

Abstract
After providing a personal description of the convoluted path leading 25 years ago to the molecular identification of the Torpedo Cl− channel ClC-0 and the discovery of the CLC gene family, I succinctly describe the general structural and functional features of these ion transporters before giving a short overview of mammalian CLCs. These can be categorized into plasma membrane Cl− channels and vesicular Cl−/H+-exchangers. They are involved in the regulation of membrane excitability, transepithelial transport, extracellular ion homeostasis, endocytosis and lysosomal function. Diseases caused by CLC dysfunction include myotonia, neurodegeneration, deafness, blindness, leukodystrophy, male infertility, renal salt loss, kidney stones and osteopetrosis, revealing a surprisingly broad spectrum of biological roles for chloride transport that was unsuspected when I set out to clone the first voltage-gated chloride channel.
Cloning of ClC-0 and identification of the CLC gene family
Exciting times dawned in the 1980s, when the first primary structures of ion channels and transporters were obtained by molecular cloning (Noda et al. 1982; Kopito & Lodish, 1985; Noda et al. 1986). Finally, we could ‘see’ the proteins underlying ion transport, precisely manipulate their function, investigate their localization and structure, and determine their role in biology and disease. I joined Harvey Lodish’s laboratory at the Whitehead Institute as postdoc in 1986 to clone the NaHCO3− cotransporter, which I had been studying previously (Jentsch et al. 1984), by homology to the erythrocyte Cl−/HCO3− exchanger that had just been cloned in his laboratory (Kopito & Lodish, 1985). We now know that both proteins are indeed distantly related, although there was no chance to clone ‘my’ cotransporter using low-stringency hybridization of phage libraries. I tried to broaden the search for anion transporters using 4,4-diisothiocyanostil- bene-2,2-disulphonate (DIDS) and 4-acetamido-4-isothiocyanostilbene-9,4’-disulphonic acid (SITS), negatively charged stilbene derivates that inhibit and covalently bind many anion transporters. I reacted several cell lines with SITS and used antibodies against SITS to identify binding proteins in western blots. Because there were far too many bands to be followed up, I thought of model systems expressing anion transporters to very high levels, such as band 3 in erythrocytes or the ACh receptor (AChR) in the electric organ of the electric fish Torpedo, from which it had been cloned a few years earlier (Noda et al. 1982). That organ also expressed an exotic, DIDS-sensitive Cl– channel (Miller & White, 1984), which had been found by Chris Miller when trying to reconstitute the AChR (White & Miller, 1979). Although having a weird ‘double-barreled’ single-channel behaviour (Miller & White, 1984), this channel appeared to represent a more worthwhile target than another anion exchanger. Neither K+, nor Cl− channels had yet been cloned by then (in 1986). Few physiological studies had addressed Cl− channels, which rather annoyed many electrophysiologists by obscuring cation currents. However, a lack of Cl− currents had been implicated in two genetic diseases: cystic fibrosis and myotonia. Hence, this uncharted territory promised to hold many novel biological insights and surprises.
In a first sample of Torpedo membranes obtained from Chris Miller’s nitrogen tank at nearby Brandeis University, I detected a major broad SITS-labelled band. It resolved into two bands in fresher samples obtained from Torpedo shipped alive from California. One of these bands could be discarded (it was the α-subunit of the Na,K-ATPase, a warning sign!) but, excitingly, the other SITS-binding band was a disulphide-linked dimer, and the Torpedo channel appeared to have two pores. After purifying the protein and obtaining both N-terminal amino-acid sequence and specific antibodies, I pulled out overlapping clones from a cDNA library. Disappointingly, sequencing revealed only one strong candidate for a transmembrane domain. Although even total RNA from electric organ generated large Cl− currents in Xenopus oocytes, nothing happened when I injected the cRNA encoding this SITS-binding protein (Jentsch et al. 1989). It might still have been a subunit of the channel, but anti-sense experiments in oocytes gave conflicting, and in the end negative, results. No homology was found to other proteins in the small DNA database available at that time, although now it appears to be a membrane-anchored glucosidase.
In the meantime, I had accepted an invitation to lead an independent research group at Hamburg University. I had been offered this 5-year position before it became clear that the SITS-binding protein was no Cl− channel after all. Quite a stressful start into an independent career. I now really had to get that channel! I swore not to touch SITS again and started from scratch using expression cloning. Expression of precisely fractionated RNA from Torpedo electric organ in Xenopus oocytes indicated that the channel was encoded by an ∼10 kb mRNA (Jentsch et al. 1990) that might encode a very large protein. Complete conversion of the mRNA into cDNA for ‘positive’ expression cloning appeared exceedingly difficult. Hence, I turned to a cumbersome hybrid depletion approach in which we searched for cDNA clones that delete Cl− channel-forming activity from electric organ RNA. By contrast to positive expression cloning, but similar to the small interfering RNA screens that we recently used to clone the volume-regulated anion channel VRAC (Voss et al. 2014), this approach allows for the identification of heteromultimeric channels if at least one subunit is not redundant. Because hybrid depletion requires a large molar excess of depleting DNA over RNA, we used very small pools of clones instead of pool sizes of many hundreds that are possible with positive expression cloning. Single-stranded DNAs derived from groups of just twelve individual clones picked from a highly size-selected cDNA library were hybridized to electric organ RNA and DNA–RNA hybrids removed by CsCl density ultracentrifugation. Reduction of Cl− currents relative to those induced by acetylcholine (the Torpedo AChR served as internal control) was examined in the oocyte expression system (Fig. 1). After ∼2 years, Klaus Steinmeyer and I finally isolated a bona fide full-length cDNA with a partial clone identified by this painstaking procedure. When injected into oocytes, cRNA derived from that clone produced large Cl− currents that showed the right kinetics and ion selectivity. Together with the hydropathy analysis of the predicted ∼100 kDa protein, these results demonstrated that we had finally cloned the first voltage-gated Cl− channel (Jentsch et al. 1990). Its primary structure did not resemble any other known protein. We later named it ClC-0 (Steinmeyer et al. 1991b) to highlight it as the founder of a Cl− Channel gene family (Footnote). The ClC-0 cDNA was sufficient to reproduce the typical double-barreled single-channel appearance (Bauer et al. 1991), suggesting that we did not lack an important ancillary subunit.
Figure 1.

Cloning of the Torpedo channel ClC-0 by hybrid depletion
Total electric organ RNA, which was hybrid-depleted with single-stranded DNA derived from pools of 12 clones from a highly size-selected cDNA library, was expressed in Xenopus oocytes. Current ‘fingerprints’ were obtained using a symmetrical voltage clamp-protocol (A, inset) and recorded by a chart recorder. After the current response had increased to steady-state magnitudes (as a result of opening of the slow gate), the response to low chloride was recorded at depolarizing potentials. Subsequent superfusion with acetylcholine (ACh) probed for the expression of the Torpedo AChR that was used as internal reference to avoid false positives as a result of RNA degradation. A, background currents in non-injected oocytes; no response to ACh. B, negative pool of clones that shows normal Cl− channel and AChR expression. C, positive pool containing a partial ClC-0 cDNA; reduction of Cl− current with normal response to ACh. D, expression of full-length ClC-0 cRNA; large Cl− currents and no response to ACh. Oocytes were measured in ND96 (in mm: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2), except for the low chloride pulse (7 mm Cl−). AChR currents were elicited by 1 mm acetylcholine in the presence of 10 μm atropine to block muscarinic receptors. Modified from Jentsch et al. (1990).
Cloning of ClC-0 opened the way to identification of CLC proteins from mammals (Table1) and many other species and phylae. The past 25 years saw many exciting discoveries concerning associated β-subunits, structure and function, and physiology, pathology and human genetic disease. I first provide a short introduction into the general structural and functional features of CLC proteins, which will be addressed in more detail by other reviews in this issue. This is followed by a concise overview of mammalian CLC proteins and their roles in physiology and disease.
Table 1.
