

Abstract
Background and Purpose
Status epilepticus is increasingly associated with cardiac injury in both clinical and animal studies. The current study examined ECG activity for up to 48 h following kainic acid (KA) seizure induction and compared the potential of atenolol and clonidine to attenuate this cardiac pathology.
Experimental Approach
Sprague-Dawley rats (male, 300–350 g) were implanted with ECG and electrocorticogram electrodes to allow simultaneous telemetric recordings of cardiac and cortical responses during and after KA-induced seizures. Animals were randomized into saline controls, and saline vehicle-, clonidine- or atenolol-pretreated KA groups.
Key Results
KA administration in the saline-pretreated group produced an immediate bradycardic response (maximal decrease of 28 ± 6%), coinciding with low-level seizure activity. As high-level seizure behaviours and EEG spiking increased, tachycardia also developed, with a maximum heart rate increase of 38 ± 7% coinciding with QTc prolongation and T wave elevation. Both clonidine and atenolol pretreatment attenuated seizure activity and reduced KA-induced changes in heart rate, QTc interval and T wave amplitude observed during both bradycardic and tachycardic phases in saline-pretreated KA animals. Clonidine, however, failed to reduce the power of EEG frequencies. Atenolol and to a lesser extent clonidine attenuated the cardiac hypercontraction band necrosis, inflammatory infiltration, and oedema at 48 h after KA, relative to the saline-KA group.
Conclusions and Implications
Severe seizure activity in this model was clearly associated with altered ECG activity and cardiac pathology. We suggest that modulation of sympathetic activity by atenolol provides a promising cardioprotective approach in status epilepticus.
Tables of Links
| TARGETS |
|---|
| GPCRs |
| α2 adrenoceptors |
| β1 adrenoceptors |
| LIGANDS |
|---|
| Atenolol |
| Clonidine |
| Kainic acid |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Seizure activity has been increasingly associated with changes in autonomic function, particularly on the cardiorespiratory system, leading to cardiac arrhythmias, apnoea and hypoxia (Jansen and Lagae, 2010). Symptoms of increased autonomic activation frequently occur during seizures, with tachycardia [>100 beats per minute (b.p.m.)] reported in the majority of clinical seizure presentations (Provini et al., 1999; Opherk et al., 2002; Surges et al., 2009). Electrocardiographic (ECG) abnormalities have been recorded in 35% of seizures (Nei et al., 2000; Surges et al., 2009), with potentially fatal changes occurring in 6–13% of seizures (Opherk et al., 2002; Devinsky, 2004). Such features as tachycardia and QTc prolongation are well-known risk factors of sudden cardiac death (Zipes and Wellens, 1998).
Status epilepticus (SE) is defined as a period of 5 minutes or more of continuous clinical and/or electrographic seizure or recurrent seizure activity without recovery (Walker, 2005; Brophy et al., 2012). Patients with SE have a higher mortality rate (21–33%) than epileptics, with death occurring within 30 days of the initial convulsant activity (Logroscino et al., 2005). Death often occurs in the absence of convulsant activity and is believed to be due to an imbalance in autonomic function, which results in altered cardiac control and electrical instability, increasing the risk of lethal cardiac arrhythmias (Metcalf et al., 2009b; Nguyen-Michel et al., 2014).
Death following SE may result as a consequence of uncontrolled tachycardia leading to malignant ventricular tachyarrhythmias (Nei et al., 2000; Jansen and Lagae, 2010). Tachycardia is most commonly reported following generalized tonic-clonic convulsions (Terndrup et al., 1994; Metcalf et al., 2009a,b,; Jansen and Lagae, 2010). Tachycardia reduces coronary blood flow, increases cardiac oxygen demand and predisposes the left ventricle to ischaemic damage, arrhythmogenesis and deterioration of cardiac function. Ictal bradycardia (<40 b.p.m.) is rarely reported, occurring in fewer than 2% of seizures (Nei et al., 2000; Leutmezer et al., 2003) and is more prevalent in seizures of temporal or left sided origin (Locatelli et al., 1999; Devinsky, 2004). Bradycardia leading to asystole lasting more than 10 s may impose severe systemic and cerebral hypoxemia (Surges et al., 2009; Nguyen-Michel et al., 2014) and therefore should not be dismissed as a contributor to seizure-related sudden cardiac death.
Systemic administration of high doses (10–12 mg·kg−1) of kainic acid (KA), an analogue of glutamate, produces strong and sustained seizure activity (Sakamoto et al., 2008; Read et al., 2014). Systemic administration of KA has been widely used as an animal model of temporal lobe epilepsy (Ben-Ari and Cossart, 2000; Sarkisian, 2001; Loscher, 2011). Seizures are induced in the hippocampus, corresponding to focalized low-level seizure behaviours such as tremors and salivation. As generalized seizures develop, animals display escalating behavioural responses such as wet dog shakes, forelimb clonus and eventually tonic-clonic convulsions (Dernovsek and Sket, 1998; Sawant et al., 2010). We have previously demonstrated that KA administration in a rodent model results in significant cortical and cardiac electrographic changes, in conjunction with the development of left ventricular perivascular and interstitial fibrosis, and myocyte vacuolization (Read et al., 2014). Earlier work with another similar seizurogenic agent, domoic acid, has also shown that seizure induction results in residual cardio haemodynamic and histological damage indicative of cardiomyopathy at 7 and 14 days after seizure (Vranyac-Tramoundanas et al., 2011).
Current clinical interventions in the treatment of seizure generally fail to address the cardiac repercussions of seizure. Furthermore, clinical evidence gained using tissue Doppler imaging has shown that the use of current anti-epileptic medications such as carbamazepine and valproate may be associated with subclinical levels of left ventricular injury (Kibar et al., 2014). Recent animal-based studies (Bealer et al., 2010; Little and Bealer, 2012; Read et al., 2014) have examined the protective effects of sympatholytic agents in cardiac responses. Atenolol, a hydrophilic β1 adrenoceptor antagonist with no reported central activity, acts directly on the heart to block the effect of sympathetic stimulation on β1 adrenoceptors (de Lange et al., 1994; Smith and Teitler, 1999). Clonidine, an agonist of pre- and post-synaptic α2 adrenoceptors and imidazoline receptors, has been shown to reduce noradrenaline and adrenaline release and to increase vagal activity, thereby simultaneously producing sympatholytic and parasympathomimetic effects (Matsukawa et al., 1995; Azevedo et al., 1999; Toader et al., 2008). As previously reported by our group, chronic administration of clonidine reduces seizure behaviours and provides cardioprotection (Read et al., 2014). The potential prophylactic effects of atenolol or clonidine on seizure activity and cardiac dysfunction were directly compared in this current study.
Methods
Animals
All animal care and experimental procedures complied with the ‘Use of Laboratory Animals’ (NIH Publication No. 85-23, 1996) and were approved by the University of Otago's Committee on Ethics in the Care and Use of Laboratory Animals. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 32 animals were used in the experiments described here. Male Sprague-Dawley rats (300–320 g) were obtained from the University of Otago Animal Resource Unit. The animals were housed on a 12 h light/dark cycle at 22°C with food and water ad libitum at least 1 week before the surgical procedure.
