

Abstract
Background and Purpose
Emerging evidence suggests a selective up-regulation of arginase I in diabetes causing coronary artery disease; however, the mechanisms behind this up-regulation are still unknown. Activated p38 MAPK has been reported to increase arginase II in various cardiovascular diseases. We therefore tested the role of p38 MAPK in the regulation of arginase I and II expression and its effect on endothelial dysfunction in diabetes mellitus.
Experimental Approach
Endothelial function was determined in septal coronary (SCA), left anterior descending coronary (LAD) and mesenteric (MA) arteries from healthy and streptozotocin-induced diabetic Wistar rats by wire myographs. Arginase activity and protein levels of arginase I, II, phospho-p38 MAPK and phospho-endothelial NOS (eNOS) (Ser1177) were determined in these arteries from diabetic and healthy rats treated with a p38 MAPK inhibitor in vivo.
Key Results
Diabetic SCA and MA displayed impaired endothelium-dependent relaxation, which was prevented by arginase and p38 MAPK inhibition while LAD relaxation was not affected. Arginase I, phospho-p38 MAPK and eNOS protein expression was increased in diabetic coronary arteries. In diabetic MA, however, increased expression of arginase II and phospho-p38 MAPK, increased arginase activity and decreased expression of eNOS were observed. All these effects were reversed by p38 MAPK inhibition.
Conclusions and Implications
Diabetes-induced activation of p38 MAPK causes endothelial dysfunction via selective up-regulation of arginase I expression in coronary arteries and arginase II expression in MA. Therefore, regional differences appear to exist in the arginase isoforms contributing to endothelial dysfunction in type 1 diabetes mellitus.
Tables of Links
| TARGETS | |
|---|---|
| Arginase I | |
| Arginase II | p38 MAPK |
| eNOS |
| LIGANDS | ||
|---|---|---|
| ABH | L-arginine | U46619 |
| ACh | Nitric oxide (NO) | Urea |
| SB203580 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Diabetes mellitus is a growing health problem associated with vascular complications including coronary and peripheral artery disease. Such complications are at present the principal causes of morbidity and mortality in patients with diabetes mellitus (Avogaro et al., 2011). Loss of the modulatory role of endothelium, known as endothelial dysfunction due to decreased nitric oxide (NO) bioavailability, is thought to be an early factor in the development of diabetic vascular diseases (De Vriese et al., 2000). Endothelial NOS (eNOS) utilizes L-arginine as a substrate to produce NO. L-arginine is also a substrate for arginase, a critical enzyme in the urea cycle. Arginase, which exists in the two distinct isoforms, arginase I and arginase II, is also expressed in the cardiovascular system including endothelial cells (Pernow and Jung, 2013). Hence, the up-regulation of arginase has emerged as an important pathophysiological factor behind the reduced bioavailability of NO due to its competition with eNOS for their common substrate.
There is emerging evidence suggesting that enhanced arginase activity contributes to the endothelial dysfunction seen in experimental animal models of diabetes (Romero et al., 2008; 2012,; Grönros et al., 2011; Patel et al., 2013; Yao et al., 2013) and patients with diabetes (Beleznai et al., 2011; Shemyakin et al., 2012; Kövamees et al., 2014). Arginase-induced impaired coronary vascular function was linked to increased arginase I expression and activity in aorta of rats with type 1 diabetes mellitus (Romero et al., 2008) and associated with increased arginase II expression in the aorta of a rat model of type 2 diabetes mellitus (Grönros et al., 2011). Interestingly, Beleznai et al. reported that the expression of arginase I is increased in human coronary arteries from patients with diabetes (Beleznai et al., 2011). However, the effect of this increased arginase I expression on coronary artery function in diabetes, and the underlying mechanism(s) leading to this up-regulation in coronary arteries in diabetes remain unknown.
Hyperglycaemia and oxidative stress have been proposed as possible mechanisms in the pathogenesis of endothelial dysfunction in diabetes (Shi and Vanhoutte, 2009; Paneni et al., 2013). Oxidative/nitrosative stress is associated with increases in arginase activity/expression (Bivalacqua et al., 2001; Chandra et al., 2012; Kiss et al., 2014) and with p38 MAPK activation (Kassan et al., 2014). In physiological states, p38 MAPK transduces extracellular signals and coordinates the cellular responses which are vital for adaptation and survival (Martin et al., 2015), whereas it triggers maladaptive responses in disease states that aggravate the disease. For instance, inhibition of p38 MAPK improves endothelium-dependent vasorelaxation in humans and in experimental animal models of disease (Widder et al., 2004; Komers et al., 2007; Cheriyan et al., 2011; Wu et al., 2012). Recently, it was reported that increased p38 MAPK is involved in eNOS uncoupling in diabetes (Kassan et al., 2014) and in increased arginase II activity/expression causing dysfunction of corpora cavernosa in angiotensin II-treated and diabetic mice (Toque et al., 2010; 2013,). It has also been proposed that arginase I is selectively up-regulated in diabetic patients with coronary dysfunction (Beleznai et al., 2011). Thus, tissue or regional differences may exist regarding the isoform of arginase that is upregulated in diabetes. Furthermore, it remains unclear whether p38 MAPK is involved in the up-regulation of arginase in coronary and peripheral arteries in diabetes. Therefore, the aims of the present study were to investigate the role of p38 MAPK in the regulation of arginase I and II in coronary and mesenteric arteries (MA), which represent two separate vascular beds, and the involvement of this effect in the endothelial dysfunction observed in a rat model of type 1 diabetes mellitus.
Methods
Ethics statement
This study was conducted in compliance with the European Directive for the Protection of Animals Used for Scientific Purposes (2010/63/EU). Animal care and all protocols in the present study were approved by the regional Ethics Committee (Stockholm Norra Djurförsöksetiska Nämnd, approval numbers approval numbers N192/12, N65/13 and N108/14) for laboratory animal experiments in Stockholm. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 57 animals were used in the experiments described below.
Induction of type 1 diabetes mellitus
Male Wistar rats were provided from Charles River Laboratories (Sulzfeld, Germany) at 6–7 weeks of age. Animals were housed at the Karolinska Institute animal care facility and maintained on standard chow and water ad libitum, until they were used for study, at 15–16 weeks of age. Type 1 diabetes mellitus was induced by a single i.v. (tail vein) injection of streptozotocin (STZ, 55 mg·kg−1, Sigma Aldrich, St. Louis, MO, USA) freshly dissolved in sterile Dulbecco's PBS (DPBS, Gibco, Life Technologies Ltd, Paisley, UK) at 7–8 weeks of age (Kiss et al., 2014). Three days later, only rats with blood glucose levels >15.0 mmol·L−1 were included in the study (n = 32). Age-matched rats that received i.v. injections of sterile DPBS were used as healthy controls (n = 25). Animals were maintained on standard chow and water ad libitum and were not supplemented with insulin or glucose-lowering agents. Blood glucose and body weight were checked weekly for 8 weeks after the injection of either STZ or DPBS.
In vivo treatment with SB203580
At the beginning of the 6th week after the injection of STZ or DPBS, healthy and diabetic rats were given the selective p38 MAPK inhibitor 4-[5-(4-fluorophenyl)-2-[4-(methylsulphonyl)phenyl]-1H-imidazol-4-yl]pyridine hydrochloride (SB203580 hydrochloride, 100 μg·kg−1·day−1 i.p.) for 2 weeks. These rats were divided into four groups: (i) healthy rats given DPBS (vehicle of SB203580 n = 8); (ii) healthy rats given SB203580 (n = 8); (iii) diabetic rats given DPBS (n = 10); (iv) diabetic rats given SB203580 (n = 10). At the end of this treatment, the animals were killed and the coronary and MA were collected and used for Western blot and arginase activity assays. In addition, MA were also used in functional experiments.
Isolation and mounting of coronary and mesenteric arteries MA
Eight weeks after the injection of either STZ or DPBS (15–16 weeks of age), diabetic and healthy rats were anaesthetized with sodium pentobarbital (50 mg·kg−1, i.p.) and killed by decapitation and exsanguination. The depth of anaesthesia was evaluated by pinching the animal's paw with forceps and all efforts were made to minimize suffering. The heart and the mesenteric bed were quickly removed and placed in cold (4°C) physiological buffer solution of the following composition (mM): 119 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 1.5 CaCl2, 24.9 NaHCO3, 0.027 EDTA and 11 glucose; bubbled with a mixture of 95% O2 and 5% CO2, resulting in a pH 7.4.