The CLC family of chloride channels and antiporters in mammals

A summary of the expression patterns of mammalian CLC proteins, their established or presumed functions, and pathologies resulting from loss-of-function mutations in humans and mice. Associated β-subunits are shown in red. Barttin and Ostm1 are obligatory β-subunits of both ClC-K isoforms and of ClC-7, respectively. Loss of barttin leads to Bartter syndrome IV, which is associated with massive renal salt loss and deafness. The glial cell adhesion molecule GlialCAM can associate with ClC-2 and change its localization and properties in glial cells. It does not qualify as an essential β-subunit. GlialCAM mutations lead to a distinct form of leukodystrophy. The HUGO gene names are CLCN1 to CLCN7, CLCNKA, CLCNKB, HEPACAM (for GlialCAM), BSND (for barttin), and OSTM1. NCL, neuronal ceroid lipofuscinosis.
General features of CLC channels and ion exchangers
CLC proteins function as dimers with one ion translocation pathway per subunit. This arrangement explains the ‘double-barreled’ appearance of ClC-0 (Miller, 1982; Bauer et al. 1991). In single-channel recordings of ClC-0, two conductances of equal magnitude are observed. They not only are gated independently by ‘protopore gates’ (also termed ‘fast gate’ in ClC-0), but also together can be closed by a common gate (‘slow gate’ in ClC-0). The dimeric nature of CLC proteins was substantiated by protein biochemistry (Middleton et al. 1994) and biophysical analysis of mutants (Ludewig et al. 1996; Middleton et al. 1996; Weinreich & Jentsch, 2001) and confirmed by X-ray structures of bacterial CLC Cl−/H+-exchangers (Dutzler et al. 2002). Because the translocation pathway is contained entirely within each subunit (Ludewig et al. 1996; Weinreich & Jentsch, 2001; Dutzler et al. 2002), a dimeric structure is, in principle, not required for ion transport. Indeed, a ‘monomerized’ ecClC-1 mutant retained ion transport activity (Robertson et al. 2010). It is not clear what advantage a dimeric structure confers to CLC transporters. It might bestow an increased spectrum of regulatory mechanisms, such as the common gating that affects both subunits in CLC channels (Miller, 1982; Bauer et al. 1991; Accardi & Pusch, 2000) and transporters (Ludwig et al. 2013), or enable the formation of heteromers with novel functions as observed in vitro with several mammalian CLC isoforms (Lorenz et al. 1996; Suzuki et al. 2006). From the perspective of pathology, the dimeric CLC structure allows for dominant negative effects with certain mutations of CLCN1 in dominant myotonia (Steinmeyer et al. 1994) and of CLCN7 in dominantly inherited osteopetrosis (Cleiren et al. 2001).
Each CLC monomer has 18 intramembrane helices, several of which do not span the membrane entirely (Dutzler et al. 2002). In eukaryotes, the transmembrane block is followed by a large, intracellular carboxyterminus containing two cystathionine-β-synthase (CBS) domains that bind each other and are in close contact with the intracellular face of the transmembrane block of the same subunit (Schmidt-Rose & Jentsch, 1997; Meyer & Dutzler, 2006; Markovic & Dutzler, 2007; Meyer et al. 2007; Feng et al. 2010). They also interact with the CBS domains from the partner monomer (Feng et al. 2010). Depending on the CLC isoform, these CBS domains can bind nucleotides such as ATP (Bennetts et al. 2007; Meyer & Dutzler, 2006; Markovic & Dutzler, 2007; Meyer et al. 2007) and may regulate the common gating (Fong et al. 1998; Estévez et al. 2004; Bennetts et al. 2007), a process that appears to be associated with a movement of these domains (Bykova et al. 2006; Ma et al. 2011). The carboxytermini of some CLC proteins can interact with other proteins such as (in the case of ClC-5) ubiquitin ligases (Schwake et al. 2001), cofilin (Hryciw et al. 2003) or KIF3B (Reed et al. 2010), although the physiological relevance of these interactions remains unclear (Rickheit et al. 2010). Moreover, several mammalian CLCs interact with transmembrane proteins that are either obligatory β-subunits (barttin for ClC-K channels: Estévez et al. 2001; Ostm1 for ClC-7: Lange et al. 2006) or may modulate their localization and function in only some tissues (GlialCAM for ClC-2: Jeworutzki et al. 2012).
Mutagenesis and crystal structures have identified the anion permeation pathway of CLC proteins. The first CLC crystal structure revealed the presence of a negatively charged glutamate side chain that apparently blocked the permeation pathway (Dutzler et al. 2002). Its position was occupied by a Cl− ion upon mutation to glutamine, suggesting that this ‘gating glutamate’ is the structural basis of the ‘protopore’ gate observed in electrophysiology (Dutzler et al. 2003). Before crystal structures were available, site-directed mutagenesis had already revealed the importance of this residue for rectification and gating (Friedrich et al. 1999; Waldegger & Jentsch, 2000) and of a Cl− co-ordinating serine (Dutzler et al. 2002) for ion selectivity and single-channel conductance (Ludewig et al. 1996). Subsequent mutations of the ‘gating glutamate’ in ClC-0 (Dutzler et al. 2003) and CLC exchangers (Leisle et al. 2011; Neagoe et al. 2010) similarly largely abolished the voltage-dependence of currents. However, channels lacking this glutamate still gate at the single-channel level (Dutzler et al. 2003; L’Hoste et al. 2013), indicating that CLC gating cannot be explained entirely by the movement of its side chain. Voltage-gating of CLC channels depends on the Cl− concentration. We proposed a reductionist model in which the gating charge is not provided by a charged intramembrane amino acid, as in many cation channels, but by the permeant anion that feels the electrical field in the pore (Pusch et al. 1995a). Chloride may compete with the negative side chain of the ‘gating glutamate’ and thereby open the channel (Chen, 2003). Although channel opening by Cl− was invoked to explain the non-equilibrium gating of ClC-0 (Richard & Miller, 1990; Chen & Miller, 1996), the current model proposes that protonation of the ‘gating glutamate’ leads to channel opening which is accompanied by a (so far unmeasurable) proton flux (Lísal & Maduke, 2008). This protonation-dependent gating fits to the role of this glutamate residue in tightly coupled Cl−/H+-exchange by other CLC members.
It came as a shock that the bacterial EcClC-1 protein is not a well-behaved Cl− channel but rather a tightly coupled Cl−/H+-exchanger (Accardi & Miller, 2004). This seminal finding was quickly followed by the demonstration that mammalian ClC-4 and ClC-5 are also Cl−/H+-exchangers (Picollo & Pusch, 2005; Scheel et al. 2005) and that plant atClC-a is a NO3−/Cl− antiporter (De Angeli et al. 2006; Bergsdorf et al. 2009). CLC anion/proton exchange is considered to rely on protonation of the ‘gating glutamate’, mutations in which convert the antiporter into a mere Cl− conductance (Accardi & Miller, 2004; Picollo & Pusch, 2005; Scheel et al. 2005; Bergsdorf et al. 2009; Leisle et al. 2011). Protons reach the ‘gating glutamate’ from the cell interior through a path diverging from that for chloride (Accardi et al. 2005). Proton transport of most, but not all (Feng et al. 2010), CLC exchangers depends on a ‘proton glutamate’ on the cytoplasmic side (Accardi et al. 2005; Zdebik et al. 2008).
Previously, the main function of vesicular CLC ‘channels’ was seen in facilitating endosomal/lysosomal acidification by neutralizing H+-ATPase currents (Günther et al. 1998; Günther et al. 2003). This notion appeared to be in doubt when mammalian vesicular CLCs were discovered to be Cl−/H+-antiporters rather than Cl− channels. Naively, it might be considered that these antiporters should rather shunt vesicular H+-gradients. However, our reductionist model calculations (Fig. 2) revealed that they may rather acidify vesicles better than channels because 2 Cl−/H+ exchange shifts luminal potentials to more negative values (Weinert et al. 2010). Furthermore, H+-driven, secondary active accumulation of Cl− into vesicles may serve important, although unknown, physiological roles (Novarino et al. 2010; Weinert et al. 2010).
Figure 2.