Experimental protocol
Combined ECG and EEG recordings were obtained using implantable radiotelemeters (Telemetry Research, Auckland, New Zealand) in conscious animals to avoid any confounding effects of anaesthetics on cardiovascular responses (Williams et al., 2006). At 7 days following telemetric implantation, animals were randomized into control-saline (no seizure induction), and saline-, atenolol- or clonidine- pretreated KA groups (n = 8 per group). Saline, atenolol (5 mg·kg−1, s.c. daily; Harris and Aston-Jones, 1993) or clonidine (0.1 mg·kg−1, twice daily, s.c.; Zhang and Cheng, 2000) calculated using a human to rat dose translation equation (Reagan-Shaw et al., 2008) were administered three days prior to KA (10 mg·kg−1, s.c.) challenge and for the duration of the experiment. Observers were not aware of either the interventions or the treatments applied.
Surgical implantation of telemetric transmitters
Animals were administered amphoprim (0.2 mL, 60 mg·mL−1, b.i.d.) and carprofen (5 mg·kg−1, sc) before surgery and once every 24 h post-operatively, for 3 days. Anaesthesia was induced with ketamine (75 mg·kg−1, s.c.), domitor (medetomidine hydrochloride; 0.5 mg·kg−1, s.c.) and atropine (0.05 mg·kg−1, s.c.). Body temperature was maintained at 37°C throughout the surgery. Transmitter implantation and electrode positioning procedures were performed as previously described (Sawant et al., 2010; Read et al., 2014). Animals were housed individually after surgery and left to recover for 7 days before seizure induction.
Seizure induction and telemetric/behavioural recordings
Rat behaviours were observed in a custom-made mirrored Perspex chamber (1 m × 0.5 m × 0.5 m) in which each rat was allowed to acclimatize for 30 min before study initiation. EEG and ECG were sampled at 2000 Hz, with receiver filters set to 0.1 Hz high pass and 1000 Hz low pass using a Powerlab 2/25 signal conditioner and LabChart v.6 Pro software (AD Instruments, Sydney, Australia). EEG, ECG and behavioural data were simultaneously recorded during a 20 min baseline period and for 3 h after saline or KA administration, with periodic recordings taken at 24 and 48 h. Seizures were induced by a single injection of KA (10 mg·kg−1, s.c., maximum volume 0.35 mL). The control animals were treated with an equivalent volume of saline s.c. Following administration of the seizurogenic agent or vehicle, rats were immediately returned to the chamber for behavioural observation. Behaviours were recorded every 15 s for 3 h with discrete changes in behavioural state additionally reported as they occurred. Behaviours were recorded by trained observers unaware of the treatment, using a 5-point scale as previously described (Sawant et al., 2010). Normal behaviours were defined as level 0, discomfort behaviours (level 1), seizure behaviours confined to the head (level 2), seizure behaviours associated with limbs or trunk such as wet dog shakes (WDS; level 3), generalized seizure behaviours (level 4) and clonic-tonic convulsion (level 5). The cumulative behavioural score was determined as the sum of the maximum score every minute over the 3 h recording period. The total number of WDS and level 4 behaviours were quantified over the 180 min recording period. Behavioural data was also expressed as the mean behavioural score every 10 min.
ECG and EEG analysis
ECG data was analysed using the LabChart6 Pro ECG Analysis module in order to assess heart rate (HR), QT intervals and T wave amplitudes. The end of the T wave was determined when the wave returned to the isoelectric line. Data were analysed every 2 min in 1 min blocks over the 3 h observation period. HR increased inversely as the RR interval decreased, resulting in a reduced QT interval. The converse was also true as HR decreased. The corrected QT (QTc) was calculated in order to adjust for rate by applying the Mitchell algorithm to the QT interval recordings, where QTc = QT0/(RR/100)1/2 (Mitchell et al., 1998). This algorithm is designed to correct for the higher HR and altered QRS-T wave morphology in rodents. An isoelectric interval was still present between the end of the T wave and initiation of the next P wave in these animals even during periods of tachycardia. EEG data were analysed using fast Fourier transformation (FFT) to quantify the frequency bands: δ (1.25–4.50 Hz), θ (4.75–6.75 Hz), α (7.00–12.50 Hz), β (12.75–35 Hz) and γ (35.5–100 Hz) over 10 min bins. These sampling periods were analysed by dividing them into half overlapping half-second epochs, using a weighted Cosine bell window (Lab Chart Pro, Spectral Analysis). Power spectral density (PSD) for each period was calculated as the sum of all epochs. Baseline PSD for each animal was determined following acclimatization and all PSD values were expressed as a percentage of baseline. High amplitude EEG spiking was defined as sharp electrographic events greater than three times the average baseline voltage fluctuations and quantified using LabChart Spike Analysis over 10 min bins. Movement artefacts were identified on the ECG trace and eliminated from the EEG analysis.
Morphological characterization of myocardial injury
Forty-eight hours following KA or saline administration, animals were anaesthetized with halothane and the heart excised. Hearts were arrested in diastole by flushing with 20 mL of ice-cold (4°C) 0.9% saline containing 20 mM of KCl. The tissue was perfusion-fixed (73.6 mmHg pressure) and maintained in 10% neutral buffered formalin (4 h). Apical ventricular tissue blocks (6 mm depth from apex) were paraffin-embedded and 5 μm thick tissue sections prepared for staining with Martius scarlet blue. Qualitative histological evaluations were randomly performed in a double-blind manner, on 10 individual digital images from each block, using an Axioplan-2 microscope and recorded with the Axiovision v.3.1 image analysis system (Carl Zeiss Ltd., Oberkochen, Germany). Semi-quantitative analysis was conducted using trained observers, unaware of the treatments, and Adobe Photoshop CS5 software in which the ratio of collagen stained (blue) or oedema (white) pixels were calculated against background in each cardiac section. Each section was also examined for evidence of hypercontracture band necrosis and myocyte vacuolization.
Data analysis
Throughout this manuscript, data are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using Prism™ v.5 (GraphPad Software Inc, San Diego, CA, USA). Behavioural data were analysed using a Kruskal–Wallis test with Bonferroni post hoc analysis. ECG variables (HR, QTc interval and T wave amplitude) were analysed using a two-way repeated measures anova with Bonferroni post hoc analysis. Statistical analyses of quantified cardiac histological staining densities were determined using a one-way anova with Dunnett post hoc analysis. The presence of contraction band necrosis and myocyte vacuolization was analysed using a Kruskal–Wallis test with Bonferroni post hoc analysis. Statistical significance was determined as P < 0.05.
Materials
All reagents were purchased from BDH (Palmerston North, New Zealand) and Sigma-Aldrich (Auckland, New Zealand) with the exception of KA (Tocris, Bristol, UK) and clonidine (Sapphire Bioscience PTY, New South Wales, Australia). Clonidine, atenolol and KA were freshly dissolved in sterile saline. Prescription animal remedies were obtained from the University of Otago Animal Welfare Office.