Intramyocardial second order branches of the left anterior descending coronary artery (LAD), segments of the septal coronary artery (SCA) and second- to third-order branches of MA were carefully dissected as previously described (Santiago et al., 2013). Arterial segments, 2 mm long, from healthy and diabetic rats were mounted in microvascular myographs (DMT, Aarhus, Denmark) and equilibrated at 37°C. For each individual artery, the internal circumference L100 corresponding to a transmural pressure of 100 mmHg for a relaxed vessel in situ was calculated. The arteries were set to an internal circumference L1 equal to 0.9 times L100, at which tension development is maximal (Climent et al., 2014).
Experimental procedures for the functional assays
At the beginning of each experiment, the ability of the preparation to develop a contraction was assessed by exposing the arteries to a high KCl solution (124 mM). Endothelium-dependent relaxations were determined by administration of cumulatively increasing concentrations of ACh (0.3 nM to 1 μM in MA; 0.1 nM to 10 μM in coronary arteries) pre-contracted with the thromboxane A2 analogue, 9,11-dideoxy-9α,11α-methanoepoxy PGF2α (U46619, 0.5–2 μM). The arteries were then washed three times every 10 min for 30 min. Following this step, concentration-response curves to ACh were determined following incubation for 60 min with the arginase inhibitor 2(S)-amino-6-boronohexanoic acid (ABH, 0.1 mM), the p38 MAPK inhibitor SB203580 (1 μM) or SB203580 plus ABH. Another set of experiments was performed in order to evaluate the effect of the in vivo inhibition of p38 MAPK for 2 weeks with SB203580 on endothelial function. Concentration-response curves to ACh (0.3 nM–1 μM) were obtained in MA from healthy and diabetic rats treated with vehicle or SB203580 in the presence and in the absence of ABH. In order to assess whether the effect of arginase inhibition is selective for endothelium-dependent relaxations, concentration-response curves to the NO donor sodium nitroprusside (SNP; 30 nM–1 μM) were determined in the presence and in the absence of ABH in endothelium-denuded arteries from healthy and diabetic rats. The endothelium was removed mechanically by passing a human hair through the vessel lumen and the absence of functional endothelium was confirmed by the lack of a relaxation response elicited by ACh (10 μM).
Determination of arginase activity
The mesenteric vascular bed was isolated, placed in a Petri dish containing chilled (4°C) physiological salt solution. The mesenteric artery was carefully dissected, placed in radio-immunoprecipitation assay (RIPA) buffer (Thermo Scientific, Waltham, MA, USA) with protease inhibitors (Roche, Diagnostics, Bromma, Sweden), homogenized and centrifuged for 20 min at 12 000× g at 4°C. Fifty microlitres of the supernatant were incubated with 75 μL of 10 mM MnCl2 in 50 mM Tris-HCl, pH 7.5 buffer to activate arginase. Next, 50 μL of 500 mM L-arginine (prepared in 50 mM Tris-HCl pH 9.7) were added. The reaction was carried out at 37°C for 1 h. The reaction was stopped by adding 400 μL of an acid solution (H2SO4–H3PO4–H2O = 1:3:7). Urea produced subsequently was assayed using 25 μL of α-isonitrosopropiophenone (9% in ethanol) followed by heating at 100°C for 1 h. The entire sample was cooled to room temperature. Arginase activity was calculated as urea (μmol·mg−1 protein·min−1) production (Kiss et al., 2014).
Western blotting
Arterial samples from both the mesenteric and coronary arterial bed were extracted in RIPA Buffer (Thermo Scientific, USA) with protease and phosphatase inhibitors (Roche), homogenized and centrifuged for 20 min at 12 000× g at 4°C. Total protein content of the extracts was quantified by using a bicinchoninic acid protein assay kit (Pierce Biotechnology, Life Technologies Ltd). The proteins were separated on 7.5% SDS gel (20 μg per lane for coronary, or 25 μg per lane for mesenteric extracts) and transferred onto PVDF membranes (Merck, Millipore, Solna, Sweden). Membranes were blocked with 5% milk for 1 h at room temperature and incubated overnight at 4°C with primary antibodies against arginase I and II (1:200, Atlas Prestige Antibody, Sigma-Aldrich), phoshorylated eNOS (p-eNOS; anti-phosphorylated Ser1177; 1:400; BD Pharmingen), total eNOS (1:500; BD Pharmingen, San Jose, CA, USA); p38 MAPK and phosphorylated p38 MAPK (1:1000; Cell Signalling, Boston, MA, USA). IRDye 800-conjugated goat anti-mouse IgG (1:10 000, LI-COR, Biosciences, Cambrige, UK), IRDye 800-conjugated goat anti-rabbit IgG (1:10 000, LI-COR Biosciences) and IRDye 680 (1:10 000) were used as secondary antibodies and bands were visualized using an infrared fluorescence scanner (IR-Odyssey, LI-COR Biosciences). Membranes were also reprobed with anti-β-actin (1:10 000, Sigma Aldrich, USA). Relative intensity for each protein was determined by comparison with the intensity of β-actin staining on blots that were stripped and then reprobed with β-actin primary antibody. Band densities were analysed with Image Studio Lite Version 3.1 (LI-COR Biosciences) and expressed as a percentage of DPBS-treated healthy rats group.
Drugs
The sources of the compounds used were as follows: ACh and SNP were obtained from Sigma Aldrich, U46619 and SB203580 hydrochloride from Tocris, Biosciences (Bristol, UK), and ABH from Enzo Clinical Labs (Farmingdale, NY, USA). Stock solutions of U46619 were dissolved in pure ethanol and further diluted in distilled water. The final volume of ethanol never exceeded 0.01% in organ baths and did not affect smooth muscle tone in control experiments. All the other compounds were dissolved in distilled water.
Statistical analysis and data presentation
In the functional experiments, mechanical responses of the arteries were measured as force and expressed as active wall tension. Results are expressed as a percentage of the constrictor response to U46619 in each artery. Data are expressed as means ± SEM; n indicates the number of animals. EC50 was the concentration of agonist giving a half maximal response (Emax) and expressed as pEC50 (−log EC50). The differences between means were analysed using one-way anova followed by a Bonferroni post-test or Student's paired or unpaired t-test when appropriate. The level of significance was set at P < 0.05. All calculations were made using a standard software package (Prism 5.0, GraphPad, San Diego, CA, USA).
Results
General parameters
Blood glucose levels were 28.6 ± 1 mmol·L−1 after eight weeks of STZ injection and 4.2 ± 1 mmol·L−1 in the healthy control group (P < 0.001; n = 12 and n = 12 respectively). Body weight was significantly lower in the diabetic group (346 ± 16 g, n = 12) than in the healthy control group (567 ± 21 g, n = 12; P < 0.001). The normalized internal lumen diameters of SCA and LDA were smaller in diabetic animals, although only in the case of LDA was this change statistically different (Supporting Information Table S1). The diameter of MA was significantly larger in the diabetic group (Supporting Information Table S1). The standard contractions evoked by a high K+ solution in all arteries were significantly smaller in the diabetic than in the healthy group (Supporting Information Table S1). Daily i.p. injections of either SB203580 or DPBS did not affect either blood glucose or body weight in comparison with the untreated groups (data not shown).
p38 MAPK and arginase inhibition prevents diabetes-induced endothelial dysfunction in coronary and MA
ACh-induced relaxations were decreased in SCA but unchanged in LAD of diabetic animals compared with healthy animals (Figure 1A,B and Table 1). Arginase inhibition with ABH increased the maximal relaxations induced by ACh in SCA and slightly improved the sensitivity of the relaxation in LAD from diabetic animals (Figure 1D,F and Table 1).
Figure 1.