Modelling vesicular acidification with Cl− channels and Cl−/H+-exchangers
Reductionist model calculations revealing differential effects of Cl− channels or 2 Cl−/H+-exchangers on the acidification of vesicles (modified from Weinert et al. (2010)). ATP is virtually added at t = 0. (−) model vesicles containing only a proton pump and a proton leak nearly instantaneously reach a high luminal potential (B) that is given by the energy supplied by ATP hydrolysis. Virtually no acidification occurs (A). (unc) model vesicles containing additionally a Cl− channel, as in the classical model of vesicular acidification and as realized in Clcn5unc/y and Clcn7unc/unc mice (Novarino et al. 2010; Weinert et al. 2010), acidify their lumen (A) and accumulate Cl− (C). They reach a more moderate inside-positive potential (B). (WT) model vesicles containing instead of a Cl− channel a 2 Cl−/H+-exchanger (CLC antiport) rather unexpectedly reach a more acidic steady-state pH than those containing a Cl− channel (A). This is related to the fact that they reach a more negative luminal potential (B). They also accumulate more Cl− (C), as expected from H+-diven uptake of Cl−. For equations and parameter used, see Weinert et al. (2010).
ClC-1: a Cl− channel electrically stabilizing the skeletal muscle membrane
Because the fish electric organ has developed from muscle, known to have a high Cl− conductance since at least the 1950’s (Hodgkin & Horowicz, 1959), we chose to isolate the first mammalian Cl− channel from skeletal muscle. The Cl− conductance of muscle, which exceeds K+ conductance, stabilizes the muscle membrane voltage and aids in the repolarization of action potentials. Early work had indicated that muscle Cl− conductance was reduced in goats (Lipicky & Bryant, 1966) and humans (Lipicky et al. 1971) with myotonia, a muscle stiffness caused by membrane hyperexcitability. Hence, a muscle Cl− channel promised to be biologically and medically important. Indeed, shortly after identifying ClC-1 by homology cloning (Steinmeyer et al. 1991b), we found that a transposon had destroyed the Clcn1 gene in myotonic adr mice (Steinmeyer et al. 1991a) and identified a CLCN1 mutation in human myotonia (Koch et al. 1992). This established myotonia congenita as one of the first known ‘channelopathies’. We identified a CLCN1 mutation in the family of Dr Thomsen (Steinmeyer et al. 1994), who suffered from, and first described (Thomsen, 1876), the less severe dominantly inherited form of the disease (Thomsen disease). Heterozygous loss of CLCN1 is not associated with myotonic symptoms. Therefore, mutant ClC-1 proteins of Thomsen disease patients must affect the function of the wild-type protein encoded by the non-mutated allele. We found that many ClC-1 mutants of patients with dominant myotonia shift the voltage-dependence of channel opening to non-physiological positive potentials and impose a similar but variable shift also on mutant/wild-type heteromers (Pusch et al. 1995b). This shift is a result of an altered voltage-dependence of the common gate (Saviane et al. 1999). It results in a loss of function because ClC-1 will now be largely closed at physiological voltages and can therefore neither repolarize action potentials, nor stabilize the resting voltage. Interestingly, abnormal splicing of CLCN1 contributes to myotonia in myotonic dystrophy (Charlet et al. 2002; Mankodi et al. 2002) and in Huntington’s disease (Waters et al. 2013), which are caused by trinucleotide repeats in different genes. A recent study reporting that CLCN1 polymorphisms may contribute to epilepsy (Chen et al. 2013) has met with skepticism because ClC-1 shows very low expression in brain (Steinmeyer et al. 1991b) and because loss of ClC-1 function leads to myotonia, but not epilepsy.
The biophysics, structure/function relationship, regula-tion and pharmacology of ClC-1 have been studied extensively by ourselves and many other groups and are only mentioned briefly here. ClC-1 is gated open by depolarization (Steinmeyer et al. 1991b) and displays the typical Cl− > I− selectivity of CLC channels. It has a low single-channel conductance of ∼1 pS (Pusch et al. 1994; Saviane et al. 1999; Weinreich & Jentsch, 2001). Similar to ClC-0, it is ‘double-barreled’ and displays fast and slow gating relaxations (Accardi & Pusch, 2000). These correspond to ‘protopore’ and common gates as in ClC-0, although both ClC-1 gates are activated by depolarization (Saviane et al. 1999). Gating of ClC-1 is modulated by anions and pH (Rychkov et al. 1996; Rychkov et al. 1998). Interestingly, it is also modulated in a redox-dependent manner by intracellular ATP (Bennetts et al. 2005; Bennetts et al. 2007; Zhang et al. 2008) and β-nicotinamide adenine dinucleotide (Bennetts et al. 2012) through binding to the CBS domains in its cytoplasmic tail. This may provide a physiologically important coupling to muscle metabolism.
ClC-2: a widely expressed Cl− channel with multiple roles
Shortly after ClC-1, we cloned ClC-2, an inwardly rectifying Cl− channel present in almost all tissues (Thiemann et al. 1992). ClC-2 opens very slowly upon hyperpolarization (beyond ∼−60 mV). Hyposmotic cell swelling or moderately acidic extracellular pH decreased or abolished the inward rectification and thereby opened the channel (Gründer et al. 1992; Jordt & Jentsch, 1997). Residues involved in the slow voltage activation were identified in the amino terminus (Gründer et al. 1992) and a positively charged intracellular loop (Jordt & Jentsch, 1997). Similar to ClC-0 and ClC-1, gating of ClC-2 is affected by extracellular (Pusch et al. 1999), and prominently also intracellular Cl− (Niemeyer et al. 2004). ClC-2 has a single-channel conductance of 2–3 pS (Weinreich & Jentsch, 2001). Although ClC-2 can be activated by cell swelling, its biophysical properties (e.g. the Cl− > I− selectivity; Thiemann et al. 1992) clearly differ from the volume-regulated anion channel VRAC that displays a I− > Cl− selectivity sequence and that we recently found to be composed of LRRC8 heteromers (Voss et al. 2014). Although ablation (Voss et al. 2014) or partial knockdown (Qiu et al. 2014) of LRRC8A blocked or diminished, respectively, cell volume regulation, salivary gland cells from Clcn2−/− mice regulated their cell volume normally (Nehrke et al. 2002).
Indications for the physiological roles of ClC-2 were gleaned from Clcn2−/− mice that display early postnatal retinal and testicular degeneration (Bösl et al. 2001). We suggested that these pathologies result from disturbed extracellular ion homeostasis in the narrow spaces surrounding photoreceptors and germ cells. Clcn2−/− mice also develop leukodystrophy, with vacuoles slowly appearing in myelin sheaths of central axons (Blanz et al. 2007). Accordingly, nerve conduction velocity in the central auditory pathway was reduced. After the retraction of a widely cited publication (Haug et al. 2009), there is no convincing evidence for a role of ClC-2 in epilepsy (Blanz et al. 2007; Depienne et al. 2013; Niemeyer et al. 2010; Niemeyer et al. 2004). Recent results show that, as in mice, human CLCN2 mutations rather result in leukodystrophy (Depienne et al. 2013) that can be associated with azoospermia (Di Bella et al. 2014).
ClC-2 was recently shown to bind to the cell adhesion molecule GlialCAM (Jeworutzki et al. 2012), which in turn binds a multiple membrane-spanning protein, Mlc1 (López-Hernández et al. 2011). Mutations in GLIALCAM or MLC1 cause megalencephalic leukoencephalopathy with subcortical cysts (Leegwater et al. 2001; López-Hernández et al. 2011), suggesting a common pathophysiology with all three genes. GlialCAM anchors ClC-2 and Mlc1 to cell–cell junctions of transfected cells (López-Hernández et al. 2011; Jeworutzki et al. 2012; Hoegg-Beiler et al. 2014). Excitingly, co-expression of GlialCAM increased ClC-2 current amplitudes and almost abolished its rectification (Jeworutzki et al. 2012). The expression pattern of GlialCAM implies that it may affect ClC-2 only in glia. GlialCAM overexpression also changes the common gates of ClC-0, ClC-1 and ClC-K channels, but not of CLC Cl−/H+ exchangers (Jeworutzki et al. 2014). This observation is of biophysical interest but lacks physiological consequences because glial expression of those channels is insignificant.