Results
Seizure activity
No difference in behavioural activity was exhibited between the treatment groups during the baseline recording period, where animals displayed exploring, grooming and resting behaviours (cumulative behavioural score of 6 ± 2, Figure 1A). Administration of KA significantly increased cumulative behavioural scores to 463 ± 30 (Figure 1A) recorded over the 180 min period. WDS were the most commonly observed seizure behaviour, occurring 164 ± 21 times during the 180 min observation period (Figure 1B) with level 4 behaviour significantly raised and maintained in these saline-KA animals (Figures 1C and 2A).
Figure 1.
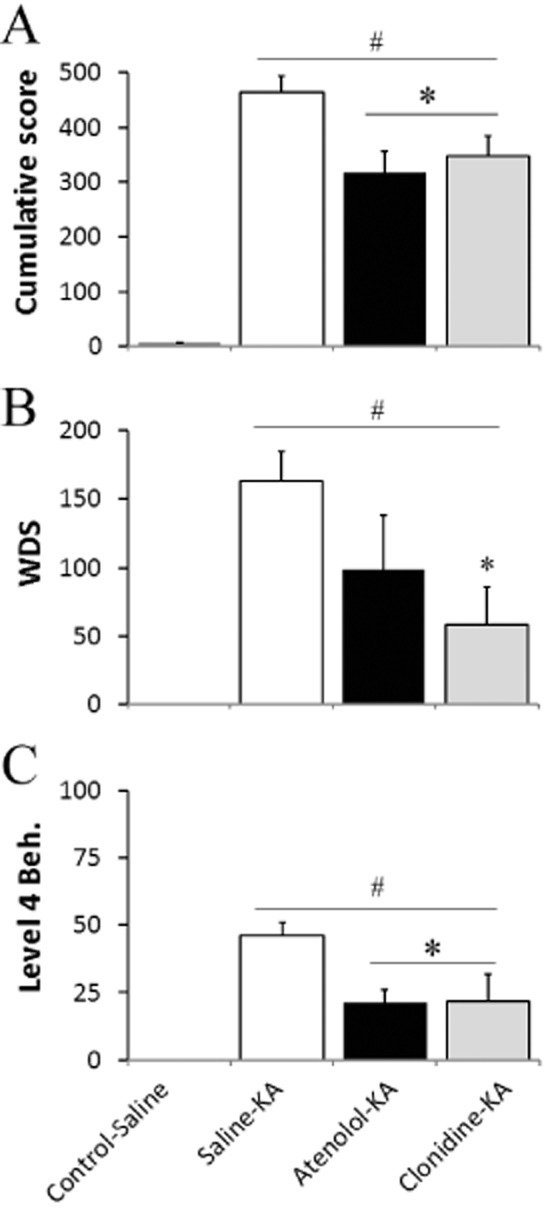
Behavioural changes during KA-induced seizure activity over 180 min. Cumulative score (A), number of wet dog shake (WDS; B), and level 4 behaviours (Beh.; C) following saline or KA (10 mg·kg−1). Values represent mean ± SEM; n = 8 per group. #P < 0.05 compared with Control-Saline. *P < 0.05 compared with Saline-KA; Kruskal–Wallis, with Bonferroni test.
Figure 2.

EEG and behavioural changes following seizure induction with KA over 180 min. (A) Mean behavioural score (mean ± SEM; n = 8) every 10 min. (B) Number of high amplitude EEG spikes (mean ± SEM) in a 10 min bin. (C) Power spectral density (PSD): Histograms represent normalized PSD (mean ± SEM; n = 8) for each 10 min bin across the δ (1.25–4.5 Hz), θ (4.75–6.75 Hz), α (7.0–12.5 Hz), β (12.75–35.0 Hz) and γ (35.0–100 Hz) frequency bands. #P < 0.05 compared with Control-Saline. *P < 0.05 compared with Saline-KA; Kruskal–Wallis, with Bonferroni test.
Significant increases in high amplitude spiking began 100 min after KA and remained elevated for the remainder of the study (maximum increase of 974 ± 153 at 180 min, Figure 2B). During the first 30 min following the KA administration, there was a significant increase in PSD in the θ (4.75–6.75 Hz) frequency band (Figure 2C) corresponding to low-level seizure behaviours such as blinking, mastication and head bobbing. As seizure severity increased, significant increases in power in the δ to β frequency band ranges were also observed (P < 0.05 compared with control-KA; Figure 2C). Behaviour scores were similarly increased over the 30–180 min after KA (P < 0.05 compared with control-saline, Figure 2C). All saline-KA animals developed level 4 behaviours (myoclonic jerks and foaming (Figures 2A and 3) while 5 of the 8 rats progressed to level 5 (tonic-clonic) seizures. Seizure behaviours and EEG activity had returned to baseline by the 24 h recording periods.
Figure 3.

Time course of ECG, EEG and behavioural changes during seizure induction. (A–D) Representative traces showing combined HR (b.p.m.; black line trace), EEG activity (mV; grey spectral trace) and behavioural score responses in individual Control-Saline (A), Saline-KA (B), Atenolol-KA (C) and Clonidine-KA (D) rats following saline vehicle or KA (dashed line) administration.
Atenolol pretreatment reduced the development of seizure progression 120–180 min after KA and decreased the cumulative score by 32% compared with the saline-KA group (P < 0.05, Figures 3). Pretreatment with atenolol was effective at reducing the development of level 4 (Figure 1C) and level 5 behaviours were only noted in 2/8 animals. The efficacy of atenolol in reducing behavioural seizure severity was supported by the significant reductions in high amplitude spiking reaching a maximum increase of only 121 ± 82 (Figure 2B) and attenuation of EEG power in the δ to β frequency bands (Figure 2C) in atenolol-treated animals relative to the saline-KA group.
Pretreatment with clonidine reduced seizure severity 150–180 min after KA and WDS development by 64% (compared with saline-KA, Figures 1B and 2A). Clonidine also reduced the development of level 4 behaviours to 22 ± 10 during the 180 min recording period (Figures 1C and 3), with only 1/8 animals exhibiting level 5 behaviours. In addition, clonidine pretreatment significantly decreased high amplitude electrographic spiking by <34% at 160–180 min after KA administration compared with the saline-KA group (P < 0.05, Figure 2B). Surprisingly, however, the clonidine-KA group exhibited significant increases in FFT power across all frequency bands, with the greatest changes recorded in the β and γ band, 80–180 min after KA (Figure 2C).
Cardiac function
The mean baseline HR (353 ± 16 b.p.m.) for the control-saline group did not significantly change during the study (Figures 3A and 4A). KA administration caused an early and pronounced reduction in HR by 28% (to 273 ± 33 b.p.m.) at 8–18 min after KA (P < 0.05 compared with control-KA). As seizure activity increased, so did HR (Figure 3A and 3B) with a maximum HR of 483 ± 12 b.p.m. recorded at 74 min (P < 0.05 compared with control-saline, Figure 4A). This HR increase coincided with prolongation of the QTc interval at the 46–180 min and at the 24 h recording periods (Figure 4B), and a progressive increase in the T wave amplitude (maximum increase of 232 ± 14%, Figure 4C).