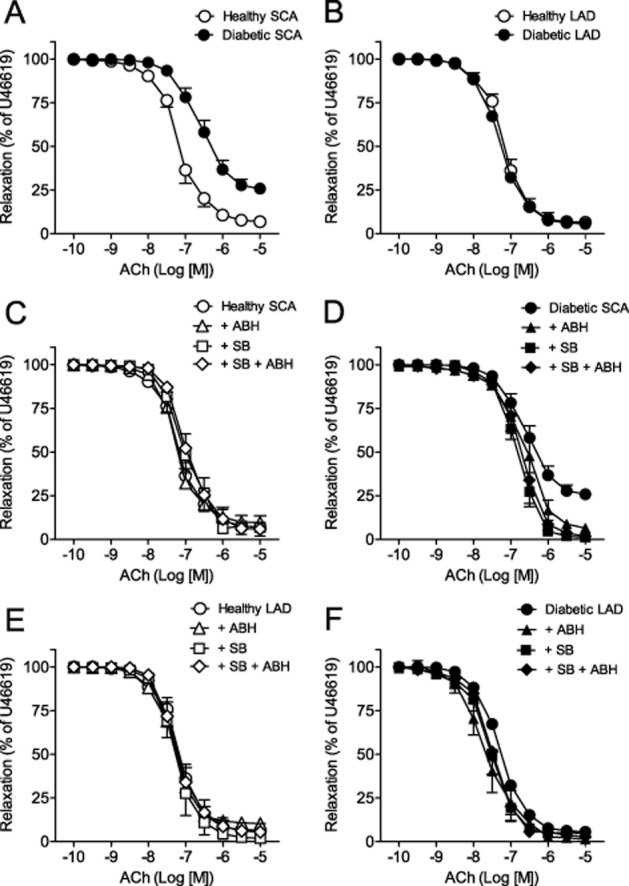
Arginase and p38 MAPK inhibition improves relaxation in diabetic coronary arteries. (A, B) Endothelium-dependent relaxation in response to ACh from healthy and diabetic SCA (A) and LAD (B). (C, D) Effect of ABH (+ABH), SB203580 (+SB) and SB203580 plus ABH (+SB+ABH) on ACh-induced relaxations of healthy and diabetic SCA (C, D) and LAD (E, F). Results (means and SEM) are expressed as a percentage of the contraction induced by U46619 (n = 5–13).
Table 1.
Effect of ABH and SB203580 (+SB) on the ACh-induced relaxation of septal and left anterior descending coronary arteries from healthy and diabetic rats
| pEC50 | Emax | n | pEC50 | Emax | n | ||
|---|---|---|---|---|---|---|---|
| Septal coronary artery (SCA) | |||||||
| H SCA | 7.10 ± 0.08 | 91 ± 2 | 12 | D SCA | 6.59 ± 0.14*** | 74 ± 3*** | 12 |
| + ABH | 7.16 ± 0.16 | 90 ± 4 | 6 | + ABH | 6.62 ± 0.16 | 93 ± 2b | 6 |
| + SB | 6.94 ± 0.14 | 92 ± 5 | 6 | + SB | 6.81 ± 0.10 | 99 ± 1c | 6 |
| SB + ABH | 6.99 ± 0.05 | 94 ± 3 | 7 | SB + ABH | 6.69 ± 0.14 | 98 ± 1b | 6 |
| Left anterior descending coronary artery (LAD) | |||||||
| H LAD | 7.19 ± 0.14 | 92 ± 3 | 12 | D LAD | 7.27 ± 0.03 | 94 ± 2 | 12 |
| + ABH | 7.29 ± 0.05 | 92 ± 4 | 6 | + ABH | 7.67 ± 0.16a | 97 ± 1 | 6 |
| + SB | 7.22 ± 0.14 | 98 ± 2 | 6 | + SB | 7.60 ± 0.11a | 95 ± 1 | 6 |
| SB + ABH | 7.18 ± 0.12 | 94 ± 5 | 8 | SB + ABH | 7.59 ± 0.14a | 97 ± 1 | 5 |
Values represent mean ± SEM. pEC50 is −logEC50. Emax is the maximal relaxation expressed as a percentage of U46619-induced pre-contraction. H SCA, healthy septal coronary artery; D SCA diabetic septal coronary artery; H LAD, healthy left anterior descending coronary artery; D LAD, diabetic left anterior descending coronary artery; ABH, 2(S)-amino-6-boronohexanoic acid; SB, SB203580. Significant differences were analysed using Student's paired t-test,
P < 0.001 D SCA versus H SCA; or one-way anova followed by a Bonferroni test for across-group comparisons;
P < 0.05,
P < 0.01 and
P < 0.001 versus D SCA, or versus D LAD.
In order to test whether p38 MAPK causes activation of arginase in coronary arteries of diabetic animals, arteries were incubated with SB203580 for 60 min in the myograph chamber. p38 MAPK inhibition improved ACh-induced relaxations in SCA and induced a very modest improvement in LAD from diabetic animals. Incubation with SB203580 plus ABH did not cause any additional effect either in SCA or in LAD (Figure 1D,F and Table 1). Arginase and p38 MAPK inhibition did not exert any significant effect on ACh-induced relaxations in LDA or SCA segments from healthy control animals (Figure 1C,E).
In diabetic MA, ACh-induced relaxations showed decreased sensitivity, but similar maximal response compared with healthy MA (Figure 2A and Table 2). Arginase inhibition with ABH improved ACh-induced relaxations whereas relaxations to SNP were not affected in diabetic MA (Figure 2C,E and Table 2). p38 MAPK inhibition increased sensitivity to ACh in MA from diabetic animals. Incubation with SB203580 plus ABH did not induce any further improvement in ACh-induced relaxations (Figure 2C and Table 2). In addition, neither ACh- nor SNP-induced relaxations were affected by arginase inhibition or by SB203580 treatment either alone or in combination in MA from healthy control animals (Figure 2B,D and Table 2). Moreover, these effects in vitro were further confirmed by studying MA from animals who received in vivo treatment with SB203580 for 2 weeks. Relaxations induced by ACh in MA were significantly enhanced in SB203580-treated rats by comparison to DPBS-treated rats with diabetes, whereas they were unaffected by SB203580 in healthy control rats (Figure 3A and Table 3). Moreover, arginase inhibition with ABH improved relaxation to ACh only in MA from DPBS-treated rats with diabetes while it did not induce any effect in arteries from SB203580-treated rats with diabetes, or in arteries from DPBS- and SB203580-treated healthy control rats (Figure 3B,C and Table 3).
Figure 2.
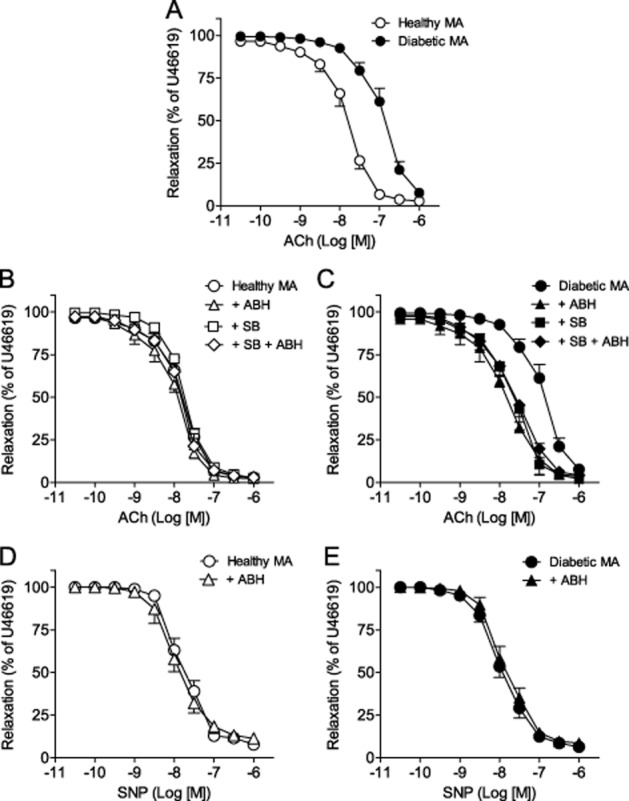
Arginase and p38 MAPK inhibition improve endothelium-dependent relaxation in diabetic MA. (A) Endothelium-dependent relaxation in response to ACh in MA from healthy and diabetic rats. (B, C) Effects of ABH (+ABH), SB203580 (+SB) and SB+ABH on ACh-induced relaxations of MA from healthy (B) and diabetic (C) animals. (D,E) Effect of ABH on relaxations induced by sodium nitroprusside (SNP) in healthy (D) and diabetic (E) endothelium-denuded MA. Results (means and SEM) are expressed as a percentage of the contraction induced by U46619 (n = 4–12).
Table 2.