In line with the in vitro data, we found that Glialcam disruption in mice affected the abundance and localization of both ClC-2 and Mlc1 in glial cells (Hoegg-Beiler et al. 2014). Unexpectedly, also Mlc1 disruption changed both GlialCAM and ClC-2 expression (Hoegg-Beiler et al. 2014; Dubey et al. 2015). Consistent with the in vitro data, deletion of GlialCAM (or of Mlc1) reduced ClC-2 currents and introduced inward rectification in oligodendrocytes but, unexpectedly, not in cerebellar Bergmann glia that prominently co-express all three proteins (Hoegg-Beiler et al. 2014). The pathology resulting from GlialCAM and Mlc1 disruption cannot be explained only by reduced ClC-2 function because mice lacking both ClC-2 and GlialCAM show more severe leukodystrophy than Clcn2−/− mice (Hoegg-Beiler et al. 2014).
ClC-2, GlialCAM and MLC1 are co-expressed at connections between oligodendrocytes and astrocytes and at astrocytic endfeet that contact blood vessels (Blanz et al. 2007; Jeworutzki et al. 2012; Hoegg-Beiler et al. 2014). This localization resembles that of the K+ channel Kir4.1 and the Cx47 gap junction protein (Blanz et al. 2007; Hoegg-Beiler et al. 2014), a lack of which also results in leukodystrophy. These proteins are assumed to have a role in K+ siphoning (Wallraff et al. 2006), a process in which K+ ions released from neurons are taken up by the glial syncytium and are equilibrated with serum at astrocytic endfeet. We postulated a similar role for ClC-2 in Cl− siphoning that may be needed to electrically compensate the movement of K+ (Blanz et al. 2007; Hoegg-Beiler et al. 2014). The linear voltage-dependence of ClC-2/GlialCAM channels allows Cl− entry into glia when they are depolarized by the rise in [K+]o during K+ siphoning.
ClC-2 is also expressed in neurons where it might lower the cytoplasmic Cl− concentration under certain circumstances (Staley et al. 1996; Földy et al. 2010; Rinke et al. 2010), although this notion has been questioned (Ratté & Prescott, 2011). A rise in [Cl−]i above its electrochemical equilibrium may open ClC-2 by shifting its voltage-dependence to more positive potentials (Pusch et al. 1999; Catalán et al. 2004) and allow Cl− to passively approach equilibrium values. When [Cl−]i is lowered below its equilibrium by the K+Cl− cotransporter KCC2, closure of ClC-2 may prevent it from counteracting the effect of KCC2 on [Cl−]i.
A role of ClC-2 in transepithelial transport is not restricted to Sertoli cells and retinal pigment epithelial cells where it was invoked to explain the testicular and retinal degeneration of Clcn2−/− mice (Bösl et al. 2001), but also has been found in the intestine. ClC-2 is expressed in the basolateral membrane of colonic enterocytes (Catalán et al. 2002; Catalán et al. 2004) where it plays a role in Cl− reabsorption (Zdebik et al. 2004; Catalán et al. 2012). This contrasts with the role of the apical Cl− channel CFTR in Cl− secretion. Indeed, we found that mice homozygous for the deleterious ΔF508 CFTR mutation survive better when ClC-2 is additionally disrupted (Zdebik et al. 2004).
ClC-K/barttin channels in renal and inner ear transepithelial ion transport
ClC-Ka and ClC-Kb (-K1 and -K2 in rodents) are highly homologous Cl− channels (Adachi et al. 1994; Kieferle et al. 1994) that require barttin, a small β-subunit with two transmembrane domains, for full functionality (Estévez et al. 2001). Barttin (encoded by the BSND gene) is required for channel activity, the transport of ClC-K to the plasma membrane (Estévez et al. 2001; Waldegger et al. 2002; Scholl et al. 2006) and for ClC-K protein stability in vivo (Rickheit et al. 2008; Nomura et al. 2011). Without barttin, only rat ClC-K1 gave currents that could be confirmed by rectification-changing mutagenesis (Waldegger & Jentsch, 2000). Exceptional within the CLC family, ClC-K channels lack a ‘gating’ glutamate and show little voltage-dependent gating. Insertion of such a glutamate introduces hyperpolarization-activated gating both in the absence and presence of barttin (Waldegger & Jentsch, 2000; L’Hoste et al. 2013). Mouse ClC-K1 has a ‘double-barreled’ appearance with a rather large single-channel conductance of ∼40 pS that is not changed by barttin co-expression (L’Hoste et al. 2013).
ClC-K proteins and barttin are almost exclusively expressed in the kidney and in the stria vascularis of the inner ear. In both renal (Uchida et al. 1995; Vandewalle et al. 1997; Kobayashi et al. 2001) and strial epithelia (Estévez et al. 2001; Rickheit et al. 2008), they reside in basolateral membranes, although ClC-K1 may additionally be apical in the thin limb of Henle’s loop (Uchida et al. 1995). Mutations inactivating ClC-Kb cause the severe salt-losing nephropathy Bartter syndrome type III in humans (Simon et al. 1997), whereas disruption of mouse ClC-K1 caused a diabetes insipidus-like phenotype (Matsumura et al. 1999). Loss-of-function mutations in BSND underlie Bartter syndrome type IV that combines severe renal salt loss with congenital deafness (Birkenhäger et al. 2001). A rather benign missense mutation in BSND may underlie non-syndromic hearing loss without renal symptoms (DFNB73; Riazuddin et al. 2009).
In the thick ascending limb (TAL), its main renal expression site, ClC-Kb/barttin provides the basolateral exit for Cl− that is taken up from urine through the NaK2Cl-cotransporter NKCC2 (SLC12A1; mutated in Bartter I) (Fig. 3A). Na+, which drives apical Cl− uptake, is extruded basolaterally by the Na,K-ATPase, whereas K+ is recycled apically through the ROMK (Kir1.1, KCNJ1) K+-channel (mutated in Bartter II). As the TAL reabsorbs the bulk of filtered NaCl, CLCNKB mutations lead to severe congenital salt and fluid loss.
Figure 3.
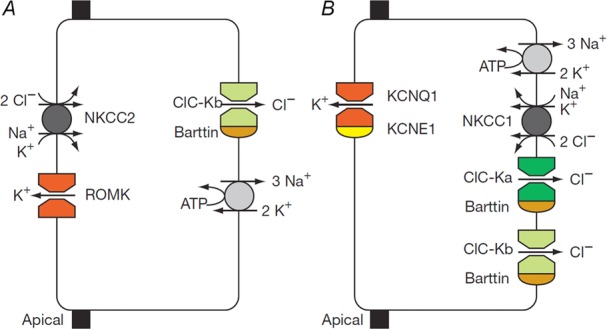
Role of ClC-K/barttin channels in transepithelial transport
Schematic diagram of NaCl reabsorption in the TAL of Henle’s loop (A) and of K+ secretion by marginal cells in the stria vascularis of the inner ear (B) (Taken from Estévez et al. (2001)).
Clcnk1−/− mice show overt nephrogenic diabetes insipidus (Matsumura et al. 1999) as a result of impaired solute accumulation in the inner medulla (Akizuki et al. 2001). Hence, ClC-K1 (-Ka) appears to be crucial for the countercurrent system that creates the strongly hypertonic environment of the renal medulla required for urine concentration. Although humans with mutations in only CLCNKA have not yet been described, there are a few patients with mutations in both CLCNKA and CLCNKB (Schlingmann et al. 2004; Nozu et al. 2008). Similar to patients with non-functional barttin, they display Bartter syndrome type IV that combines deafness with a particularly severe renal phenotype.
The stria vascularis is a multilayered epithelium in the lateral wall of the cochlea. It generates a positive potential and a high K+ concentration in the scala media, both of which are crucial for mechanotransduction currents in hair cells. Transport of ions across the most apical cell layer of the stria, the marginal cells, involves basolateral uptake of K+ by the Na,K-ATPase and the NKCC1 NaK2Cl cotransporter (Fig. 3B). K+ is secreted through apical KCNQ1/KCNE1 K+ channels (the loss of which causes deafness with cardiac arrhythmia), whereas Cl− ions accumulated though NKCC1 are recycled basolaterally through ClC-Ka/barttin and ClC-Kb/barttin Cl− channels (Rickheit et al. 2008). Disruption of only one of the ClC-K isoforms is compatible with hearing, although loss of both, or of their essential β-subunit barttin, leads to deafness (Birkenhäger et al. 2001; Schlingmann et al. 2004). Although constitutive barttin disruption entails early postnatal lethality due to renal salt and fluid loss (Rickheit et al. 2008), inner ear-specific Bsnd disruption revealed a breakdown of the endocochlear potential (Rickheit et al. 2008). Sensory outer hair cells showed anatomic degeneration over the first few postnatal weeks but had already shown functional impairment before these cells died, resulting in a hearing loss that was present from birth onward, as in humans with Bartter IV.
