

Abstract
This unapologetically subjective essay recalls the Torpedo Cl– channel in the years when it had neither a molecular identity nor proper name (ClC-0), and membership in a large superfamily. I discuss the circumstances surrounding its discovery and subsequent research through the 1980s that revealed its unusual molecular architecture and other strange mechanistic characteristics.
The CLC superfamily of anion-transporting proteins is nearly ubiquitous among organisms and the cell-types within them. As highlighted in this special issue of The Journal of Physiology, these membrane proteins operate for an immense range of biological purposes calling for gated (or constitutive) conductance (or H+-coupled flux) of Cl–, NO3–, F−, and, if recent experience is a faithful guide (Baker et al. 2012; Brammer et al. 2014), other anions as yet undocumented. I wonder, for instance, if any CLC out there is specifically purposed to handle HCO3– in some as yet unstudied epithelium, or if the Br– ions used for post-translational modification of tryptophan residues in certain snail venom peptides hitch a ride into the venom-sac secretory cells on a conventional CLC Cl– channel. We’ve known for over a decade that the superfamily is split into two mechanistically antipodal subtypes: thermodynamically passive anion channels and energy-utilizing H+-coupled antiporters (Accardi & Miller, 2004; Picollo & Pusch, 2005; Scheel et al. 2005). Although all bacterial CLCs so far studied have turned out to be antiporters, the two subclasses are roughly equally populated in vertebrate organisms, channels residing in plasma membranes and antiporters in acidifying cytoplasmic vesicles such as endosomes, lysosomes and synaptic vesicles.
The CLC superfamily offers problems to engage scientists from diverse disciplines. The family’s remarkable division into two distinct subtypes gives evolutionary biologists opportunities to probe the provenance of and linkages between channels and transporters. These proteins provide translational researchers with insights into various human physiologies since several of the nine human CLC genes have been found to be disrupted in certain diseases. In addition the three CLC antiporter homologues with high-resolution crystal structures (no channels yet!) allow close-up molecular investigation of ion transport mechanisms (Dutzler et al. 2002; Feng et al. 2010; Jayaram et al. 2011).
But it was not always thus. The CLC family had humble beginnings. As happens so often in research, the experiments that led to ClC-0, the family’s founding member, were designed for a completely different purpose: to record nicotinic acetylcholine receptor (AChR) channels in planar lipid bilayers. I had begun my independent career in 1976 developing and refining a method for reconstituting ion channels by fusing native membrane fragments into these electrically accessible ‘model membranes,’ and I’d had my first publishable success with a K+-selective cation channel from mammalian sarcoplasmic reticulum (SR) (Miller, 1978). In those days, the only ion channel thought to reside in SR was the then-mysterious ‘Ca2+ release channel’ (now the ryanodine receptor), so most of my established colleagues considered the ‘SR K+ channel’ that I was working on simply an artifact of this ‘non-physiological’ planar bilayer system. In those days, this journal explicitly prohibited papers that employed such systems as outrages to proper physiology – or so the rumour-mill had it – so my manuscripts went mainly to its less persnickety American counterpart, J Gen Physiol. My colleagues’ skepticism was a welcome boon to me, since it provided elbowroom to work without much competition during my career’s launch-phase, but I nevertheless did worry that this new technique had bagged a channel that wasn’t supposed to be present in its source-membrane, while failing to observe the Ca2+ channels that were certainly present there.
So in 1978, after settling into my new lab at Brandeis, I began to look around for a second ion channel to reconstitute. The AChR, a cation-selective channel from the electric ray Torpedo californica was the obvious choice. The electric organ of this fish is essentially a 50 V, high-current battery fashioned from muscle end-plates gone wild – stacks of coin-shaped cells in which one side of the coin is densely packed with AChR in its plasma membrane, and the other is analogous to a resting skeletal muscle membrane, specially loaded with a high density of Na+/K+-ATPase pumps to sustain the ion gradients as the fish zaps its prey. Torpedo electroplax membrane vesicles are thus an exceedingly abundant biochemical source of AChRs (Weill et al. 1974), and so it seemed to me that recording AChR channels by this method would be a can’t-fail positive control that would certify the planar bilayer system as physiologically kosher. Accordingly I had a fish flown in, first-class, from Venice Beach, California, immediately dissected the electric organ to prepare membrane vesicles, and added these to planar bilayers under fusion conditions. It was exciting to see in the very first experiments packages of conductance appearing in discrete insertion events, and then depressing to realize that these currents were completely unresponsive to AChR agonists or antagonists. Worse, a NaCl gradient revealed the current to be ideally selective to anions! I’d again stumbled upon a channel unknown in its source membrane, and had failed to observe the channel known to be there. We now know that this ‘Torpedo Cl– channel’ lives in the non-innervated-face membrane of the electrocyte along with the Na+ pumps and that it provides the high voltage and low internal resistance that the electric organ needs to electrocute prey.