Effects of ABH and SB203580 (+SB) on the ACh- and SNP-induced relaxations of MA from healthy and diabetic rats
| pEC50 | Emax | n | pEC50 | Emax | n | ||
|---|---|---|---|---|---|---|---|
| ACh | |||||||
| H MA | 7.79 ± 0.08 | 97 ± 1 | 9 | D MA | 6.91 ± 0.08*** | 96 ± 1 | 12 |
| + ABH | 8.09 ± 0.09 | 97 ± 2 | 4 | + ABH | 7.67 ± 0.22a | 97 ± 1 | 4 |
| + SB | 7.72 ± 0.10 | 99 ± 1 | 5 | + SB | 7.70 ± 0.17b | 97 ± 2 | 6 |
| SB + ABH | 7.8 ± 0.07 | 97 ± 1 | 5 | SB + ABH | 7.64 ± 0.09b | 96 ± 1 | 8 |
| SNP | |||||||
| H MA | 7.80 ± 0.10 | 91 ± 2 | 5 | D MA | 7.96 ± 0.11 | 94 ± 2 | 4 |
| + ABH | 7.96 ± 0.13 | 89 ± 2 | 5 | + ABH | 7.88 ± 0.06 | 91 ± 2 | 4 |
Values represent the mean ± SEM of the number n of individual arteries. pEC50 is −logEC50. Emax is the maximal relaxation expressed as a percentage of U46619-induced pre-contraction. H MA, healthy mesenteric artery; D MA, diabetic mesenteric artery; ABH, 2(S)-amino-6-boronohexanoic acid; SB, SB203580; significant differences were analysed using Student's paired t-test;
P < 0.001 H MA versus D MA; or one-way anova followed by Bonferroni test for multiple comparisons;
P < 0.01,
P < 0.001 versus D MA.
Figure 3.
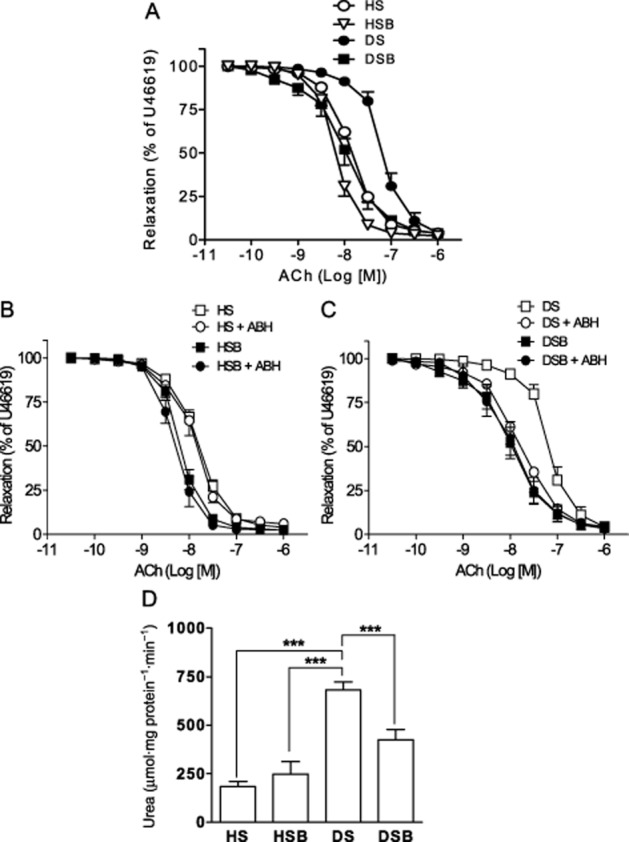
In vivo inhibition of p38 MAPK prevents arginase-induced endothelial dysfunction and decreases arginase activity of diabetic MA. (A) Endothelium-dependent relaxations in response to ACh in mesenteric segments from DPBS-treated healthy (HS), SB203580-treated healthy (HSB), DPBS-treated diabetic (DS) and SB203580-treated diabetic (DSB) MA. (B, C) Effects of ABH on ACh-induced relaxations in MA from HS and HSB rats (B) and from DS and DSB rats (C). Data are shown as means and SEM of 8–10 arteries. (D) Arginase activity of MA from HS, HSB, DS and DSB rats. Data are shown as means and SEM of 8–10 animals. Significant differences were analysed using Student's unpaired t-test or one-way anova followed by a Bonferroni test for multiple comparisons. ***P < 0.001.
Table 3.
Effects of in vivo inhibition of p38 MAPK on the ACh-induced relaxations of MA from healthy and diabetic rats in the presence or in the absence of ABH
| pEC50 | Emax | n | pEC50 | Emax | n | ||
|---|---|---|---|---|---|---|---|
| ACh | |||||||
| HS | 7.84 ± 0.04a | 96 ± 2 | 6 | DS | 7.10 ± 0.15 | 96 ± 2 | 9 |
| + ABH | 7.91 ± 0.12 | 94 ± 2 | 6 | + ABH | 7.72 ± 0.14** | 96 ± 2 | 9 |
| HSB | 8.19 ± 0.05a | 98 ± 1 | 6 | DSB | 8.02 ± 0.13a | 95 ± 2 | 7 |
| + ABH | 8.25 ± 0.09 | 98 ± 1 | 6 | + ABH | 7.98 ± 0.16 | 97 ± 1 | 7 |
Values represent the mean ± SEM. pEC50 is −logEC50. Emax is the maximal relaxation expressed as a percentage of U46619-induced pre-contraction. HS, DPBS-treated healthy mesenteric artery; DS, DPBS-treated diabetic mesenteric artery; HSB, SB203580-treated healthy mesenteric artery; DSB, SB203580-treated diabetic mesenteric artery; ABH, 2(S)-amino-6-boronohexanoic acid; SB, SB203580. Significant differences were analysed using Student's paired t-test;
P < 0.01 versus DS; or one-way anova followed by Bonferroni test for multiple comparisons;
P < 0.001 versus DS.
Activated p38 MAPK mediates the diabetes-induced increase in arginase activity in mesenteric arteries
Arginase activity was measured in MA in order to determine the role of p38 MAPK as an upstream activator of arginase in type 1 diabetes. A ∼ 2.5–fold increase in arginase activity was observed in MA from DPBS-treated diabetic rats compared with DPBS- and SB203580-treated healthy rats. This increase was attenuated after inhibition of p38 MAPK in vivo (Figure 3D).
p38 MAPK inhibition prevents diabetes-induced increased expression of arginase I in coronary and of arginase II in mesenteric arteries
Arginase I but not arginase II protein expression was increased in coronary arteries from DPBS-treated diabetic rats compared with DPBS- and SB203580-treated healthy rats. This increase in arginase I expression was abolished by SB203580 in rats with diabetes (Figure 4A,B). Moreover, the expression of phospho-p38 MAPK normalized to total p38 MAPK was increased ∼1.3-fold in coronary arteries from DPBS-treated rats with diabetes compared with DPBS- and SB203580-treated healthy rats, and this increase was abolished in diabetic animals treated with SB203580 (Figure 4C).
Figure 4.
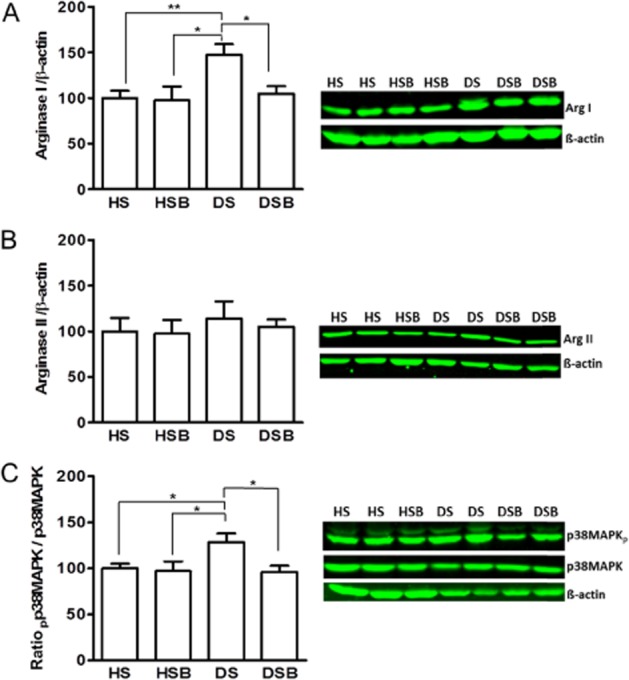
p38 MAPK activation increases arginase I expression in diabetic coronary arteries. Western blot analysis of arginase I (A), arginase II (B) and ratio of phospho-p38 MAPK/total p38MAPK (C) in coronary arteries of DPBS-treated healthy (HS), SB203580-treated healthy (HSB), DPBS-treated diabetic (DS) and SB203580-treated diabetic (DSB) rats. Data are shown as means and SEM from 8–10 animals and expressed as a percentage of HS group. Significant differences were analysed using one-way anova followed by a Bonferroni test. *P < 0.05; **P < 0.01.