ClC-3: a highly controversial endosomal Cl−/H+-exchanger
ClC-3 is the most controversial member of the CLC family. Several different plasma membrane Cl− currents with mutually incompatible characteristics have been assigned to it. This prominently includes a purported role as volume-regulated anion channel VRAC (Duan et al. 1997), a claim sometimes repeated even today. However, VRAC currents were unaffected in our Clcn3−/− mice (Stobrawa et al. 2001), a finding confirmed in two other Clcn3−/− mouse models (Arreola et al. 2002; Gong et al. 2004). It might be hoped that the identification of LRRC8 proteins as essential VRAC components (Qiu et al. 2014; Voss et al. 2014) may finally convince the last proponents of the VRAC = ClC-3 hypothesis.
ClC-3 is expressed in almost all tissues. It is mainly found on endosomes (Stobrawa et al. 2001; Hara-Chikuma et al. 2005b; Suzuki et al. 2006) where it co-localizes partially with the late endosomal/lysosomal protein lamp1 (Stobrawa et al. 2001). It was also found on synaptic vesicles (SVs) (Stobrawa et al. 2001; Salazar et al. 2004) and synaptic-like microvesicles (Maritzen et al. 2008), although a significant presence of ClC-3 on synaptic vesicles has been questioned recently (Schenck et al. 2009). The ClC-3B splice variant displaying a C-terminal PDZ-binding motif (Ogura et al. 2002) might localize to the Golgi (Gentzsch et al. 2003).
Most plasma membrane currents reported upon ClC-3 overexpression are probably carried by channels endogenous to the host cells. However, some studies (Li et al. 2002; Picollo & Pusch, 2005; Matsuda et al. 2008; Guzman et al. 2013) reported small strongly outwardly-rectifying Cl− currents upon ClC-3 overexpression. They strongly resembled those of ClC-4 and ClC-5 (Friedrich et al. 1999; Steinmeyer et al. 1995) and were similarly affected by mutations of the gating glutamate (Li et al. 2002; Matsuda et al. 2008). Hence, they probably represent genuine ClC-3 currents. ClC-3 is most probably an intracellular voltage-dependent, electrogenic 2Cl−/H+-exchanger (Jentsch, 2008; Guzman et al. 2013). However, because of its low transport rates at the plasma membrane, this could not be shown as convincingly as for ClC-4 through ClC-7 (Picollo & Pusch, 2005; Scheel et al. 2005; Neagoe et al. 2010; Leisle et al. 2011).
Similar to ClC-5 in renal proximal tubules (Piwon et al. 2000; Günther et al. 2003; Hara-Chikuma et al. 2005a; Novarino et al. 2010), ClC-3 may be important for endosomal acidification and chloride accumulation (Hara-Chikuma et al. 2005b). Unlike ClC-5, however, ClC-3 deletion did not impair renal endocytosis (Rickheit et al. 2010). Synaptic vesicles of Clcn3−/− mice showed impaired acidification in vitro (Stobrawa et al. 2001). Their reduced uptake of glutamate could be explained by reduced expression of the vesicular glutamate transporter vGlut1 (Stobrawa et al. 2001). It was suggested recently that SVs express only a low amount of ClC-3 (Schenck et al. 2009) and that vGlut1 itself provides a Cl− conductance (Schenck et al. 2009; Preobraschenski et al. 2014). The impaired acidification of SVs from Clcn3−/− mice was therefore attributed to the reduction of vGlut1 in Clcn3−/− mice (Schenck et al. 2009), which may result from their severe neurodegeneration (Stobrawa et al. 2001). Amplitudes of miniature postsynaptic currents, which reflect the neurotransmitter contents of SVs, may reveal changes in vesicular neurotransmitter uptake which depends on the electrochemical potential of SVs as driving force. However, there is disagreement on whether miniature postsynaptic current amplitudes are changed in Clcn3−/− neurons (Stobrawa et al. 2001; Riazanski et al. 2011; Guzman et al. 2014). More work is needed to sort out these contradictory results.
Disruption of ClC-3 in mice leads to neuronal degeneration in the retina and brain that eventually leads to a dramatic loss of the hippocampus (Stobrawa et al. 2001) and degeneration of other brain areas (Stobrawa et al. 2001; Dickerson et al. 2002; Yoshikawa et al. 2002). Signs of pathological lysosomal storage were found in one study (Yoshikawa et al. 2002), although this was much milder than in Clcn6−/− (Poët et al. 2006) or Clcn7−/− mice (Kasper et al. 2005). The mechanism by which ClC-3 disruption leads to neurodegeneration remains unclear. It should also be noted that Clcn3−/− mice are systemically sick and display, for example, reduced body weight. Possible effects on, for example, insulin secretion might therefore not be a β-cell intrinsic consequence of ClC-3 disruption and require cautious interpretation (Maritzen et al. 2008; Jentsch et al. 2010).
ClC-4: an endosomal Cl−/H+ exchanger with a possible role in brain development
ClC-4 is widely expressed in many tissues (Jentsch et al. 1995; van Slegtenhorst et al. 1994). ClC-4 is a strongly voltage-dependent 2Cl−/H+-exchanger (Picollo & Pusch, 2005; Scheel et al. 2005) that is present on endosomes and, in heterologous expression, to a small degree on the plasma membrane (Mohammad-Panah et al. 2003; Suzuki et al. 2006). However, other studies have reported its localization in the endoplasmic reticulum (Okkenhaug et al. 2006). Unfortunately, no knock-out controlled immunohistochemistry is available for ClC-4. ClC-4 was suggested to function in endosomal acidification and trafficking (Mohammad-Panah et al. 2003). However, although Clcn5y/− mice display impaired proximal tubular endocytosis (Piwon et al. 2000) (see below), no such defect is seen in Clcn4−/− mice that also lack other obvious phenotypes (Rickheit et al. 2010).
Human genetics suggests that a loss of ClC-4 impacts on brain function. Although Clcn4 resides on chromosome 7 in inbred house mice, human CLCN4 is located on the X chromosome (Rugarli et al. 1995). A patient carrying a deletion of the X-chromosome encompassing CLCN4 and many other genes displayed severe psychomotor delay (Meindl et al. 1993) and a de novo loss-of-function mutation (G544R) was identified in a patient displaying severe early-onset epilepsy and delayed development (Veeramah et al. 2013). More convincingly, five families with different CLCN4 mutations were identified in a screen for genes underlying X-linked intellectual disability (Hu et al. 2015). These mutations reduced ClC-4 currents in heterologous expression. Cultured neurons derived from Clcn4−/− mice, or primary neurons subjected to Clcn4 shRNA knockdown, displayed moderately reduced neurite outgrowth and branching (Hur et al. 2013; Hu et al. 2015). Defective intracellular trafficking may underlie these disturbances.
ClC-5: an endosomal Cl−/H+ exchanger crucial for renal endocytosis
CLCN5 was discovered in the search for genes underlying Dent’s disease (Fisher et al. 1994), an X-linked renal disorder associated with low molecular weight proteinuria and a variable presence of kidney stones, nephrocalcinosis, and renal failure (Wrong et al. 1994). ClC-5 is a 2Cl−/H+-exchanger (Picollo & Pusch, 2005; Scheel et al. 2005) that is most highly expressed in renal and other epithelia (Vandewalle et al. 2001; Steinmeyer et al. 1995). Similar to ClC-4, upon heterologous expression, ClC-5 partially reaches the plasma membrane where it can be studied electrophysiologically (Steinmeyer et al. 1995; Friedrich et al. 1999). Both transporters activate almost instantaneously upon strong depolarization (∼+30 mV) and lack measurable tail currents. Although a small proportion of ClC-5 can be detected in the apical brush-border membrane of renal proximal tubular cells, the majority of the protein is located in apical endosomes in renal (Günther et al. 1998; Wartosch et al. 2009) and intestinal epithelia (Vandewalle et al. 2001). The plasma membrane expression of ClC-5 can be increased by mutating a PY motif located between both CBS domains that can interact with ubiquitin ligases (Schwake et al. 2001; Hryciw et al. 2004). However, disrupting this motif had no effect in vivo (Rickheit et al. 2010).