What to do in this circumstance – continue searching for a known ion channel to dispel the widespread view of the planar bilayer system as a reliable artifact-generator? I was tempted to keep trying for AChR channels, but the properties of this Cl– conductance were intriguing in themselves, and their novelty was appealing. The conductance showed an unfamiliar ‘reversed’ voltage dependence that slowly turned off with depolarization, a strong Cl– selectivity and bell-shaped pH dependence, and, most exciting of all in those days before gigaseal patch-clamping, single-channel fluctuations slow enough to record (Fig.1) on chart-paper with the crude home-built amplifier I was using. (I was probably the only electrophysiologist in those days with recordings low-pass filtered at 500 mHz.) Mike White, who had just joined the lab as my third graduate student, got to work characterizing this channel (White & Miller, 1979). We never did manage to record AChR channels, and the Torpedo Cl– channel remained a back-burner project in the lab until my first sabbatical, when everything changed.
Figure 1.
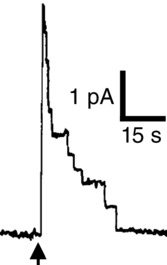
Torpedo vesicle channel fusion event (arrow) at +50 mV
This vesicle contained approximately 10 channels open at the moment of fusion into the bilayer, which then proceeded to inactivate upon fusion into the planar bilayer. Adapted from White (1979).
In 1981 my wife, a then-unemployed gypsy-scholar of Russian literature, was offered a 1 year job at Cornell University, and since my own sabbatical was coming up, we moved to Ithaca, New York for the academic year 1981–82. Efraim Racker, my postdoc advisor 5 years before, gave me a home in his lab for this visit. I mostly puttered around for the first few months, writing up some papers on SR K+ channels using an amazing new invention, a ‘word-processor’ that eliminated the tedium of retyping pages and the mess of White-out fluid on my hands and shirts, playing around with giant liposomes in Watt Webb’s lab with his graduate student David Tank (Tank et al. 1982), and being entirely useless for the project on α-adrenergic receptors that Ef had suggested I might like to join for the duration. The electrophysiological breakthrough of gigaseal patch-recording had just been published (Hamill et al. 1981) by Neher and Sakmann’s group at Göttingen, with much justifiable fanfare. That method depended critically on recent time-response improvements of the high-sensitivity amplifiers necessary for single-channel experiments. I spent some time that fall in Ithaca building a primitive version of Fred Sigworth’s fast patch amplifier and installing it into the planar bilayer setup I’d brought to Cornell, but I’d only diddled around optimizing the electronics and had not used it for any actual experiments.
I probably would have continued to shilly-shally in this way for the rest of the sabbatical if I had not received an invitation from Richard Keynes to speak at a Royal Society meeting on anion transport in the approaching spring. I was amazed that anyone had noticed my two or three papers on this obscure fish channel, and it was thrilling to imagine myself delivering a lecture at the birthplace of modern scientific discourse. But what to talk about? I did not want to throw together some sort of ‘review’ by rehashing the few published findings on the channel’s conduction properties and its mystifying voltage-dependent gating; what more could be said beyond the observations themselves? So I decided to speak about some new, unpublished observations for no reason other than personal vanity: a terror of appearing boring before that august body in London. But there was a problem – I didn’t have any new results. I did have a slight inkling, though, that something worth recording might be lurking in the millisecond time domain; the single channels routinely recorded on chart paper for the past 2 years were observable as clean 20 pS fluctuations only at depolarized voltages such as +50 mV, as in Fig.1. But switching to −100 mV, a physiologically familiar voltage for a channel in a muscle-like cell, would evoke only garbage: noisy, uninterpretable hash – one of those dirty little secrets that shrinks from the light of publication. The pessimist might conclude from this that the channel was electrophysiologically intractable at such voltages, while to the optimist, some sort of interesting voltage-dependent gating might be occurring, too fast for chart paper to capture.