In contrast to coronary arteries, the expression of arginase II was increased ∼2.5-fold in MA from DPBS-treated diabetic rats compared with DPBS- and SB203580-treated healthy rats (Figure 5A). Arginase I expression in MA was similar in the four groups of animals (Figure 5B). Two weeks of in vivo administration of the p38 MAPK inhibitor prevented the diabetes-induced increase in arginase II expression in MA (Figure 5A). In addition, the expression of phospho-p38 MAPK normalized to total p38 MAPK was significantly increased in MA from DPBS-treated diabetic rats compared with DPBS- and SB203580-treated healthy rats, and this increase was abolished by SB203580 treatment (Figure 5C).
Figure 5.
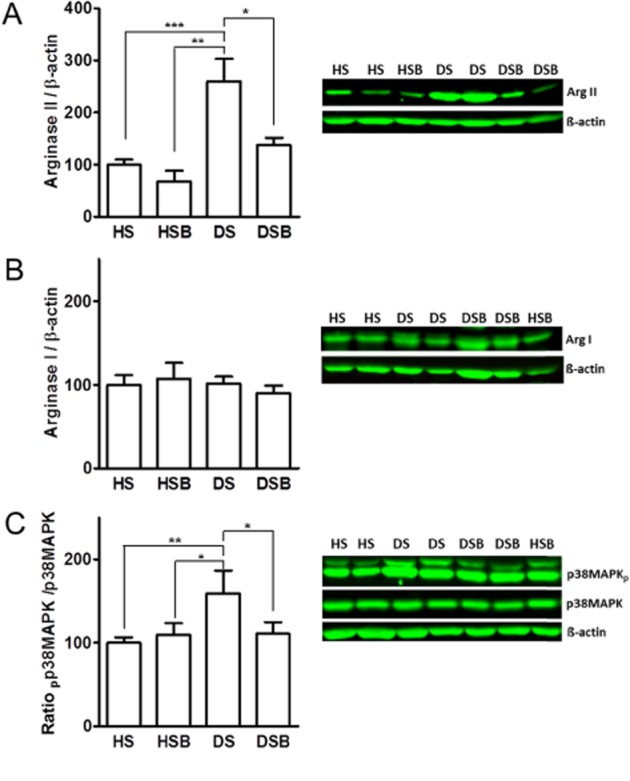
p38 MAPK activation increases arginase II expression in MA. Western blot analysis of arginase II (A), arginase I (B) and ratio of phospho-p38 MAPK/total p38 MAPK (C) in MA of DPBS-treated healthy (HS), SB203580-treated healthy (HSB), DPBS-treated diabetic (DS) and SB203580-treated diabetic (DSB). Data are shown as means ± SEM from 8–10 animals and expressed as a percentage of HS group. Significant differences were analysed using one-way anova followed by a Bonferroni test. *P < 0.05; **P < 0.01 and ***P < 0.001.
eNOS expression is increased in coronary arteries and decreased in MA from diabetic animals and SB203580 prevented these changes
In coronary samples, both total and phosphorylated (Ser1177) eNOS protein levels were higher in DPBS-treated diabetic rats than in DPBS- and SB203580-treated healthy rats. p38 MAPK inhibition decreased both eNOS and phospho-eNOS (Ser1177) expression in arteries from diabetic animals to values similar to those in the DPBS- and SB203580-treated healthy group (Figure 6A).
Figure 6.
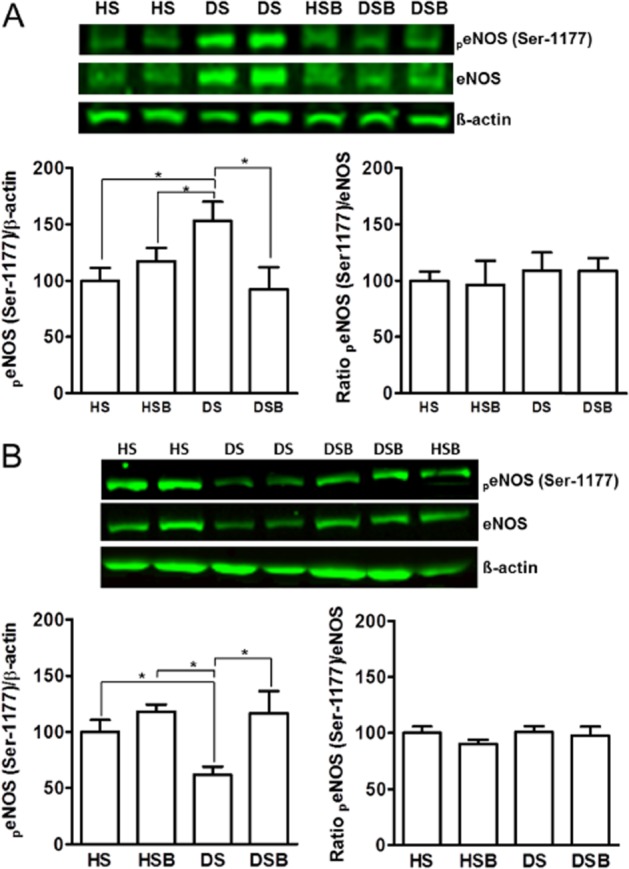
Effect of p38 MAPK inhibition on eNOS protein expression in mesenteric and coronary arteries. Western blot analysis of total eNOS and phospho-eNOS (Ser1177) from coronary (A) and mesenteric (B) arteries of DPBS-treated healthy (HS), SB203580-treated healthy (HSB), DPBS-treated diabetic (DS) and SB203580-treated diabetic (DSB) rats. Data are shown as means and SEM from 8–10 animals and expressed as a percentage of HS group. Significant differences were analysed using one-way anova followed by a Bonferroni test. *P < 0.05.
Total and phosphorylated (Ser1177) eNOS protein expression was reduced in MA from DPBS-treated diabetic rats compared with DPBS- and SB203580-treated healthy rats. Treatment with the inhibitor of p38 MAPK in diabetic animals prevented the reduction in both eNOS and phospho-eNOS (Ser1177) protein expression in MA (Figure 6B).
The phospho-eNOS (Ser1177) protein level normalized to the total eNOS protein was unchanged in both coronary and mesenteric samples (Figure 6). This means that the absolute amount of phospho-eNOS (Ser1177) was increased proportionally with the increased expression of the eNOS in coronary arteries and decreased proportionally with the decreased expression of the eNOS in MA (Okon et al., 2005).
Discussion
The major findings of the present study were (i) type 1 diabetes mellitus was associated with impaired endothelium-dependent relaxations in SCA and MA but not in LAD; (ii) inhibition of arginase and p38 MAPK prevented diabetes-induced endothelial dysfunction in SCA and MA; (iii) expression of arginase I, phospho-p38 MAPK and eNOS was increased in coronary arteries from type 1 diabetic rats; (iv) expression of arginase II, arginase activity and expression of phospho-p38 MAPK was increased and expression of eNOS was decreased in diabetic MA, all favouring endothelial dysfunction; and (v) inhibition of p38 MAPK in vivo reversed diabetes-induced up-regulation of arginase and development of endothelial dysfunction. Taken together, our results show that activation of p38 MAPK, by selectively up-regulating arginase I in coronary arteries and arginase II in MA, is an upstream mediator of diabetes-induced endothelial dysfunction.
There is emerging evidence suggesting that the up-regulation of arginase is a key factor contributing to the development of vascular dysfunction in diabetes mellitus by its reciprocal regulation of NO bioavailability (Bagi et al., 2013; Durante, 2013; Pernow and Jung, 2013). Accordingly, arginase inhibition has been shown to improve vascular function in experimental and clinical studies of diabetes (Romero et al., 2008; Beleznai et al., 2011; Grönros et al., 2011; Shemyakin et al., 2012). The underlying signalling mechanisms resulting in the up-regulation of arginase in diabetes remain poorly defined. Arginase I expression is stimulated by ROS and nitrogen species such as H2O2 in healthy coronary arteries (Thengchaisri et al., 2006) and peroxynitrite in cultured endothelial cells (Chandra et al., 2012), and these species have also been shown to activate p38 MAPK (Evans et al., 2002). It is in this context that p38 MAPK has been proposed as the upstream mediator of arginase II up-regulation in corpora cavernosa of angiotensin II-treated mice and diabetic mice (Toque et al., 2010; 2013,). Based on these observations, we hypothesized that p38 MAPK regulates coronary and peripheral vascular arginase expression and activity in diabetes. Accordingly, we observed that arginase I expression was up-regulated in coronary arteries from diabetic rats via a p38 MAPK-dependent mechanism. Furthermore, inhibition of arginase and p38 MAPK both improved endothelial function in SCA and caused a slight improvement in LAD. In MA, the expression of arginase II, but not arginase I, was increased by diabetes via a p38 MAPK-dependent mechanism. Arginase inhibition improved endothelium-dependent but not endothelium-independent relaxation and both in vivo and in vitro inhibition of p38 MAPK prevented arginase-induced endothelial dysfunction. Hence, we can infer that arginase is up-regulated via a signalling mechanism involving p38 MAPK and that this signalling cascade underlies the development of mesenteric and coronary endothelial dysfunction in type 1 diabetes mellitus.