The pathogenesis of Dent’s disease has been determined by knock-out (Piwon et al. 2000; Wang et al. 2000; Günther et al. 2003) and knock-in (Novarino et al. 2010; Rickheit et al. 2010) mouse models. These mice display proteinuria resulting from impaired proximal tubular endocytosis (Piwon et al. 2000; Wang et al. 2000), which is a cell-autonomous consequence of Clcn5 disruption (Piwon et al. 2000; Novarino et al. 2010). Both receptor-mediated and fluid-phase endocytosis is affected, and the endocytic retrieval of apical membrane proteins such as NaPi-IIa is slowed (Piwon et al. 2000; Novarino et al. 2010). The decreased expression of the apical scavenger receptor megalin (Piwon et al. 2000), which may result from impaired recycling, exacerbates the urinary loss of ligands such as vitamin D binding protein or parathyroid hormone (PTH). The ensuing increased tubular PTH concentration may stimulate apical PTH receptors and thereby reduce the apical expression of the resorptive phosphate transporter NaPi-IIa (SLC34A1) (Piwon et al. 2000). The resultant phosphaturia contributes to the formation of kidney stones in Dent’s disease. Many patients with CLCN5 mutations display proteinuria but lack hypercalciuria and kidney stones (Sekine et al. 2014). Similarly, the Clcn5y/− mice of (Wang et al. 2000), but not those generated in our laboratory (Piwon et al. 2000), displayed hypercalciuria. We attributed these different outcomes to the opposing effects of Clcn5 disruption on vitamin D levels. In Clcn5y/− proximal tubules, increased luminal PTH stimulates the conversion of the precursor 1(OH)-D3 into the active form 1,25(OH)2-D3, but the levels of both the precursor and the active form are reduced by urinary loss. This delicate balance can result in a decrease or increase of serum 1,25(OH)2-D3, which may decrease or increase, respectively, the intestinal absorption of Ca2+ and its subsequent renal excretion (Piwon et al. 2000). Hence, hyperphosphaturia, hypercalciuria and kidney stones are secondary to a primary defect in renal endocytosis.
We hypothesized that impaired endocytosis is caused by impaired acidification of endosomes (Günther et al. 1998). Endosomal acidification was indeed reduced in vesicle preparations (Günther et al. 2003; Novarino et al. 2010) or cell cultures (Hara-Chikuma et al. 2005a) from Clcn5y/− kidneys. However, Clcn5y/unc mice (Novarino et al. 2010), in which we uncoupled Cl− from H+-countertransport by the E211A ‘gating glutamate’ mutation (Scheel et al. 2005), displayed normal proximal tubular endosomal acidification but impaired tubular endocytosis, which was as severe as that in Clcn5y/− mice (Novarino et al. 2010). A similar uncoupling mutation (E211Q) was recently identified in a patient with Dent’s disease (Sekine et al. 2014). Hence, impaired endocytosis in Clcn5y/− and Clcn5y/unc mice can be attributed not only to reduced endosomal acidification, but also to changes of additional parameters such as luminal Cl− concentration or transmembrane voltage of endosomes (Weinert et al. 2010). Furthermore, the normal expression and localization of the mutant ClC-5 protein in Clcn5y/unc mice (Novarino et al. 2010) suggests that, in Clcn5y/− mice, a lack of interactions of ClC-5 with other proteins is not a major pathogenic factor.
ClC-6: a mainly neuronal late endosomal Cl−/H+-exchanger
Together with ClC-7, ClC-6 forms the third branch of the CLC family (Brandt & Jentsch, 1995). Although Clcn6 mRNA is found in many tissues (Brandt & Jentsch, 1995), the ClC-6 protein is predominantly expressed in the nervous system (Poët et al. 2006). ClC-6 resides in endosomes of cultured cells (Suzuki et al. 2006; Ignoul et al. 2007) and of neurons in situ (Poët et al. 2006) and partially co-localizes with the late endosomal/lysosomal protein lamp1. Subcellular fractionation of brain membranes revealed that ClC-6, similar to ClC-3, is present in endosomal rather than lysosomal fractions (Poët et al. 2006).
The late endosomal localization of ClC-6 precluded its biophysical characterization for many years. Only recently was it shown that a GFP-ClC-6 fusion protein reaches the plasma membrane. It mediates electrogenic Cl−/H+-exchange as confirmed by point mutations in the ‘gating glutamate’ and an ion-selectivity changing mutation in the Cl−co-ordinating serine (Neagoe et al. 2010).
Clcn6−/− mice display a peculiar form of lysosomal storage disease in which intracellular deposits localize mainly at axon hillocks (Poët et al. 2006). Unlike Clcn7−/− mice, this storage disease progresses slowly, is not associated with significant neuronal cell loss, and causes little microglial activation (Poët et al. 2006; Pressey et al. 2010). The loss of ClC-6 did not change lysosomal pH. Clcn6−/− mice show mild behavioural abnormalities (Poët et al. 2006). This includes a reduction in pain sensitivity that correlates with strong lysosomal storage disease in dorsal root ganglion neurons. We considered CLCN6 as a candidate gene for mild forms of human neuronal ceroid lipofuscinosis, but we found only two heterozygous missense mutations in 2 out of 75 neuronal ceroid lipofuscinosis patients (Poët et al. 2006). It may just be a matter of time until convincing CLCN6 mutations are found in human neuronal ceroid lipofuscinosis.
ClC-7/Ostm1: a lysosomal Cl−/H+-antiporter crucial for brain and bone integrity
ClC-7 needs Ostm1, a highly glycosylated type I transmembrane protein, for ion transport activity (Leisle et al. 2011) and protein stability in vivo (Lange et al. 2006). ClC-7 is the only lysosomal CLC protein, as demonstrated by subcellular fractionation (Poët et al. 2006) and immunohistochemistry of transfected and native cells (Kornak et al. 2001; Kasper et al. 2005; Lange et al. 2006; Suzuki et al. 2006). In bone-degrading osteoclasts, ClC-7/Ostm1 and the H+-ATPase are inserted by lysosomal exocytosis into the ruffled border membrane that faces the acidic resorption lacuna (Kornak et al. 2001; Lange et al. 2006). Although ClC-7 traffics to lysosomes also without Ostm1, Ostm1 needs ClC-7 for lysosomal targeting, processing and stability (Lange et al. 2006).
The lysosomal localization of ClC-7/Ostm1 complicated the characterization of its transport properties. Isolated lysosomes display Cl−/H+-exchange activity (Graves et al. 2008; Weinert et al. 2010), which was reduced in the absence of ClC-7 (Weinert et al. 2010). The identification of sorting signals in the ClC-7 N-terminus allowed the engineering of point mutants partially localizing to the plasma membrane (Stauber & Jentsch, 2010), where they can be analysed biophysically (Leisle et al. 2011). The mutant also reaches the plasma membrane without Ostm1 but needs the β-subunit Ostm1 for ion transport activity (Leisle et al. 2011). ClC-7/Ostm1 rectifies as strongly in the outward direction as ClC-4 through ClC-6 but, in contrast to those transporters, it activates very slowly (within seconds) upon depolarization. Deactivation of ClC-7/Ostm1 is also slow, permitting the measurement of tail currents. These reveal that macroscopic current rectification is caused by voltage gating of an electrogenic exchange process with an almost linear intrinsic voltage-dependence (Leisle et al. 2011). Reversal potentials of tail currents established a 2Cl−/1H+ exchange stoichiometry for ClC-7. Gating of ClC-7/Ostm1 involves a common gate (Ludwig et al. 2013). Intriguingly, several CLCN7 mutations found in human osteopetrosis accelerate ClC-7/Ostm1 gating, suggesting that the slow gating of ClC-7 is physiologically important (Leisle et al. 2011).