I confess to an instinctive pessimism about (among other things) discovering interesting results, but the looming Royal Society meeting propelled me to try out the goosed-up bilayer system on Torpedo vesicles. Luckily, I didn’t have to order a fish or do any preps, since George Hess, a Cornell biochemist, was studying AChRs and had a nearby freezer full of electroplax vesicles. As soon as I could beg the Applied Physics Department for an oscilloscope (something Racker had declined to buy me when I was his postdoc, and for which his lab had no use), I set up to look for single Torpedo Cl– channels in the only quiet area in Ef’s lab – the musty radioactivity room. The experiment worked right away. Now, over three decades later, the exhilaration of first seeing the channel at high time resolution remains vivid in memory. A familiar single channel appeared in the bilayer under the usual fusion conditions at +50 mV, and just as on chart paper, the 20 pS channel was clean and quiet before it closed after a few seconds. Then, switching to −90 mV, I was stunned by an amazing sight: the channel opened and closed in a stochastic, millisecond-timescale dance among three well-defined ‘substates,’ which at once named themselves ‘Up,’ ‘Middle’ and ‘Down’ (Fig.2). Substate M looked to be about 10 pS, half the conductance of U, and D’s current was close to zero, so a picture of a channel built like a double-barrelled shotgun popped into my head without any cognitive intervention on my part.
Figure 2.
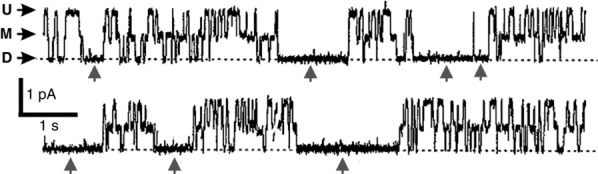
Double-barrelled gating of single Torpedo channel at −90 mV
Substates are marked and inactivated intervals are indicated by arrows. Scale bars: 1 pA, 1 s. Adapted from Richard & Miller (1990).
This single Cl– channel looked like two channels gating independently in parallel, but the obvious possibility that two separate channels had inserted into the bilayer was nullified by an additional feature of the record: a long-lived non-conducting state persisting for seconds that separated ‘bursts’ of the three-substate dance. If these substates represented two separate channel proteins in the bilayer, it would be impossibly unlikely that both of them would close and open simultaneously. Instead, the two presumed Cl– channel pores had to be tightly coupled together, by whatever process caused these long-lived non-conducting intervals, a process I called ‘inactivation,’ since it became increasingly prominent at depolarized voltage. Simple logic thus located these two pores, each with its own fast gate, within the same protein complex. Upon hyperpolarization, the M and D states within the bursts became more likely, while at depolarized voltages, the fluctuations tended increasingly towards the U state; at voltages positive to about −30 mV, the burst became fully dominated by the U state, thus explaining the clean, 20 pS chart-paper-accessible channels at +50 mV.
The very first glimpses of this channel at higher time resolution, then, revealed a completely unexpected picture: a double-barrelled, voltage-gated channel in which each of the two parallel pores, gating independently on the millisecond timescale, were driven open by depolarization, while some sort of slow inactivation process acting obligatorily on both pores would kick in at depolarized voltages, according to the scheme of Fig.3. It was clear that there would be many fun experiments to do over the next few years, but the Royal Society manuscript was due in just a few weeks. So some simple tests would be needed to refute or support this double-barrelled channel picture. That would be the story to tell at the meeting, however it turned out.
Figure 3.
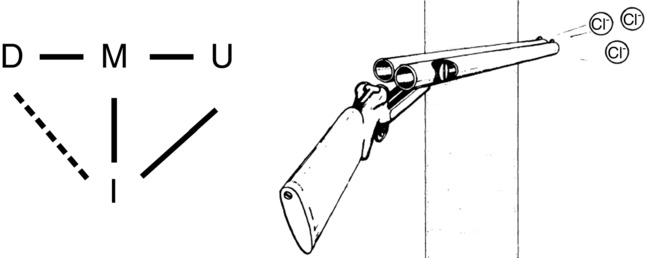
Double-barrelled gating model
Left: observable states of double-barrelled gating, with observable transitions shown by continuous lines and silent transition by dashed line. Right: diagram adapted from Miller (1982).