Figure 4.
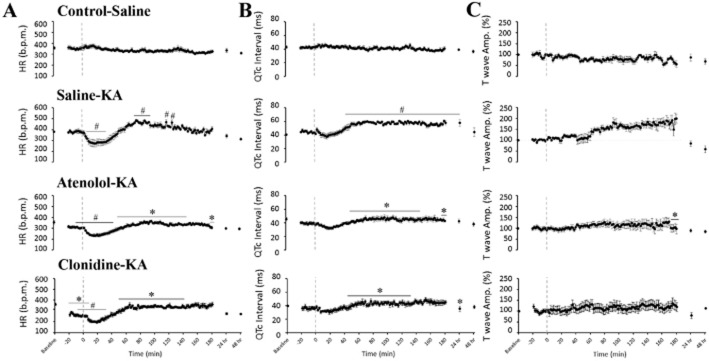
Effects of atenolol and clonidine pretreatment on ECG changes. The figure shows the individual group responses (mean ± SEM; n = 8 per group) to treatment in (A) HR (b.p.m.); (B) corrected QT interval in seconds (QTc = QT/(RR/100)1/2); and (C) T wave elevation (normalized to baseline) over a 48 h recording period following seizure induction with KA (indicated using a dashed line). Significance is expressed against the corresponding time point; #P < 0.05 compared with Control-Saline, *P < 0.05 compared with Saline-KA; two-way repeated measures anova with Bonferroni test
Examination of the apical-midplane sections of left ventricular myocardium showed evidence of sub-endocardial damage at 48 h after KA-induced seizure in the saline pretreated animals (Figure 5B, C, D). There was significant evidence of ventricular oedema (31 ± 5%, Figure 5B and 5G) and myocyte vacuolization with nuclear displacement, indicative of early reversible ischaemic damage (Table 1). Multifocal lesions evidenced by hypercontraction band necrosis and muscle fibre degeneration were also found in 62% of micrographs assessed. Inflammatory cell infiltration was observed in the sub-endocardial interfibril spaces of all saline-KA animals. Clear evidence of dense collagen deposition (fibrosis) encapsulating necrotic cells was also observed disseminated along the sub-endocardium with diminished levels in the endocardium of apical sections isolated from saline-KA animals (0.9 ± 0.3% Figure 5C, D and H). A clear correlation was observed between the extent of fibrosis and HR elevation during the initial 180 min following KA (R2 = 0.8164, data not shown).
Figure 5.

Representative micrographs of left ventricular subendocardium collected at 48 h following (A) saline administration to control animals showing normal representative myocardium; (B–F) Effects of KA administration showing: (B) myocyte vacuolization (asterisks indicate intracellular vacuoles displacing nuclei) and oedema; (C) Coagulative myocytolysis illustrated by hypercontraction band necrosis associated with fibre derangement (arrowheads) and fibrosis; (D) Inflammatory necrosis encapsulated by collagen deposition (blue fibres) indicative of early restorative fibrosis. Hearts from atenolol (E) and clonidine (F) pretreated animals showing preserved normal cardiac morphology. The percentage of tissue positively stained for oedema (G) and fibrosis (H) recorded in 10 sections taken 6 mm from the apex. *P < 0.05 compared with Control-Saline, #P < 0.05 compared with Saline-KA; one-way anova with Dunnett test
Table 1.
Relative extent of morphological features in left ventricular subendocardial regions at 48 h post-seizure onset
| Treatment pathological feature | Control-saline | Saline-KA | Atenolol-KA | Clonidine-KA |
|---|---|---|---|---|
| Hypercontracture band necrosis (positive fields/10) | 0.0 ± 0.0 | 6.2 ± 1.1# | 0.5 ± 0.3* | 2.6 ± 0.4* |
| Myocyte vacuolization (positive fields/10) | 0.2 ± 0.2 | 7.7 ± 1.1# | 1.5 ± 0.7*† | 4.3 ± 0.3* |
Quantification of each feature was conducted in a double-blind manner on 10 random fields from each ventricular section. Contraction band necrosis in each field was positively graded on the presence of ≥3 myocardial cells containing contraction bands. Reversible nuclear vacuolization was graded on the presence of ≥2 vacuolized nucleus in each field.
P < 0.05 compared with control-saline.
P < 0.05 compared with saline-KA;
P < 0.05 compared with clonidine-KA; Kruskal–Wallis, with Bonferroni test.
Pretreatment with atenolol decreased HR to 308 ± 10 b.p.m. (10%) during the baseline period (Figure 4A). Atenolol administration reduced the extent of HR changes (HR ranged from 233–362 b.p.m. after KA) and prevented the development of tachycardia seen in the saline-KA group (P < 0.05 at 50–140 min compared with saline-KA). Atenolol was successful in preventing QTc prolongation induced by KA seizures, with a maximum increase reduced to 17% (compared with 39% in the saline-KA group, P < 0.05, Figure 4B). Pretreatment with atenolol reduced T wave elevation at the 132–180 min compared with the saline-KA group (P < 0.05, Figure 4C). Atenolol treatment also preserved normal cardiac morphology as seen 48 h after KA (Figure 5E). The appearance of the myocardial basement membrane remained intact in this group with no significant evidence of oedema or fibrosis (Figure 5G and 5H). Atenolol treatment significantly reduced the development of hypercontracture band necrosis and ischaemic injury myocyte vacuolization by 94 and 74%, respectively (Table 1).
Clonidine pretreatment reduced baseline HR from 335 ± 7 to 267 ± 13 b.p.m. (P < 0.05, Figure 4A). Subsequent administration of KA in this clonidine group resulted in a further decrease in HR to 197 ± 12 b.p.m. at 18 min. Clonidine pretreatment significantly prevented the tachycardia recorded in the saline-KA group during high-level seizure behaviours (50–140 min after KA, P < 0.05 Figures 3 and 4). Clonidine treatment also significantly prevented QTc prolongation and reduced the extent of T wave elevation 148 to 180 min after KA administration (P < 0.05 compared with saline-KA), with maximum increases of 30–40% above baseline. Pretreatment with clonidine significantly reduced the extent of cardiac damage compared with the saline pretreated group. Clonidine prevented the development of early fibrotic deposition in the left ventricular myocardium by 83% compared with the saline-KA animals (P < 0.05; Figure 5H). Oedema was still prevalent in the clonidine-KA animals (24% compared with 7.5% in the control-saline animals); however, clonidine reduced the extent of hypercontracture band necrosis and myocyte vacuolization (58 and 44%, respectively) compared with the saline pretreated animals (Table 1).
Discussion
This study demonstrates that seizure activity is clearly associated with the development of ECG changes and injury. Pretreatment with atenolol reduced seizure severity and ECG changes and preserved cardiac morphology in the KA-induced seizure model. While clonidine also significantly reduced high-level seizure behaviours, no effect was seen with this sympatholytic on EEG activity. Clonidine did however reduce tachycardia, QTc prolongation and T wave elevation and attenuated the cardiac damage observed in the saline-KA animals.