The physiological roles of ClC-7 became apparent from knock-out mice (Kornak et al. 2001) that display severe osteopetrosis, retinal degeneration and neurodegeneration associated with lysosomal storage (Kasper et al. 2005). Grey-lethal mice that carry a mutation in the β-subunit Ostm1 display an almost indistinguishable phenotype (Chalhoub et al. 2003; Lange et al. 2006; Pressey et al. 2010). Similarly, total loss of either ClC-7 or Ostm1 function leads to severe infantile osteopetrosis in humans (Kornak et al. 2001; Chalhoub et al. 2003), which is probably associated with neurodegeneration as well (Frattini et al. 2003). Certain CLCN7 missense mutations cause autosomal dominant osteopetrosis that is clinically more benign and lacks the involvement of the CNS (Cleiren et al. 2001; Frattini et al. 2003).
Clcn7−/− mice die at ∼6 weeks of age and show neuronal cell loss predominantly in the hippocampus (Kornak et al. 2001). Mice with forebrain-specific Clcn7 disruption live much longer and almost completely lose neurons in areas lacking ClC-7 (Wartosch et al. 2009). In vivo pulse-chase experiments revealed that protein degradation in proximal tubular cells lacking ClC-7 is impaired in a cell-intrinsic manner (Wartosch et al. 2009). The enlargement of lamp1-positive compartments in tubular cells lacking ClC-7, however, is not a result of protein accumulation, as is evident from cells in which protein uptake was impaired by ClC-5 disruption (Wartosch et al. 2009). This observation suggests a role for ClC-7 in vesicular trafficking or fusion processes. It is tempting to speculate that impaired protein degradation and lysosomal storage resulted from a less acidic lysosomal pH. However, careful ratiometric measurements showed a normal lysosomal pH in mice lacking ClC-7/Ostm1 repeatedly (Kasper et al. 2005; Lange et al. 2006; Weinert et al. 2010). This can be rationalized by the presence of a lysosomal cation conductance that obviates the need for the ClC-7/Ostm1 conductance in neutralizing H+-ATPase currents (Steinberg et al. 2010; Weinert et al. 2010). As expected for a pH-gradient driven transport of Cl− by ClC-7, the lysosomal Cl− concentration was decreased in knock-out mice (Weinert et al. 2010).
Unlike the normal pH of Clcn7−/− lysosomes (Kasper et al. 2005; Lange et al. 2006; Weinert et al. 2010), the resorption lacuna of cultured Clcn7−/− osteoclasts was less acidic (Kornak et al. 2001). This fits with osteopetrosis because an acidic pH is required both for the dissolution of inorganic bone material and the activity of secreted proteases. Indeed, mutations in the a3 subunit of the lysosomal H+-ATPase also cause osteopetrosis (Kornak et al. 2000; Scimeca et al. 2000). The failure of Clcn7−/− osteoclasts to acidify the resorption lacuna may be attributed to the lack of neutralizing currents or to the underdevelopment of the ruffled border observed in electron microscopy (Kornak et al. 2001; Weinert et al. 2014). This underdevelopment may be a consequence of reduced lysosomal exocytosis.
Two other Clcn7 mouse models were generated aiming to determine the respective biological roles of ClC-7 ion transport and protein––protein interactions. Similar to Clcn5y/unc mice (Novarino et al. 2010), Clcn7unc/unc mice (Weinert et al. 2010) carry a ‘gating glutamate’ mutation that converts ClC-7 into a mere anion conductance, whereas, in Clcn7td/td mice (Weinert et al. 2014), ion transport was abolished by a mutation in the ‘proton glutamate’ (Leisle et al. 2011) (td: transport-deficient). Similar to Clcn7−/− mice, both mouse models had unchanged lysosomal pH but decreased lysosomal Cl− concentration. Both new mouse models were osteopetrotic and displayed lysosomal storage (Weinert et al. 2010; Weinert et al. 2014), although with different severities. Compared to the total knock-out, osteopetrosis was less severe in Clcn7unc/unc mice and neurodegeneration was less severe in Clcn7td/td mice. We concluded that ClC-7 Cl−/H+-exchange cannot be functionally replaced by a mere Cl− conductance, and that the ClC-7unc Cl− conductance partially rescues the osteopetrotic phenotype (Weinert et al. 2010). Although the mere presence of the (non-transporting) ClC-7td protein ameliorates neurodegeneration, a lysosomal Cl− conductance appears to be detrimental (Weinert et al. 2014). Intriguingly, Clcn7−/− and Ostm1−/− mice, but not Clcn7unc/unc or Clcn7td/td mice, display grey fur in an agouti background (Weinert et al. 2014). This suggests that ClC-7 protein–protein interactions, rather than ClC-7 ion transport, are needed for melanocyte function.
Outlook
Twenty-five years after the discovery of ClC-0, we look back on the many exciting discoveries concerning their structure, function and amazingly diverse physiological and pathological roles, as described in more than two thousand papers. Cl− channels have emerged from the dark ages and we now appreciate their diverse functions in the cell and the organism. Moreover, Cl− channels have provided refreshing insights into the diverse ways of building ion channels and transporters and the fine line separating them. More surprises will probably follow.
Footnote
We proposed to indicate, if appropriate, the species by prefixes and individual isoforms by suffixes after a dash (e.g. hClC-1 for the human skeletal muscle Cl− channel; atClC-a and ecClC-1 for particular CLCs from Arabidopsis thaliana and Escherichia coli, respectively), although this nomenclature is not followed by everybody in the field. The official human gene nomenclature (HUGO) is CLCN1, CLCN2, etc.
Acknowledgments
I thank my many talented and dedicated co-workers over the past 25 years, beginning with Klaus Steinmeyer and Michael Pusch, followed by many other equally gifted postdocs and students, many of whom have continued to make substantial contributions to CLCs or other channels in their own laboratories.
Glossary
- ACh
acetylcholine
- AChR
ACh receptor
- CBS
cystathionine-β-synthase
- DIDS
4,4′-diisothio-cyanostilbene-2,2′-disulphonate
- PTH
parathyroid hormone
- SITS
4-acetamido-4′-isothiocyanostilbene-9,4′-disulo-phonic acid
- SV
synaptic vesicle
- TAL
thick ascending limb
Biography
Thomas J. Jentsch received both his PhD in physics (1982) and his MD (1984) from the Freie Universität Berlin. After characterizing Na+-coupled bicarbonate transport with Michael Wiederholt at the Institute for Clinical Physiology in Berlin, he joined (in 1986) Harvey Lodish's laboratory at theWhitehead Institute (MIT) as a postdoc. After his first attempt to clone a Cl- channel in Harvey’s laboratory was a failure, he succeeded in molecularly identifying the first voltage-gated Cl- channel in his own group at the Centre for Molecular Neurobiology (ZMNH) of Hamburg University in 1990. This opened the door to the CLC gene family of Cl- channels and transporters, which he subsequently characterized using a broad array of biophysical, cell biological and genetic techniques. He has discovered several human diseases related tomutations in CLC and other channels and extensively uses genetic mouse models to study the physiological and pathological roles of ion transport proteins. In 2006, he moved from Hamburg back to Berlin where he was co-appointed by the Leibniz-Institut für Molekulare Pharmakologie (FMP), the Max-Delbrück-Centrum (MDC) and the Charit’e University Medicine. Although best known for his discovery and characterization of CLC transport proteins, he also made seminal contributions to the field of KCNQ K+ channels and KCC K+-Cl- cotransporters.Most recently, his laboratory discovered that LRRC8 heteromers constitute the long-sought volume-regulated anion channel VRAC.

Additional information
Competing interests
The author declares that there are no competing interests.