If the three-substate dance really represented two identical pores opening and closing independently, each between a single open and a single closed state, four classes of quantitative demands would have to be satisfied. First, the M-state current amplitude would have to be precisely half that of the U state, and the D-state current would have to be non-conducting. Second, the 3-substate dance within the bursts would have to follow a binomial distribution, such that the overall open probability p, determined directly by integrating the current trace, would predict, without any adjustable parameters, the frequencies fi of observing each of the three substates within the bursts:
| 1 |
Third, all transitions between D and U would have to proceed through M; D–U transitions would be strictly forbidden, after accounting for missed events. Fourth, the transition kinetics within the burst would have to obey quantitatively prescribed behaviour: that the dwell times in each substate should be exponentially distributed, with time constants τi given in terms of the individual pore’s rate constants of opening α and closing β:
| 2 |
and
| 3 |
Thus, measurement of only two parameters – p and any one of the substate time constants – would completely determine five additional measurable quantities: all three substate frequencies and the two other time constants.
It was exciting that this simple model made such a powerful set of demands that could be quantitatively scrutinized by single-channel analysis. Despite the aesthetic appeal of the double-barrelled shotgun, I thought it unlikely that the model would pass this battery of severe tests. But it did, without any shoehorning whatsoever. Over those frantic weeks at Cornell, I accumulated many single-channel records at voltages in the range −100 to −30 mV, ran them out on a commandeered tape recorder, played back the tape at slow speed into a chart recorder, measured the substate dwell-times using a plastic ruler as A-to-D converter, and calculated the time-distributions. The results still strike me as astonishing: full quantitative agreement with all four predictions (Miller, 1982; Hanke & Miller, 1983). Of course, this did not prove the two-pore model; alternative proposals envisioning a single pore fluctuating among three different states that coincidentally follow these predictions kept a robust controversy going for almost 20 years (Fahlke et al. 1998), until a projection structure of a bacterial CLC (Mindell et al. 2001), followed soon thereafter by a high-resolution crystal structure (Dutzler et al. 2002) settled the matter. I enjoyed participating in this controversy as a two-pore partisan, since a single-pore channel adhering so strictly to the binomial behaviour – as was eventually repeated over many years by many hands in many labs – struck me as just too much of a coincidence to swallow.
An additional experiment from that early time – now forgotten but still one of my favourites – further supported the double-barrelled channel model. In 1983, when Mike White visited the lab for a few months in a hiatus from his postdoctoral work at CalTech, we decided to follow up an observation he had made as a graduate student: irreversible inhibition by DIDS. In those days, this stilbene disulfonate was commonly considered a specific ‘anion transport inhibitor,’ although it is in fact just a greasy, anionic electrophile that reacts promiscuously with lysine and cysteine sidechains to wreak havoc on many proteins; indeed, this molecule is now known to form more potent breakdown products upon storage in aqueous solution (Matulef et al. 2008), and the site of DIDS action on ClC-0 has yet to be identified. Back in the chart-recorder days, I’d observed that DIDS at micromolar concentrations produces irreversible inhibition of macroscopic Torpedo Cl– channel current over a few minutes. It therefore seemed worthwhile to re-examine DIDS action at the improved single-channel level, on the off-chance that inhibition might proceed in two hits, one on each putative pore. And we found that this is exactly how the inhibitor kills the channel. A few minutes after adding the compound to a single channel, the U state suddenly disappears, leaving the channel opening to only a single 10 pS M state (Miller & White, 1984); then, after another minute or so, the channel disappears entirely. Moreover, during the interval between the first and second hit, the gating kinetics of the single pore matches up quantitatively with the opening and closing rates deduced from the two-pore behaviour before the first hit, as in eqn 2, further indicating the independence of the individual pores. This kind of ‘chemical biology’ experiment would be reprised years later in a more sophisticated way to definitively confirm the molecular architecture and quaternary structure of genetically manipulable ClC-0 channels (Ludewig et al. 1996; Middleton et al. 1996), but it was the DIDS-inhibition experiment that allowed me to sleep peacefully at night through the ‘80s with a double-barrelled shotgun under my pillow.
