurons of amygdala by presynaptic GABAb receptor. Neuron 61, 917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC & Pare D (2010). Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90, 419–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radian R, Ottersen OP, Storm‐Mathisen J, Castel M & Kanner BI (1990). Immunocytochemical localization of the GABA transporter in rat brain. J Neurosci 10, 1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineki C, Shionoya K, Sander K & Sullivan RM (2009). Ontogeny of odor‐LiCl vs. odor‐shock learning: similar behaviors but divergent ages of functional amygdala emergence. Learn Mem 16, 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau AR, Ariwodola OJ & Weiner JL (2014). Presynaptic adenosine A1 receptors modulate excitatory transmission in the rat basolateral amygdala. Neuropharmacology 77, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV & LeDoux JE (1997). Fear conditioning induces associative long‐term potentiation in the amygdala. Nature 390, 604–607. [DOI] [PubMed] [Google Scholar]
- Romanski LM, Clugnet MC, Bordi F & LeDoux JE (1993). Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behav Neurosci 107, 444–450. [DOI] [PubMed] [Google Scholar]
- Rudy JW (1993). Contextual conditioning and auditory cue conditioning dissociate during development. Behav Neurosci 107, 887–891. [DOI] [PubMed] [Google Scholar]
- Ryan SJ, Ehrlich DE & Rainnie DG (2015). Morphology and dendritic maturation of developing principal neurons in the rat basolateral amygdala. Brain Struct Funct DOI: 10.1007/s00429-014-0939-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M & Power J (2003). The amygdaloid complex: anatomy and physiology. Physiol Rev 83, 803–834. [DOI] [PubMed] [Google Scholar]
- Sangha S, Narayanan RT, Bergado‐Acosta JR, Stork O, Seidenbecher T & Pape HC (2009). Deficiency of the 65 kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear memory. J Neurosci 29, 15713–15720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S & Fendt M (2006). Effects of the mGluR8 agonist (S)‐3,4‐DCPG in the lateral amygdala on acquisition/expression of fear‐potentiated startle, synaptic transmission, and plasticity. Neuropharmacology 50, 154–164. [DOI] [PubMed] [Google Scholar]
- Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, Barbieri S, van der Putten H, Kaupmann K, Bettler B & Luthi A (2006). Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat Neurosci 9, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Shin RM, Tsvetkov E & Bolshakov VY (2006). Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron 52, 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumyatsky GP, Tsvetkov E, Malleret G, Vronskaya S, Hatton M, Hampton L, Battey JF, Dulac C, Kandel ER & Bolshakov VY (2002). Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 111, 905–918. [DOI] [PubMed] [Google Scholar]
- Stork O, Ji FY & Obata K (2002). Reduction of extracellular GABA in the mouse amygdala during and following confrontation with a conditioned fear stimulus. Neurosci Lett 327, 138–142. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Landers M, Yeaman B & Wilson DA (2000). Good memories of bad events in infancy. Nature 407, 38–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szinyei C, Heinbockel T, Montagne J & Pape HC (2000). Putative cortical and thalamic inputs elicit convergent excitation in a population of GABAergic interneurons of the lateral amygdala. J Neurosci 20, 8909–8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E & Bolshakov VY (2007). Norepinephrine enables the induction of associative long‐term potentiation at thalamo‐amygdala synapses. Proc Natl Acad Sci USA 104, 14146–14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vislay RL, Martin BS, Olmos‐Serrano JL, Kratovac S, Nelson DL, Corbin JG & Huntsman MM (2013). Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J Neurosci 33, 7548–7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Neubauer FB, Luscher HR & Thurley K (2010). GABAB receptor‐dependent modulation of network activity in the rat prefrontal cortex in vitro. Eur J Neurosci 31, 1582–1594. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Ikegaya Y, Saito H & Abe K (1995). Roles of GABAA, NMDA and muscarinic receptors in induction of long‐term potentiation in the medial and lateral amygdala in vitro. Neurosci Res 21, 317–322. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG & LeDoux JE (1999). Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol 81, 930–934. [DOI] [PubMed] [Google Scholar]
- Woodruff AR & Sah P (2007). Networks of parvalbumin‐positive interneurons in the basolateral amygdala. J Neurosci 27, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Saitow F, Satake S, Kiyohara T & Konishi S (1999). GABAB receptor‐mediated presynaptic inhibition of glutamatergic and GABAergic transmission in the basolateral amygdala. Neuropharmacology 38, 1743–1753. [DOI] [PubMed] [Google Scholar]
- Yap CS & Richardson R (2007). Extinction in the developing rat: an examination of renewal effects. Dev Psychobiol 49, 565–575. [DOI] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG (2002). Short‐term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]