An interesting finding in the present study is that regional differences appear to exist regarding the isoform of arginase that is stimulated, as p38 MAPK up-regulates arginase I in coronary arteries and arginase II in MA. Previous studies have resulted in inconclusive results regarding the isoform of arginase that is up-regulated in conditions of hyperglycaemia. Thus, some studies have demonstrated an up-regulation of arginase I (Romero et al., 2008; Beleznai et al., 2011), whereas others detected an increase in arginase II (Grönros et al., 2011) or both isoforms (Dhar et al., 2012). Our data show that the arginase isoform activated in diabetes varies between different vascular regions, that this activation is dependent on p38 MAPK activation and that both arginase I and II contribute to endothelial dysfunction.
The mechanism behind the actions of p38 MAPK on vascular arginase seems complex and has not been fully elucidated. Transcriptional regulation of arginase I in cultured endothelial cells in response to thrombin and angiotensin II has been demonstrated to occur through activator protein-1 (AP-1) consensus site, via a p38 MAPK-dependent activation of activating transcription factor 2 and c-Jun transcription factor (Zhu et al., 2010; Shatanawi et al., 2015). Furthermore, p38 MAPK was reported to cause the degradation of histone deacetylases (HDACs) in chondrocytes (Zhou et al., 2015) while activation of HDACs constrains arginase II transcription in cultured endothelial cells (Pandey et al., 2014b). On the other hand, the fact that in the present study, in vitro incubation with the p38 MAPK inhibitor SB203580 for 60 min restored endothelial function in diabetic arteries, suggests effects on arginase activity rather than on protein levels. These data are in agreement with the observations that short-term inhibition of p38 MAPK reduced arginase activity and enhanced ACh-induced relaxations of corpora cavernosa from diabetic mice (Toque et al., 2013). Arginase may be activated by the Rho/ROCK pathway (Ming et al., 2004) promoting translocation of the enzyme from the mitochondria to the cytosol (Pandey et al., 2014a). Furthermore, ROCK activation has been reported to be upstream of p38 MAPK in mediating diabetes-induced elevation of corpora cavernosa arginase activity and expression (Toque et al., 2013). Thus, p38 MAPK may increase arginase activity via non-transcriptional effects in diabetes but the exact mechanism behind this requires further consideration.
The mechanism underlying endothelial dysfunction induced by increased arginase involves impaired NO bioavailability due to a reduced NO production and/or increased NO inactivation by ROS (Pernow and Jung, 2013; Yang and Ming, 2013). Increased expression of arginase II and arginase activity is linked to the down-regulation of eNOS activity and expression in cavernosal tissue from diabetic patients (Bivalacqua et al., 2001) and from angiotensin II-treated mice (Toque et al., 2010). Moreover, inhibition of p38 MAPK prevented the arginase-induced alteration in cavernosal tissue relaxation of angiotensin-II-treated mice (Toque et al., 2010). In the present study, expression of active phospho-eNOS (Ser1177) was decreased while levels of arginase II were increased in diabetic MA. In vivo p38 MAPK inhibition restored phospho-eNOS expression and normalized arginase II levels. In addition, NO-dependent relaxation is decreased in MA from type 1 diabetic rats (Leo et al., 2011). Therefore, our results suggest that activated p38 MAPK, via increasing arginase II expression, decreases eNOS activity, leading to endothelial dysfunction in diabetic MA. Interestingly, eNOS expression has been reported to be increased in diabetic coronary arteries (Kajikuri et al., 2009), in diabetic myocardial tissue (Desrois et al., 2010; Pechánová et al., 2014) and in coronary arteries from pre-diabetic rats (Contreras et al., 2011). In the present study, both eNOS and arginase I expression were increased in diabetic coronary arteries and p38 MAPK inhibition normalized not only arginase I but also eNOS protein levels. Previous data indicate that p38 MAPK exerts dual, beneficial and detrimental, effects on coronary artery function, whereby global inhibition of this kinase in disease states can lead to detrimental effects (Weinbrenner et al., 1997; Evans et al., 2002; Fujimoto et al., 2004; Yun et al., 2009; Kassan et al., 2014; Martin et al., 2015). Therefore, such effects might explain the current findings. In the present study, p38 MAPK activation in coronary arteries seems to be the upstream mediator of the arginase I-induced endothelial dysfunction whereas this kinase could also exert a compensatory effect increasing eNOS expression as shown in aorta of diabetic female rats (Taguchi et al., 2012). Although the mechanism underlying the ability of p38 MAPK to differentially regulate eNOS expression in coronary versus mesenteric arteries in diabetes remains unclear, one might speculate whether activation of different p38 MAPK isoforms may result in different eNOS regulation.
Vasomotor dysfunction in diabetes has been repeatedly and consistently demonstrated in different vascular beds (Shi and Vanhoutte, 2009; Howangyin and Silvestre, 2014). However, contradictory results exist regarding the coronary circulation in humans with diabetes and in animal models showing impaired (Nitenberg et al., 1993; Koltai et al., 1997; Tawfik et al., 2006; Romero et al., 2008; Beleznai et al., 2011; Grönros et al., 2011), preserved (Sanz et al., 2003; Knudson et al., 2007; Villalba et al., 2009; Contreras et al., 2011; Climent et al., 2014) or even augmented (Gebremedhin et al., 1988; Szerafin et al., 2006) coronary endothelium-dependent dilatation. Coronary vessels, unlike vascular beds of the periphery, have been proposed as being capable of developing compensatory mechanisms, which aim to reduce the detrimental effects of diabetes (Szerafin et al., 2006; Bagi et al., 2009; Koller et al., 2013). In the present study, endothelium-dependent relaxation was decreased in diabetic MA, whereas different responses within the coronary arteries of diabetic rats were observed. Coronary relaxation in response to ACh was impaired in segments of the SCA while it was preserved in segments of the LAD. These results are in agreement with previous data demonstrating that endothelium-dependent NO-mediated relaxation in LAD is preserved (Sanz et al., 2003; Kajikuri et al., 2009; Villalba et al., 2009) while impaired in SCA from diabetic rats (Tawfik et al., 2006; Romero et al., 2008). However, to date, no comparative study exploring regional differences regarding endothelial function in both types of coronary arteries has been performed. Therefore, conclusions based on data obtained in one artery may not be applicable to the overall coronary circulation due to regional differences. Our data demonstrate that the presence and level of endothelial dysfunction is not uniform in all vessel types. Indeed, the occurrence of endothelial dysfunction not only differs between the peripheral circulation versus coronary circulation, as previously proposed (Szerafin et al., 2006; Bagi et al., 2009), but also between regions within the coronary circulation.
There are certain limitations with the present study that deserve consideration. It should be noted that the arginase inhibitors available are not selective for particular arginase isoforms. Hence, the functional role of arginase II in the development of vasomotor dysfunction in coronary arteries, as well as the role of arginase I in MA, cannot be entirely excluded in the present study. Furthermore, the slight improvement in endothelial function after inhibition of arginase and p38 MAPK observed in LAD, together with the preserved relaxation reported in this artery, suggests that unknown compensatory mechanism(s) contribute to enhanced endothelium-dependent relaxation despite arginase I up-regulation in this artery. The nature of this is at present unclear, but it is of interest that it was recently suggested that the production of hydrogen peroxide by eNOS induces endothelium-dependent relaxations in a mouse model of tetrahydrobiopterin deficiency (Chuaiphichai et al., 2014).
In conclusion, the present study demonstrates regional differences in arginase isoforms contributing to endothelial dysfunction in an experimental model of type 1 diabetes mellitus. p38 MAPK mediates endothelial dysfunction via selective up-regulation of arginase I in coronary arteries and arginase II in MA. Vascular arginase might therefore represent a promising therapeutic target for the treatment of vascular dysfunction in diabetes. Given the differential expression of arginase I and II between the coronary and non-coronary circulation, the development of potent isoform-selective arginase inhibitors is of particular interest.