In this study, bolus administration of KA resulted in an immediate period of hypoactivity in the animals coinciding with bradycardia before progressing to head tremors and WDS. This finding is similar to previous studies where animals exhibited staring behaviours and tremors before progressing to limbic seizures (Lothman and Collins, 1981; Read et al., 2014). During the 40 min following KA administration, EEG activity in the θ and α frequency bands increased, coinciding with the development of mild seizure behavioural activity. Activation of these lower frequencies has been previously associated with neurological diseases, specifically temporal lobe epilepsy (Zaveri et al., 2001). As in previous studies (Read et al., 2014), these low-level seizure behaviours coincided with a bradycardic period where HR dropped by 28%. Similar effects have been reported by Ferrari et al. (2008) where HR decreased by 22% within 30 min of KA administration. In that study, the bradycardia was not attenuated by adrenalectomy, suggesting that the change in HR was most likely due to enhanced parasympathetic activity as opposed to decreased sympathetic stimulation. This hypothesis was supported by Sakamoto et al. (2008) who showed that KA administration (10–12 mg·kg−1) through either i.p., i.a. or i.v. routes in urethane-anesthetized rats produced a sustained increase in vagal nerve activity with 41% of animals dying due to profound bradycardia or arrhythmia, such as AV block.
In the ensuing 60–180 min following KA administration in this study, there was a progressive increase in EEG activity and seizure behaviours, consistent with the development of SE, as behavioural scores progressed to level 4 and 5 with little or no interictal recovery periods (Figure 3B). This generalized seizure activity coincided with the development of tachycardia, QTc prolongation and T wave elevation. Interestingly, there appeared to be a slight ‘phase discrepancy’ between the onset and persistence of electrographic spiking and tachycardia. As our study was limited to implanting the EEG electrode in the cortex, it was not possible to determine the pattern or extent of electrographic spiking occurring specifically within the CNS cardiac (sympathetic) control centres. Tachycardia is commonly reported in animal and clinical studies during generalized seizure activity (Terndrup et al., 1994; Powell et al., 2008; Metcalf et al., 2009a). Hotta et al. (2009) reported a 17% increase in HR and a twofold increase in sympathetic nerve activity following KA administration in an anaesthetized animal model. Metcalf et al. (2009a) showed that SE, induced in rats by lithium and pilocarpine, caused significant increases in HR, QT prolongation and elevated troponin I levels. This QTc prolongation has also been speculated to occur as a consequence of decreased expression of the Kv4.2 potassium channels following SE (Bealer et al., 2010). In the present study, the QTc interval remained prolonged at the 24 h recording period suggesting the development of these changes in ventricular depolarization and repolarization in a clinical setting may predispose the patient to ventricular fibrillation and sudden cardiac death (Shimizu and Antzelevitch, 1998). In an earlier report by Speerschneider and Thomsen, (2013), both cardiac ischaemia and β-adrenergic stimulation with isoproterenol were capable of producing positive T wave changes. ECG analysis in our seizure animals similarly demonstrated an increase in T wave amplitude. This electrophysiological remodelling may reflect the combined effects of a catecholamine-induced increase in HR and ion channel activity and the consequences of micro-ischaemic zones leading to cellular depolarization and conduction block (Speerschneider and Thomsen, 2013).
Deterioration in cardiac morphology was evident 48 h following a SE event. The saline-KA hearts showed evidence of perivascular inflammatory cell infiltration and nuclear vacuolization, a marker of reversible ischaemic damage and hypercontraction band necrosis, indicative of elevated catecholamine levels (Boggs et al., 1993; Kloster and Engelskjon, 1999; Jansen and Lagae, 2010). In the present study, the hearts from animals with seizures showed a significant increase in myocardial fibrosis correlated with tachycardia, suggesting that these hearts may be more susceptible to arrhythmia. The cardiac pathology observed in this study is similar to previously reported animal (Metcalf et al., 2009a) and clinical studies (Kloster and Engelskjon, 1999; Stollerberger and Finsterer, 2004). Natelson et al. (1998) found evidence of irreversible perivascular and interstitial fibrosis, as well as reversible myocyte vacuolization in hearts from epileptic patients. Repeated hypoxaemia and increased catecholamines can cause structural heart damage making the heart more susceptible to fatal arrhythmias, increasing the risk of sudden cardiac death. Haemodynamic changes have also been reported following SE with deterioration of cardiac output due to decreased HR and mean arterial pressure, causing gradual cardiac decompensation and myocardial injury due to elevated endogenous catecholamine release (Manno et al., 2005). Hypercontraction band necrosis is a marker of excessive catecholamine levels and has been reported at post-mortem in 73% of patients who died consequent to SE (Manno et al., 2005). Boggs et al. (1993) reported a mortality rate of 25% in SE patients with 10% of the SE patients suffering myocardial infarction as shown by ECG and confirmed by elevated cardiac enzyme markers. The presence of myocyte vacuolization and fibrosis in these hearts provides support for tachycardia-induced ventricular injury. The development of micro-infarcts may be implicated in sudden cardiac death by altering electrical conduction and increasing the risk of ventricular arrhythmias. Therefore, preventing the development of tachycardia with atenolol or clonidine may preserve cardiac function during seizure (Figure 6).
Figure 6.
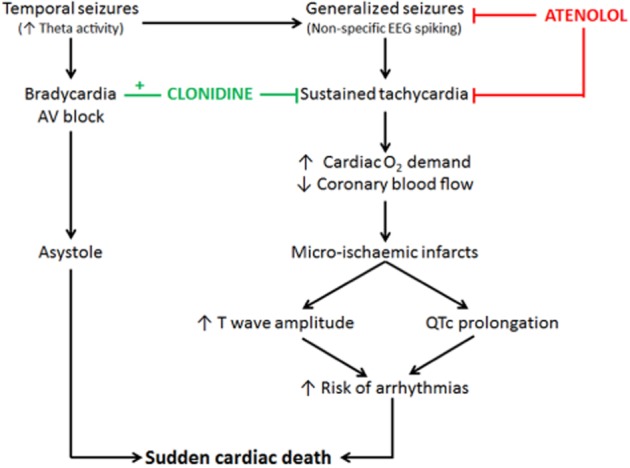
Diagram summarizing the EEG, ECG and cardiac morphological changes contributing to the cardiac injury seen in this seizure study and the cardio-protective effects of atenolol and clonidine.
Pretreatment with atenolol attenuated seizure severity and progression as well as EEG activity following KA administration. As atenolol is a hydrophilic drug reported to have a low blood brain barrier permeability (brain: plasma ratio of 0.2), this central effect was unexpected (Patel and Turner, 1981; Drayer, 1987). In a previous study, Little and Bealer (2012) indicated that a single bolus dose of atenolol (1 mg·kg−1, i.v.) given immediately before seizure induction had no influence on EEG activity or seizure behaviours in rats with electrically induced SE. It is possible that the higher dose and chronic administration of atenolol used in the present study allowed atenolol to cross into the brain where it could inhibit neuronal β1 adrenoceptors in the hippocampus (Mueller and Dunwiddie, 1983) and inhibit their proconvulsant effect. An alternative explanation is that by preventing the development of tachycardia, atenolol reduced the extent of cerebral hypoxia and thereby decreased consequent cerebral hyperexcitabilty, preventing generalized seizure activity (Morady et al., 1985; Kobari et al., 1992; Ocon et al., 2009).