For the next 6 years my lab mostly worked on K+ channels. Cl− channel work continued at low burn, almost always on single channels, since double-barrelled behaviour averages out to invisibility in macroscopic currents. Years of staring at bursts of substates had revealed an oddity – that the recordings are asymmetric in time. Most bursts begin in the U state and end from the M state. This violation of microscopic reversibility is pregnant with fundamental meaning: the gating process cannot be at thermodynamic equilibrium. In other words, gating had to be somehow coupled to a source of free energy, and the only source that we could imagine was the Cl– gradient itself (Richard & Miller, 1990). Identification of Cl− as the culprit turned out to be wrong. Merritt Maduke much later showed (Lisal & Maduke, 2008) that the energy source is instead a H+ gradient, a point that made sense once the H+-coupled antiporters were discovered (Accardi & Miller, 2004; Miller, 2006; Traverso et al. 2006).
The ‘electrophysiology-only’ era abruptly ended in 1990 when Thomas Jentsch, in a brilliant tour-de-force of expression-cloning by hybrid depletion, identified the gene for the Torpedo Cl– channel, and gave it a name, ClC-0 (Jentsch et al. 1990). An abundant harvest of this breakthrough came fast and furious. It soon became clear that CLC proteins are everywhere, first seen in muscle-like contexts, putting a molecular face on the unusual Cl– conductance that was classically known to dominate resting muscle membranes (Hodgkin & Horowicz, 1960; Hutter & Warner, 1967; Palade & Barchi, 1977), and then observed in a wide swath of other physiologies: kidney, bone and lysosome functions, metal homeostasis in yeast, NO3– uptake in plants. Accompanying these discoveries were the CLC mutations underlying genetic diseases that provided exciting opportunities for grant proposals. At the protein-biochemical level, the cloning of ClC-0 was the root from which enhanced mechanistic understanding of CLCs grew. Simply knowing the amino acid sequence opened the way for Rich Middleton’s immunoaffinity purification of the channel from Torpedo electroplax, which established its completely unexpected quaternary architecture as a homodimer and demanded a non-symmetrical pore worming its way across each subunit (Middleton et al. 1994). A few years later, Merritt Maduke found and characterized the first prokaryotic CLC (Maduke et al. 1999), and although we failed to observe single-channel behaviour for this protein, its Cl− selectivity and overall architectural similarity to ClC-0 led us to declare it a bacterial Cl− channel. Within 2 years, Raimund Dutzler, Rod MacKinnon and colleagues managed to crystalize this homologue, providing the first high-resolution view of a CLC protein (Dutzler et al. 2002). Gazing upon this beautiful crystal structure with 18 membrane-embedded helices, we all thought that we were looking at a ClC-0-like Cl– channel, despite the conspicuous absence of an aqueous pore. But we all were wrong: 2 years later, Alessio Accardi proved that this bacterial CLC is not a channel at all, but instead an energy-utilizing H+-coupled antiporter, and suggested that the CLC family might generally be split in two, such that some of the familiar mammalian CLCs living in intracellular membrane compartments might also be H+/Cl– antiporters (Accardi & Miller, 2004). This idea was quickly confirmed for human CLC-4 and CLC-5 (Picollo & Pusch, 2005; Scheel et al. 2005). The janus-faced character of the entire superfamily is now a universal and almost unique feature that confers deep mechanistic richness on our growing understanding of how CLC proteins – both channels and transporters – operate in their biological roles, and how they work as membrane macromolecules.
But those stories are for other contributors to this issue to tell.
Biography
Chris Miller received his undergraduate education in Physics at Swarthmore College and a PhD in Molecular Biology from the University of Pennsylvania. After 2 years of postdoctoral work with Efraim Racker at Cornell University, he launched his own lab at Brandeis University in 1976, where he has remained throughout his career. He became a HowardHughesMedical Institute Investigator in 1988 and currently also holds an appointment as Visiting Professor of Biochemistry at the University of Oxford. Dr Miller's research seeks to understand basic mechanisms of ion channels and membrane transporters. His abiding terror of facing the complexities of intact cellular systems has driven his work towards reduced systems for mechanistic analysis: membrane protein reconstitution, electrical recording of single channels in model membranes, and X-ray crystallography.

Additional information
Competing interests
None declared.
Funding
None declared.