Acknowledgments
We thank Marita Wallin for excellent technical assistance. This study was supported by Swedish Research Council (10857), the Swedish Heart and Lung Foundation, the Stockholm County Council (ALF), Karolinska Institutet/Stockholm County Council Strategic Cardiovascular Programme, King Gustav V and Queen Victoria Foundation, Diabetes Wellness Foundation and Novo Nordisk Foundation. B. C. was supported by a grant from the Gästforskarstipendier Program of the Wenner-Gren Foundation, Stockholm, Sweden.
Glossary
- ABH
arginase inhibitor 2(S)-amino-6-boronohexanoic acid
- eNOS
endothelial NOS
- LAD
left anterior descending coronary artery
- MA
mesenteric arteries
- ROS
reactive oxygen species
- SB203580
4-[5-(4-fluorophenyl)-2-[4-(methylsulphonyl)phenyl]-1H-imidazol-4-yl]pyridine hydrochloride
- SCA
septal coronary
- SNP
sodium nitroprusside
- STZ
streptozotocin
- U46619
9,11-dideoxy-9α,11α-methanoepoxy PGF2α
Author contributions
B. C., A. K. and Y. T. performed the research. B. C. and J. P. designed the research study. A. K., Y. T., B. C. and J. P. contributed essential reagents or tools. B. C., A. K., Y. T. and J. P. analysed the data. B. C., A. K., Y. T. and J. P. wrote the manuscript.
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.13242
Table S1 Normalized internal lumen diameters and standard contractions evoked by a high K+ solution of septal coronary, left anterior descending coronary and mesenteric arterial segments from healthy and diabetic rats.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avogaro A, Albiero M, Menegazzo L, Kreutzenberg S, Fadini G. Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care. 2011;34:S285–S290. doi: 10.2337/dc11-s239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagi Z, Feher A, Beleznai T. Preserved coronary arteriolar dilatation in patients with type 2 diabetes mellitus: implications for reactive oxygen species. Pharmacol Rep. 2009;61:99–104. doi: 10.1016/s1734-1140(09)70011-8. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Feher A, Dou H, Broskova Z. Selective up-regulation of arginase-1 in coronary arteries of diabetic patients. Front Immunol. 2013;4:293. doi: 10.3389/fimmu.2013.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beleznai T, Feher A, Spielvogel D, Lansman S, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol. 2011;300:777–783. doi: 10.1152/ajpheart.00831.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivalacqua T, Hellstrom W, Kadowitz P, Champion H. Increased expression of arginase II in human diabetic corpus cavernosum: in diabetic-associated erectile dysfunction. Biochem Biophys Res Commun. 2001;283:923–927. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]
- Chandra S, Romero M, Shatanawi A, Alkilany M, Caldwell R, Caldwell R. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol. 2012;165:506–519. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheriyan J, Webb A, Sarov-Blat L, Elkhawad M, Wallace S, Mäki-Petäjä K, et al. Inhibition of p38 mitogen-activated protein kinase improves nitric oxide-mediated vasodilatation and reduces inflammation in hypercholesterolemia. Circulation. 2011;123:515–523. doi: 10.1161/CIRCULATIONAHA.110.971986. [DOI] [PubMed] [Google Scholar]
- Chuaiphichai S, McNeill E, Douglas G, Crabtree M, Bendall J, Hale A, et al. Cell-autonomous role of endothelial GTP cyclohydrolase 1 and tetrahydrobiopterin in blood pressure regulation. Hypertension. 2014;64:530–540. doi: 10.1161/HYPERTENSIONAHA.114.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent B, Moreno L, Martínez P, Contreras C, Sánchez A, Pérez-Vizcaíno F, et al. Upregulation of SK3 and IK1 channels contributes to the enhanced endothelial calcium signaling and the preserved coronary relaxation in obese Zucker rats. PLoS ONE. 2014;9:e109432. doi: 10.1371/journal.pone.0109432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras C, Sánchez A, García-Sacristán A, Martínez M, Andriantsitohaina R, Prieto D. Preserved insulin vasorelaxation and up-regulation of the Akt/eNOS pathway in coronary arteries from insulin resistant obese Zucker rats. Atherosclerosis. 2011;217:331–339. doi: 10.1016/j.atherosclerosis.2011.03.036. [DOI] [PubMed] [Google Scholar]
- De Vriese A, Verbeuren T, Van de Voorde J, Lameire N, Vanhoutte P. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrois M, Clarke K, Lan C, Dalmasso C, Cole M, Portha B, et al. Upregulation of eNOS and unchanged energy metabolism in increased susceptibility of the aging type 2 diabetic GK rat heart to ischemic injury. Am J Physiol Heart Circ Physiol. 2010;299:H1679–H1686. doi: 10.1152/ajpheart.00998.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar I, Dhar A, Wu L, Desai K. Arginine attenuates methylglyoxal- and high glucose-induced endothelial dysfunction and oxidative stress by an endothelial nitric-oxide synthase-independent mechanism. J Pharmacol Exp Ther. 2012;342:196–204. doi: 10.1124/jpet.112.192112. [DOI] [PubMed] [Google Scholar]
- Durante W. Role of arginase in vessel wall remodeling. Front Immunol. 2013;4:111. doi: 10.3389/fimmu.2013.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J, Goldfine I, Maddux B, Grodsky G. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, et al. Carbon monoxide protects against cardiac ischemia–reperfusion injury in vivo via MAPK and Akt–eNOS pathways. Arterioscler Thromb Vasc Biol. 2004;24:1848–1853. doi: 10.1161/01.ATV.0000142364.85911.0e. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Kaldunski M, Jacobs E, Harder D, Roman R. Influence of experimental diabetes on the mechanical responses of canine coronary arteries: role of endothelium. Cardiovasc Res. 1988;22:537–544. doi: 10.1093/cvr/22.8.537. [DOI] [PubMed] [Google Scholar]
- Grönros J, Jung C, Lundberg J, Cerrato R, Östenson C, Pernow J. Arginase inhibition restores in vivo coronary microvascular function in type 2 diabetic rats. Am J Physiol Heart Circ Physiol. 2011;300:H1174–H1181. doi: 10.1152/ajpheart.00560.2010. [DOI] [PubMed] [Google Scholar]
- Howangyin K, Silvestre J. Diabetes mellitus and ischemic diseases: molecular mechanisms of vascular repair dysfunction. Arterioscler Thromb Vasc Biol. 2014;34:1126–1135. doi: 10.1161/ATVBAHA.114.303090. [DOI] [PubMed] [Google Scholar]
- Kajikuri J, Watanabe Y, Ito Y, Ito R, Yamamoto T, Itoh T. Characteristic changes in coronary artery at the early hyperglycaemic stage in a rat type 2 diabetes model and the effects of pravastatin. Br J Pharmacol. 2009;158:621–632. doi: 10.1111/j.1476-5381.2009.00348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassan M, Choi S, Galán M, Lee Y, Trebak M, Matrougui K. Enhanced p22phox expression impairs vascular function through p38 and ERK1/2 MAP kinase-dependent mechanisms in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2014;306:H972–H980. doi: 10.1152/ajpheart.00872.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill I, Emerson M, Altman D. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss A, Tratsiakovich Y, Gonon A, Fedotovskaya O, Lanner J, Andersson D, et al. The role of arginase and Rho Kinase in cardioprotection from remote ischemic perconditioning in non-diabetic and diabetic rat in vivo. PLoS ONE. 2014;9:e104731. doi: 10.1371/journal.pone.0104731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson J, Dincer U, Bratz I, Sturek M, Dick G, Tune J. Mechanisms of coronary dysfunction in obesity and insulin resistance. Microcirculation. 2007;14:317–338. doi: 10.1080/10739680701282887. [DOI] [PubMed] [Google Scholar]
- Koller A, Balasko M, Bagi Z. Endothelial regulation of coronary microcirculation in health and cardiometabolic diseases. Intern Emerg Med. 2013;8:1–6. doi: 10.1007/s11739-013-0910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltai M, Hadhazy P, Posa I, Kocsis E, Wickler G, Rosen P, et al. Characteristics of coronary endothelial dysfunction in experimental diabetes. Cardiovasc Res. 1997;34:157–163. doi: 10.1016/s0008-6363(97)00050-3. [DOI] [PubMed] [Google Scholar]