Atenolol successfully prevented the development of tachycardia, QTc prolongation and T wave elevation contributing to the preserved cardiac morphology seen in these animals at 48 h. There was very little evidence of myocyte vacuolization and no hypercontraction band necrosis observed in these treated hearts. Atenolol also prevented the development of fibrosis and reduced the extent of oedema in these hearts. Little and Bealer (2012) found that atenolol treatment preserved cardiac output and reduced the cardiac damage caused by SE (Little and Bealer, 2012). In fact, atenolol has been applied following head trauma to prevent an elevation in cardiac enzymes, ECG abnormalities and arrhythmias (Cruickshank et al., 1987).
Clonidine pretreatment altered the seizure activity and effectively reduced high amplitude spiking and WDS; however, clonidine did not reduce the power of the EEG frequencies. Clonidine has been previously reported to decrease WDS, protect against limbic seizures and prevent neurochemical changes (Kleinrok and Turski, 1980; Ohno et al., 1987; Yoshioka et al., 2000; Read et al., 2014). The findings in the present study are in agreement with earlier research suggesting that clonidine has anticonvulsant effects which appear to be mediated by the α2 adrenoreceptor. Other α2 adrenoceptor agonists such as dexmedetomidine effectively reduce the development, generalization and severity of KA-induced seizures while selective α2 antagonists such as yohimbine and atipamezole, have a pro-convulsant effect and are associated with increased mortality (Ohno et al., 1987; Halonen et al., 1995). Clonidine additionally provided significant cardiac protection as observed by the prevention of tachycardia, QTc prolongation and T wave elevation. Clonidine is known act pre-synaptically to reduce the release of noradrenaline at cardiac adrenergic synapses resulting in an attenuation of the sympathetic response and consequent reduction in HR (Matsukawa et al., 1995; Zhang and Cheng, 2000; Champeroux et al., 2010). Clonidine reduced the degree of seizure-induced tachycardia, thereby preserving cardiac morphology and protecting the heart from the development of microinfarcts as observed by the absence of fibrotic deposition and myocyte vacuolization. The anticonvulsant effects of clonidine are not without controversy, as some studies have shown that clonidine can exert both age- and dose-dependent pro-convulsant effects (Szot et al., 2004; Feron et al., 2008). Szot et al. (2004) hypothesized that pre-synaptic α2A autoreceptors mediated clonidine's pro-convulsant effects while post-synaptic α2A receptors were responsible for the anti-convulsant effects (Szot et al., 2004). Clonidine has a 10 times higher affinity for pre-synaptic α2 adrenoreceptors than it does for the post-synaptic receptors (Maura et al., 1985) suggesting that a high dose of clonidine may be required to achieve the anticonvulsant effects. Chronic high doses of clonidine were used in the current study, and while this produced a significant reduction in seizure severity, animals appeared sedated and less responsive following the clonidine injections. Sedation and dry mouth for up to 12 h following a high dose of clonidine (Keranen et al., 1978; Hall et al., 2001) are also commonly reported clinically and these adverse effects may result in reduced compliance if clonidine is to be used prophylactically in epileptic patients.
This study clearly demonstrates that sustained seizure activity results in prolonged tachycardia leading to microinfarct formation, which may be implicated in the development of cardiac pathology and mortality in epilepsy (Figure 6). This study also highlights the importance of protecting the heart against sympathetic overdrive during the early stages of SE. Prophylactic treatment with either atenolol or clonidine was effective at protecting the heart during seizure. Both therapies reduced seizure progression, ECG abnormalities and histopathological evidence of cardiac injury. Prophylactic treatment with clonidine in epileptic patients would require clear adherence to exclusion criteria with age, dose and compliance strictly monitored. Atenolol on the other hand appears to be better tolerated clinically and proved more effective than clonidine at reducing the extent of ischaemic damage seen at 48 h. While it is paramount that sympatholytic intervention remains contraindicated in patients at risk of bradycardia and bradyarrhythmia, these data strongly suggest that atenolol can add cardioprotective value to current antiepileptic treatment strategies conducted in patients suffering frequent seizures. Intervention trials with atenolol should include serial assessments of cardiac function using echocardiography and injury markers in order to provide much needed information on the pathology and validation of this pharmacological approach. Furthermore, prophylactic treatment with atenolol may also have a role in patients presenting with a number of risk factors for sudden unexpected death in epilepsy (Surges et al., 2009). The results from this study demonstrate that atenolol has the potential to enhance cardiac protection in epileptic patients and should be considered as a combination therapy with antiepileptic agents.
Acknowledgments
The authors greatly appreciate the technical assistance of Amanda Fisher (University of Otago Histology Unit) with the histological studies.
Glossary
- b.p.m
beats per minute
- HR
heart rate
- KA
kainic acid
- PSD
power spectral density
- SE
status epilepticus
- WDS
wet dog shakes
Author contributions
M. I. R., J. C. H., D. S. K. and I. A. S. conceived and designed the experiments. M. I. R. performed the experiments, analysed the data and drafted the paper. J. C. H., D. S. K. & I. A. S. supervised the study, and edited and revised manuscript. All authors approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo ER, Newton GE, Parker JD. Cardiac and systemic sympathetic activity in response to clonidine in human heart failure. J Am Coll Cardiol. 1999;33:186–191. doi: 10.1016/s0735-1097(98)00524-5. [DOI] [PubMed] [Google Scholar]
- Bealer SL, Little JG, Metcalf CS, Brewster AL, Anderson AE. Autonomic and cellular mechanisms mediating detrimental cardiac effects of status epilepticus. Epilepsy Res. 2010;91:66–73. doi: 10.1016/j.eplepsyres.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Boggs JG, Painter JA, De Lorenzo RJ. Analysis of electrocardiographic changes in status epilepticus. Epilepsy Res. 1993;14:87–94. doi: 10.1016/0920-1211(93)90077-k. [DOI] [PubMed] [Google Scholar]
- Brophy GM, Bell R, Claassen J, Alldredge B, Glauser T, Laroche SM, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17:3–23. doi: 10.1007/s12028-012-9695-z. [DOI] [PubMed] [Google Scholar]