References
- Accardi A. Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- Baker JL, Sudarsan N, Weinberg Z, Roth A, Stockbridge RB. Breaker RR. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012;335:233–235. doi: 10.1126/science.1215063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brammer AE, Stockbridge RB. Miller C. F−/Cl− selectivity in CLCF-type F−/H+ antiporters. J Gen Physiol. 2014;144:129–136. doi: 10.1085/jgp.201411225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT. MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Fahlke C, Rhodes TH, Desai RR. George AL. Pore stoichiometry of a voltage-gated chloride channel. Nature. 1998;394:687–690. doi: 10.1038/29319. [DOI] [PubMed] [Google Scholar]
- Feng L, Campbell EB, Hsiung Y. MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B. Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hanke W. Miller C. Single chloride channels from Torpedo electroplax: activation by protons. J Gen Physiol. 1983;82:25–45. doi: 10.1085/jgp.82.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL. Horowicz P. The effect of sudden changes in ionic concentrations on the membrane potential of single muscle fibres. J Physiol. 1960;153:370–385. doi: 10.1113/jphysiol.1960.sp006540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter OF. Warner AE. The pH sensitivity of the chloride conductance of frog skeletal muscle. J Physiol. 1967;189:403–425. doi: 10.1113/jphysiol.1967.sp008176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H, Robertson JL, Wu F, Williams C. Miller C. Structure of a slow CLC Cl–/H+ antiporter from a cyanobacterium. Biochemistry. 2011;50:788–794. doi: 10.1021/bi1019258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Steinmeyer K. Schwarz G. Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature. 1990;348:510–514. doi: 10.1038/348510a0. [DOI] [PubMed] [Google Scholar]
- Lisal J. Maduke M. The ClC-0 chloride channel is a ‘broken’ Cl−/H+ antiporter. Nat Struct Mol Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig U, Pusch M. Jentsch TJ. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 1996;383:340–343. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]
- Maduke M, Pheasant DJ. Miller C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J Gen Physiol. 1999;114:713–722. doi: 10.1085/jgp.114.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulef K, Howery AE, Tan L, Kobertz WR, Du Bois J. Maduke M. Discovery of potent CLC chloride channel inhibitors. ACS Chem Biol. 2008;3:419–428. doi: 10.1021/cb800083a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton RE, Pheasant DJ. Miller C. Purification, reconstitution, and subunit composition of a voltage-gated chloride channel from Torpedo electroplax. Biochemistry. 1994;33:13189–13198. doi: 10.1021/bi00249a005. [DOI] [PubMed] [Google Scholar]
- Middleton RE, Pheasant DJ. Miller C. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- Miller C. Voltage-gated cation conductance channel from fragmented sarcoplasmic reticulum: steady-state electrical properties. J Membr Biol. 1978;40:1–23. doi: 10.1007/BF01909736. [DOI] [PubMed] [Google Scholar]
- Miller C. Open-state substructure of single chloride channels from Torpedo electroplax. Philos Trans R Soc Lond B Biol Sci. 1982;299:401–411. doi: 10.1098/rstb.1982.0140. [DOI] [PubMed] [Google Scholar]
- Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:484–489. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- Miller C. White MM. Dimeric structure of Cl− channels from Torpedo electroplax. Proc Natl Acad Sci U S A. 1984;81:2772–2775. doi: 10.1073/pnas.81.9.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Maduke M, Miller C. Grigorieff N. Projection structure of a ClC-type chloride channel at 6.5 Å resolution. Nature. 2001;409:219–223. doi: 10.1038/35051631. [DOI] [PubMed] [Google Scholar]
- Palade PT. Barchi RL. Characteristics of the chloride conductance in muscle fibers of the rat diaphragm. J Gen Physiol. 1977;69:325–342. doi: 10.1085/jgp.69.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picollo A. Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- Richard EA. Miller C. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 1990;247:1208–1210. doi: 10.1126/science.2156338. [DOI] [PubMed] [Google Scholar]
- Scheel O, Zdebik AA, Lourdel S. Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Tank DW, Miller C. Webb WW. Isolated-patch recording from liposomes containing functionally reconstituted chloride channels from Torpedo electroplax. Proc Natl Acad Sci U S A. 1982;79:7749–7753. doi: 10.1073/pnas.79.24.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverso S, Zifarelli G, Aiello R. Pusch M. Proton sensing of ClC-0 mutant E166D. J Gen Physiol. 2006;127:51–66. doi: 10.1085/jgp.200509340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill CL, McNamee MG. Karlin A. Affinity-labeling of purified acetylcholine receptor from Torpedo californica. Biochem Biophys Res Commun. 1974;61:997–1003. doi: 10.1016/0006-291x(74)90254-x. [DOI] [PubMed] [Google Scholar]
- White MM. Miller C. A voltage-gated anion channel from electric organ of Torpedo californica. J Biol Chem. 1979;254:10161–10166. [PubMed] [Google Scholar]