- Komers R, Schutzer W, Xue H, Oyama T, Lindsley J, Anderson S. Effects of p38 mitogen-activated protein kinase inhibition on blood pressure, renal hemodynamics, and renal vascular reactivity in normal and diabetic rats. Transl Res. 2007;150:343–349. doi: 10.1016/j.trsl.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Kövamees O, Shemyakin A, Pernow J. Effect of arginase inhibition on ischemia-reperfusion injury in patients with coronary artery disease with and without diabetes mellitus. PLoS ONE. 2014;9:e103260. doi: 10.1371/journal.pone.0103260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo C, Hart J, Woodman O. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br J Pharmacol. 2011;162:365–377. doi: 10.1111/j.1476-5381.2010.01023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Bassi R, Marber M. p38 mitogen-activated protein kinase in cardioprotection – are we there yet? Br J Pharmacol. 2015;172:2101–2113. doi: 10.1111/bph.12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X, Barandier C, Viswambharan H, Kwak B, Mach F, Mazzolai L, et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- Nitenberg A, Valensi P, Sachs R, Dali M, Aptecar E, Attali J. Impairment of coronary vascular reserve in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes. 1993;42:18–21. doi: 10.2337/diab.42.7.1017. [DOI] [PubMed] [Google Scholar]
- Okon E, Chung A, Rauniyar P, Padilla E, Tejerina T, McManus B, et al. Compromised arterial function in human type 2 diabetic patients. Diabetes. 2005;54:2415–2423. doi: 10.2337/diabetes.54.8.2415. [DOI] [PubMed] [Google Scholar]
- Pandey D, Bhunia A, Oh Y, Chang F, Bergman Y, Kim J, et al. OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ Res. 2014a;115:450–459. doi: 10.1161/CIRCRESAHA.115.304262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey D, Sikka G, Bergman Y, Kim J, Ryoo S, Romer L, et al. Transcriptional regulation of endothelial arginase 2 by histone deacetylase 2. Arterioscler Thromb Vasc Biol. 2014b;34:1556–1566. doi: 10.1161/ATVBAHA.114.303685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paneni F, Beckman J, Creager M, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013;34:2436–2443. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C, Rojas M, Narayanan S, Zhang W, Xu Z, Lemtalsi T, et al. Arginase as a mediator of diabetic retinopathy. Front Immunol. 2013;4:173. doi: 10.3389/fimmu.2013.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechánová O, Varga Z, Cebová M, Giricz Z, Pacher P, Ferdinandy P. Cardiac nitric oxide signalling in metabolic syndrome. Br J Pharmacol. 2014;172:1415–1433. doi: 10.1111/bph.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res. 2013;98:334–343. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- Romero M, Platt D, Tawfik H, Labazi M, El-Remessy A, Bartoli M, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res. 2008;102:95–102. doi: 10.1161/CIRCRESAHA.107.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero M, Iddings J, Platt D, Ali M, Cederbaum S, Stepp D, et al. Diabetes-induced vascular dysfunction involves arginase I. Am J Physiol Heart Circ Physiol. 2012;302:H159–H166. doi: 10.1152/ajpheart.00774.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago E, Contreras C, García-Sacristán A, Sánchez A, Rivera L, Climent B, et al. Signaling pathways involved in the H2O2-induced vasoconstriction of rat coronary arteries. Free Radic Biol Med. 2013;60:136–146. doi: 10.1016/j.freeradbiomed.2013.02.014. [DOI] [PubMed] [Google Scholar]
- Sanz E, Fernández N, Monge L, Martínez M, Climent B, Diéguez G, et al. Effects of diabetes on the vascular response to nitric oxide and constrictor prostanoids: gender and regional differences. Life Sci. 2003;72:1537–1547. doi: 10.1016/s0024-3205(02)02444-x. [DOI] [PubMed] [Google Scholar]
- Shatanawi A, Lemtalsi T, Yao L, Patel C, Caldwell R, Caldwell R. Angiotensin II limits NO production by upregulating arginase through a p38 MAPK–ATF-2 pathway. Eur J Pharmacol. 2015;746:106–114. doi: 10.1016/j.ejphar.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemyakin A, Kövamees O, Rafnsson A, Böhm F, Svenarud P, Settergren M, et al. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation. 2012;126:2943–2950. doi: 10.1161/CIRCULATIONAHA.112.140335. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vanhoutte P. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. J Diabetes. 2009;1:151–162. doi: 10.1111/j.1753-0407.2009.00030.x. [DOI] [PubMed] [Google Scholar]
- Szerafin T, Erdei N, Fülöp T, Pasztor E, Edes I, Koller A, et al. Increased cyclooxygenase-2 expression and prostaglandin-mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ Res. 2006;99:e12–e17. doi: 10.1161/01.RES.0000241051.83067.62. [DOI] [PubMed] [Google Scholar]
- Taguchi K, Morishige A, Matsumoto T, Kamata K, Kobayashi T. Enhanced estradiol-induced vasorelaxation in aortas from type 2 diabetic mice may reflect a compensatory role of p38 MAPK-mediated eNOS activation. Pflugers Arch. 2012;464:205–215. doi: 10.1007/s00424-012-1131-x. [DOI] [PubMed] [Google Scholar]
- Tawfik H, El-remessy A, Matragoon S, Ma G, Caldwell R, Caldwell R, et al. Simvastatin improves diabetes-induced coronary endothelial dysfunction. J Pharmacol Exp Ther. 2006;319:386–395. doi: 10.1124/jpet.106.106823. [DOI] [PubMed] [Google Scholar]
- Thengchaisri N, Hein T, Wang W, Xu X, Li Z, Fossum T, et al. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- Toque H, Romero M, Tostes R, Shatanawi A, Chandra S, Carneiro Z, et al. p38 mitogen-activated protein kinase (MAPK) increases arginase activity and contributes to endothelial dysfunction in corpora cavernosa from angiotensin-II treated mice. J Sex Med. 2010;7:3857–3867. doi: 10.1111/j.1743-6109.2010.01996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toque H, Nunes K, Yao L, Liao J, Webb R, Caldwell R, et al. Activated Rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: possible involvement of p38 MAPK activation. J Sex Med. 2013;10:1502–1515. doi: 10.1111/jsm.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba N, Martínez P, Bríones A, Sánchez A, Salaíces M, García-Sacristán A, et al. Differential structural and functional changes in penile and coronary arteries from obese Zucker rats. Am J Physiol Heart Circ Physiol. 2009;297:H696–H707. doi: 10.1152/ajpheart.01308.2008. [DOI] [PubMed] [Google Scholar]
- Weinbrenner C, Liu G, Cohen M, Downey J. Phosphorylation of tyrosine 182 of p38 mitogen-activated protein kinase correlates with the protection of preconditioning in the rabbit heart. J Mol Cell Cardiol. 1997;29:2383–2391. doi: 10.1006/jmcc.1997.0473. [DOI] [PubMed] [Google Scholar]
- Widder J, Behr T, Fraccarollo D, Hu K, Galuppo P, Tas P, et al. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc Res. 2004;63:161–167. doi: 10.1016/j.cardiores.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Wu Z, Xiong Y, Gajanayake T, Ming X, Yang Z. p38 mitogen-activated protein kinase is required for glucosamine-induced endothelial nitric oxide synthase uncoupling and plasminogen-activator inhibitor expression. Circ J. 2012;76:2015–2022. doi: 10.1253/circj.cj-12-0016. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ming X. Arginase: the emerging therapeutic target for vascular oxidative stress and inflammation. Front Immunol. 2013;4:149. doi: 10.3389/fimmu.2013.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Chandra S, Toque H, Bhatta A, Rojas M, Caldwell R, et al. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc Res. 2013;97:509–519. doi: 10.1093/cvr/cvs371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Rocic P, Pung Y, Belmadani S, Carrao A, Ohanyan V, et al. Redox-dependent mechanisms in coronary collateral growth: the redox window hypothesis. Antioxid Redox Signal. 2009;11:1961–1974. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Li P, Chen Q, Wei X, Zhao T, Wang Z, et al. Mitogen-activated protein kinase p38 induces HDAC4 degradation in hypertrophic chondrocytes. Biochim Biophys Acta. 2015;1853:370–376. doi: 10.1016/j.bbamcr.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Chandrasekharan U, Bandyopadhyay S, Morris S, DiCorleto P, Kashyap V. Thrombin induces endothelial arginase through AP-1 activation. Am J Physiol Cell Physiol. 2010;298:C952–C960. doi: 10.1152/ajpcell.00466.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Normalized internal lumen diameters and standard contractions evoked by a high K+ solution of septal coronary, left anterior descending coronary and mesenteric arterial segments from healthy and diabetic rats.