- Champeroux P, Ouille A, Martel E, Fowler JS, Maurin A, Jude S, et al. Interferences of the autonomic nervous system with drug induced QT prolongation: a point to consider in non-clinical safety studies. J Pharmacol Toxicol Methods. 2010;61:251–263. doi: 10.1016/j.vascn.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Cruickshank JM, Neil-Dwyer G, Degaute JP, Hayes Y, Kuurne T, Kytta J, et al. Reduction of stress/catecholamine-induced cardiac necrosis by beta 1-selective blockade. Lancet. 1987;2:585–589. doi: 10.1016/s0140-6736(87)92984-9. [DOI] [PubMed] [Google Scholar]
- Dernovsek MZ, Sket D. The effects of kainic acid in rats with spontaneous recurrent seizures. Gen Pharmacol. 1998;31:447–449. doi: 10.1016/s0306-3623(98)00015-9. [DOI] [PubMed] [Google Scholar]
- Devinsky O. Effects of seizures on autonomic and cardiovascular function. Epilepsy Curr. 2004;4:43–46. doi: 10.1111/j.1535-7597.2004.42001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayer DE. Lipophilicity, hydrophilicity, and the central nervous system side effects of beta blockers. Pharmacotherapy. 1987;7:87–91. doi: 10.1002/j.1875-9114.1987.tb04029.x. [DOI] [PubMed] [Google Scholar]
- Feron FJ, Hendriksen JG, Nicolai J, Vles JS. New-onset seizures: a possible association with clonidine? Pediatr Neurol. 2008;38:147–149. doi: 10.1016/j.pediatrneurol.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Ferrari MF, Fior-Chadi DR, Chadi G. Effects of bilateral adrenalectomy on systemic kainate-induced activation of the nucleus of the solitary tract. Regulation of blood pressure and local neurotransmitters. J Mol Histol. 2008;39:253–263. doi: 10.1007/s10735-008-9161-6. [DOI] [PubMed] [Google Scholar]
- Hall JE, Uhrich TD, Ebert TJ. Sedative, analgesic and cognitive effects of clonidine infusions in humans. Br J Anaesth. 2001;86:5–11. doi: 10.1093/bja/86.1.5. [DOI] [PubMed] [Google Scholar]
- Halonen T, Kotti T, Tuunanen J, Toppinen A, Miettinen R, Riekkinen PJ. Alpha 2-adrenoceptor agonist, dexmedetomidine, protects against kainic acid-induced convulsions and neuronal damage. Brain Res. 1995;693:217–224. doi: 10.1016/0006-8993(95)00744-b. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Beta-adrenergic antagonists attenuate withdrawal anxiety in cocaine- and morphine-dependent rats. Psychopharmacology (Berl) 1993;113:131–136. doi: 10.1007/BF02244345. [DOI] [PubMed] [Google Scholar]
- Hotta H, Koizumi K, Stewart M. Cardiac sympathetic nerve activity during kainic acid-induced limbic cortical seizures in rats. Epilepsia. 2009;50:923–927. doi: 10.1111/j.1528-1167.2008.01860.x. [DOI] [PubMed] [Google Scholar]
- Jansen K, Lagae L. Cardiac changes in epilepsy. Seizure. 2010;19:455–460. doi: 10.1016/j.seizure.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Keranen A, Nykanen S, Taskinen J. Pharmacokinetics and side-effects of clonidine. Eur J Clin Pharmacol. 1978;13:97–101. doi: 10.1007/BF00609752. [DOI] [PubMed] [Google Scholar]
- Kibar AE, Unver O, Oflaz MB, Guven AS, Balli S, Ece I, et al. Effect of antiepilepsy drug therapy on ventricular function in children with epilepsy: a tissue Doppler imaging study. Pediatr Cardiol. 2014;35:280–288. doi: 10.1007/s00246-013-0771-8. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinrok Z, Turski L. Kainic acid-induced wet dog shakes in rats. The relation to central neurotransmitters. Naunyn Schmiedebergs Arch Pharmacol. 1980;314:37–46. doi: 10.1007/BF00498429. [DOI] [PubMed] [Google Scholar]
- Kloster R, Engelskjon T. Sudden unexplained death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurosurgical Psychiatry. 1999;67:439–444. doi: 10.1136/jnnp.67.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobari M, Fukuuchi Y, Tomita M, Tanahashi N, Shinohara T, Yamawaki T, et al. Cerebral microcirculatory changes during and following transient ventricular tachycardia in cats. J Neurol Sci. 1992;111:153–157. doi: 10.1016/0022-510x(92)90063-q. [DOI] [PubMed] [Google Scholar]
- de Lange EC, Danhof M, de Boer AG, Breimer DD. Critical factors of intracerebral microdialysis as a technique to determine the pharmacokinetics of drugs in rat brain. Brain Res. 1994;666:1–8. doi: 10.1016/0006-8993(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Leutmezer F, Schernthaner C, Lurger S, Potzelberger K, Baumgartner C. Electrocardiographic changes at the onset of epileptic seizures. Epilepsia. 2003;44:348–354. doi: 10.1046/j.1528-1157.2003.34702.x. [DOI] [PubMed] [Google Scholar]
- Little JG, Bealer SL. Beta adrenergic blockade prevents cardiac dysfunction following status epilepticus in rats. Epilepsy Res. 2012;99:233–239. doi: 10.1016/j.eplepsyres.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Locatelli ER, Varghese JP, Shuaib A, Potolicchio SJ. Cardiac asystole and bradycardia as a manifestation of left temporal lobe complex partial seizure. Ann Intern Med. 1999;130:581–583. doi: 10.7326/0003-4819-130-7-199904060-00018. [DOI] [PubMed] [Google Scholar]
- Logroscino G, Hesdorffer DC, Cascino GD, Hauser WA, Coeytaux A, Galobardes B, et al. Mortality after a first episode of status epilepticus in the United States and Europe. Epilepsia. 2005;46:46–48. doi: 10.1111/j.1528-1167.2005.00409.x. [DOI] [PubMed] [Google Scholar]
- Loscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20:359–368. doi: 10.1016/j.seizure.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Collins RC. Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981;218:299–318. doi: 10.1016/0006-8993(81)91308-1. [DOI] [PubMed] [Google Scholar]
- Manno EM, Pfeifer EA, Cascino GD, Noe KH, Wijdicks EF. Cardiac pathology in status epilepticus. Ann Neurol. 2005;58:954–957. doi: 10.1002/ana.20677. [DOI] [PubMed] [Google Scholar]
- Matsukawa K, Wilson LB, Wall PT, Mitchell JH. Differential effects of clonidine on renal sympathetic nerve activity and heart rate at onset of static exercise. J Auton Nerv Syst. 1995;51:205–212. doi: 10.1016/0165-1838(94)00132-4. [DOI] [PubMed] [Google Scholar]
- Maura G, Gemignani A, Raiteri M. Alpha 2-adrenoceptors in rat hypothalamus and cerebral cortex: functional evidence for pharmacologically distinct subpopulations. Eur J Pharmacol. 1985;116:335–339. doi: 10.1016/0014-2999(85)90173-6. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf CS, Poelzing S, Little JG, Bealer SL. Status epilepticus induces cardiac myofilament damage and increased susceptibility to arrhythmias in rats. AJP-Heart Circ Physiol. 2009a;297:120–127. doi: 10.1152/ajpheart.00724.2009. [DOI] [PubMed] [Google Scholar]
- Metcalf CS, Radwanski PB, Bealer SL. Status epilepticus produces chronic alterations in cardiac sympathovagal balance. Epilepsia. 2009b;50:747–754. doi: 10.1111/j.1528-1167.2008.01764.x. [DOI] [PubMed] [Google Scholar]
- Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–H751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- Morady F, Shen EN, Bhandari A, Schwartz AB, Scheinman MM. Clinical symptoms in patients with sustained ventricular tachycardia. West J Med. 1985;142:341–344. [PMC free article] [PubMed] [Google Scholar]
- Mueller AL, Dunwiddie TV. Anticonvulsant and proconvulsant actions of alpha- and beta-noradrenergic agonists on epileptiform activity in rat hippocampus in vitro. Epilepsia. 1983;24:57–64. doi: 10.1111/j.1528-1157.1983.tb04866.x. [DOI] [PubMed] [Google Scholar]
- Natelson BH, Suarez RV, Terrence CF, Turizo R. Patients with epilepsy who die suddenly have cardiac disease. Arch Neurol. 1998;55:857–860. doi: 10.1001/archneur.55.6.857. [DOI] [PubMed] [Google Scholar]
- Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia. 2000;41:542–548. doi: 10.1111/j.1528-1157.2000.tb00207.x. [DOI] [PubMed] [Google Scholar]
- Nguyen-Michel VH, Adam C, Dinkelacker V, Pichit P, Boudali Y, Dupont S, et al. Characterization of seizure-induced syncopes: EEG, ECG, and clinical features. Epilepsia. 2014;55:146–155. doi: 10.1111/epi.12482. [DOI] [PubMed] [Google Scholar]
- Ocon AJ, Medow MS, Taneja I, Clarke D, Stewart JM. Decreased upright cerebral blood flow and cerebral autoregulation in normocapnic postural tachycardia syndrome. AJP-Heart Circ Physiol. 2009;297:H664–H673. doi: 10.1152/ajpheart.00138.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Yamamoto T, Ueki S. Clonidine attenuates wet-dog shaking induced by hippocampal stimulation in rats. Eur J Pharmacol. 1987;137:161–166. doi: 10.1016/0014-2999(87)90217-2. [DOI] [PubMed] [Google Scholar]
- Opherk C, Cocomilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res. 2002;52:117–127. doi: 10.1016/s0920-1211(02)00215-2. [DOI] [PubMed] [Google Scholar]
- Patel L, Turner P. Central actions of beta-adrenoceptor blocking drugs in man. Med Res Rev. 1981;1:387–410. doi: 10.1002/med.2610010405. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell KL, Ng C, O'Brien TJ, Xu SH, Williams DA, Foote SJ, et al. Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats. Epilepsia. 2008;49:1686–1695. doi: 10.1111/j.1528-1167.2008.01593.x. [DOI] [PubMed] [Google Scholar]
- Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy: a clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122:1017–1031. doi: 10.1093/brain/122.6.1017. [DOI] [PubMed] [Google Scholar]
- Read MI, Andreianova AA, Harrison JC, Goulton CS, Sammut IA, Kerr DS. Cardiac electrographic and morphological changes following status epilepticus: effect of clonidine. Seizure. 2014;23:55–61. doi: 10.1016/j.seizure.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Saito T, Orman R, Koizumi K, Lazar J, Salciccioli L. Autonomic consequence of kainic acid-induced limbic cortical seizures in rats: peripheral autonomic nerve activity, acute cardiovascular changes and death. Epilepsia. 2008;73:982–996. doi: 10.1111/j.1528-1167.2008.01545.x. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR. Overview of the current animal models for humans and epileptic disorders. Epilepsy & Behavior. 2001;2:201–216. doi: 10.1006/ebeh.2001.0193. [DOI] [PubMed] [Google Scholar]
- Sawant PM, Mountfort DO, Kerr DS. Spectral analysis of electrocorticographic activity during pharmacological preconditioning and seizure induction by intrahippocampal domoic acid. Hippocampus. 2010;20:994–1002. doi: 10.1002/hipo.20698. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circ. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- Smith C, Teitler M. Beta-blocker selectivity at cloned human beta 1- and beta 2-adrenergic receptors. Cardiovasc Drugs Ther. 1999;13:123–126. doi: 10.1023/a:1007784109255. [DOI] [PubMed] [Google Scholar]
- Speerschneider T, Thomsen MB. Physiology and analysis of the electrocardiographic T wave in mice. Acta Physiol. 2013;209:262–271. doi: 10.1111/apha.12172. [DOI] [PubMed] [Google Scholar]
- Stollerberger C, Finsterer J. Cardiorespiratory findings in sudden unexplained/unexpected death in epilepsy. Epilepsy Res. 2004;59:51–60. doi: 10.1016/j.eplepsyres.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Surges R, Thijs RD, Tan HJ, Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nature Rev Neurololgy. 2009;5:492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- Szot P, Lester M, Laughlin ML, Palmiter RD, Liles LC, Weinshenker D. The anticonvulsant and proconvulsant effects of alpha2-adrenoreceptor agonists are mediated by distinct populations of alpha2A-adrenoreceptors. Neurosci. 2004;126:795–803. doi: 10.1016/j.neuroscience.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Terndrup TE, Starr F, Fordyce WE. A piglet model of status epilepticus: comparison of cardiorespiratory and metabolic changes with two methods of pentylenetetrazol administration. Ann Emerg Med. 1994;23:470–479. doi: 10.1016/s0196-0644(94)70065-6. [DOI] [PubMed] [Google Scholar]
- Toader E, Cividjian A, Rentero N, McAllen RM, Quintin L. Cardioinhibitory actions of clonidine assessed by cardiac vagal motoneuron recordings. J Hypertens. 2008;26:1169–1180. doi: 10.1097/HJH.0b013e3282fd10e0. [DOI] [PubMed] [Google Scholar]
- Vranyac-Tramoundanas A, Harrison JC, Sawant PM, Kerr DS, Sammut IA. Ischemic cardiomyopathy following seizure induction by domoic acid. Am J Pathol. 2011;179:141–154. doi: 10.1016/j.ajpath.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC. Status epilepticus: an evidence based guide. BMJ. 2005;331:673–677. doi: 10.1136/bmj.331.7518.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P, White A, Ferraro D, Clark S, Staley K, Dudek FE. The use of radiotelemetry to evaluate electrographic seizures in rats with kainate-induced epilepsy. J Neurosci Methods. 2006;155:39–48. doi: 10.1016/j.jneumeth.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Yoshioka S, Mitani H, Maeda K, Takeo S, Matsuda K, Katayama S, et al. Age-specific effects of noradrenergic alpha-2 agonist clonidine on the development of amygdaloid kindling in developing rats. Brain Res Dev Brain Res. 2000;119:283–288. doi: 10.1016/s0165-3806(99)00179-0. [DOI] [PubMed] [Google Scholar]
- Zaveri HP, Duckrow RB, de Lanerolle NC, Spencer SS. Distinguishing subtypes of temporal lobe epilepsy with background hippocampal activity. Epilepsia. 2001;42:725–730. doi: 10.1046/j.1528-1157.2001.00500.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cheng Z. Sympathetic inhibition with clonidine prolongs survival in experimental chronic heart failure. Int J Cardiol. 2000;73:157–162. doi: 10.1016/s0167-5273(00)00213-8. [DOI] [PubMed] [Google Scholar]
- Zipes DP, Wellens HJ. Sudden cardiac death. Circ. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]