References
- Accardi A. Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- Accardi A. Pusch M. Fast and slow gating relaxations in the muscle chloride channel CLC-1. J Gen Physiol. 2000;116:433–444. doi: 10.1085/jgp.116.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi A, Walden M, Nguitragool W, Jayaram H, Williams C. Miller C. Separate ion pathways in a Cl−/H+ exchanger. J Gen Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi S, Uchida S, Ito H, Hata M, Hiroe M, Marumo F. Sasaki S. Two isoforms of a chloride channel predominantly expressed in thick ascending limb of Henle’s loop and collecting ducts of rat kidney. J Biol Chem. 1994;269:17677–17683. [PubMed] [Google Scholar]
- Akizuki N, Uchida S, Sasaki S. Marumo F. Impaired solute accumulation in inner medulla of Clcnk1−/− mice kidney. Am J Physiol Renal Physiol. 2001;280:F79–F87. doi: 10.1152/ajprenal.2001.280.1.F79. [DOI] [PubMed] [Google Scholar]
- Arreola J, Begenisch T, Nehrke K, Nguyen HV, Park K, Richardson L, Yang B, Schutte BC, Lamb FS. Melvin JE. Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl− channel gene. J Physiol. 2002;545.1:207–216. doi: 10.1113/jphysiol.2002.021980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CK, Steinmeyer K, Schwarz JR. Jentsch TJ. Completely functional double-barreled chloride channel expressed from a single Torpedo cDNA. Proc Natl Acad Sci U S A. 1991;88:11052–11056. doi: 10.1073/pnas.88.24.11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetts B, Parker MW. Cromer BA. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J Biol Chem. 2005;282:32780–32791. doi: 10.1074/jbc.M703259200. [DOI] [PubMed] [Google Scholar]
- Bennetts B, Rychkov GY, Ng HL, Morton CJ, Stapleton D, Parker MW. Cromer BA. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J Biol Chem. 2007;280:32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- Bennetts B, Yu Y, Chen TY. Parker MW. Intracellular beta-nicotinamide adenine dinucleotide inhibits the skeletal muscle ClC-1 chloride channel. J Biol Chem. 2012;287:25808–25820. doi: 10.1074/jbc.M111.327551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsdorf EY, Zdebik AA. Jentsch TJ. Residues impor-tant for nitrate/proton coupling in plant and mammalian CLC transporters. J Biol Chem. 2009;284:11184–11193. doi: 10.1074/jbc.M901170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NVAM, Antignac C, Sudbrack R, Kispert A. Hildebrandt F. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hübner CA. Jentsch TJ. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci. 2007;27:6581–6589. doi: 10.1523/JNEUROSCI.0338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösl MR, Stein V, Hübner C, Zdebik AA, Jordt SE, Mukhophadhyay AK, Davidoff MS, Holstein AF. Jentsch TJ. Male germ cells and photoreceptors, both depending on close cell-cell interactions, degenerate upon ClC-2 Cl−-channel disruption. EMBO J. 2001;20:1289–1299. doi: 10.1093/emboj/20.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt S. Jentsch TJ. ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 1995;377:15–20. doi: 10.1016/0014-5793(95)01298-2. [DOI] [PubMed] [Google Scholar]
- Bykova EA, Zhang XD, Chen TY. Zheng J. Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat Struct Mol Biol. 2006;13:1115–1119. doi: 10.1038/nsmb1176. [DOI] [PubMed] [Google Scholar]
- Catalán M, Cornejo I, Figueroa CD, Niemeyer MI, Sepúlveda FV. Cid LP. ClC-2 in guinea pig colon: mRNA, immunolabeling, and functional evidence for surface epithelium localization. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1004–G1013. doi: 10.1152/ajpgi.00158.2002. [DOI] [PubMed] [Google Scholar]
- Catalán MA, Flores CA, González-Begne M, Zhang Y, Sepúlveda FV. Melvin JE. Severe defects in absorptive ion transport in distal colons of mice that lack ClC-2 channels. Gastroenterology. 2012;142:346–354. doi: 10.1053/j.gastro.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán M, Niemeyer MI, Cid LP. Sepúlveda FV. Basolateral ClC-2 chloride channels in surface colon epithelium: Regulation by a direct effect of intracellular chloride. Gastroenterology. 2004;126:1104–1114. doi: 10.1053/j.gastro.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Chalhoub N, Benachenhou N, Rajapurohitam V, Pata M, Ferron M, Frattini A, Villa A. Vacher J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med. 2003;9:399–406. doi: 10.1038/nm842. [DOI] [PubMed] [Google Scholar]
- Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA. Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- Chen TY( Coupling gating with ion permeation in ClC channels. Sci STKE. 2003;2003:pe23. doi: 10.1126/stke.2003.188.pe23. [DOI] [PubMed] [Google Scholar]
- Chen TT, Klassen TL, Goldman AM, Marini C, Guerrini R. Noebels JL. Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology. 2013;80:1078–1085. doi: 10.1212/WNL.0b013e31828868e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY. Miller C. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J Gen Physiol. 1996;108:237–250. doi: 10.1085/jgp.108.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleiren E, Benichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, deVernejoul MC. Van Hul W. Albers-Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet. 2001;10:2861–2867. doi: 10.1093/hmg/10.25.2861. [DOI] [PubMed] [Google Scholar]
- De Angeli A, Monachello D, Ephritikhine G, Frachisse JM, Thomine S, Gambale F. Barbier-Brygoo H. The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature. 2006;442:939–942. doi: 10.1038/nature05013. [DOI] [PubMed] [Google Scholar]
- Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, van Berkel C, Polder E, Tollard E, Darios F, Brice A, de Die-Smulders CE, Vles JS, Vanderver A, Uziel G, Yalcinkaya C, Frints SG, Kalscheuer VM, Klooster J, Kamermans M, Abbink TE, Wolf NI, Sedel F. van der Knaap MS. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12:659–668. doi: 10.1016/S1474-4422(13)70053-X. [DOI] [PubMed] [Google Scholar]
- Di Bella D, Pareyson D, Savoiardo M, Farina L, Ciano C, Caldarazzo S, Sagnelli A, Bonato S, Nava S, Bresolin N, Tedeschi G, Taroni F. Salsano E. Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology. 2014;83:1217–1218. doi: 10.1212/WNL.0000000000000812. [DOI] [PubMed] [Google Scholar]
- Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC, Nehrke K, Williamson RA. Lamb FS. Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res. 2002;958:227–250. doi: 10.1016/s0006-8993(02)03519-9. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR. Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Dubey M, Bugiani M, Ridder MC, Postma NL, Brouwers E, Polder E, Jacobs JG, Baayen JC, Klooster J, Kamermans M, Aardse R, de Kock CP, Dekker MP, van Weering JR, V MH, Abbink TE, Scheper GC, Boor I, Lodder JC, Mansvelder HD. van der Knaap MS. Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Annals Neurol. 2015;77:114–131. doi: 10.1002/ana.24307. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT. MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB. MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- Estévez R, Boettger T, Stein V, Birkenhäger R, Otto M, Hildebrandt F. Jentsch TJ. Barttin is a Cl−-channel β-subunit crucial for renal Cl−-reabsorption and inner ear K+-secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- Estévez R, Pusch M, Ferrer-Costa C, Orozco M, Jentsch TJ. Functional and structural conservation of CBS domains from CLC chloride channels. J Physiol. 2004;557:363–378. doi: 10.1113/jphysiol.2003.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Campbell EB, Hsiung Y. MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SE, Black GC, Lloyd SE, Hatchwell E, Wrong O, Thakker RV. Craig IW. Isolation and partial characterization of a chloride channel gene which is expressed in kidney and is a candidate for Dent’s disease (an X-linked hereditary nephrolithiasis) Hum Mol Genet. 1994;3:2053–2059. [PubMed] [Google Scholar]
- Földy C, Lee SH, Morgan RJ. Soltesz I. Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat Neurosci. 2010;13:1047–1049. doi: 10.1038/nn.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong P, Rehfeldt A. Jentsch TJ. Determinants of slow gating in ClC-0, the voltage-gated chloride channel of Torpedo marmorata. Am J Physiol. 1998;274:C966–C973. doi: 10.1152/ajpcell.1998.274.4.C966. Cell Physiol. [DOI] [PubMed] [Google Scholar]
- Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM, Mihci E, Notarangelo LD, Ramenghi U, Teti A, Van Hove J, Vujic D, Young T, Albertini A, Orchard PJ, Vezzoni P. Villa A. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res. 2003;18:1740–1747. doi: 10.1359/jbmr.2003.18.10.1740. [DOI] [PubMed] [Google Scholar]
- Friedrich T, Breiderhoff T. Jentsch TJ. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem. 1999;274:896–902. doi: 10.1074/jbc.274.2.896. [DOI] [PubMed] [Google Scholar]
- Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH. Riordan JR. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with CFTR-interacting PDZ proteins. J Biol Chem. 2003;278:6440–6449. doi: 10.1074/jbc.M211050200. [DOI] [PubMed] [Google Scholar]
- Gong W, Xu H, Shimizu T, Morishima S, Tanabe S, Tachibe T, Uchida S, Sasaki S. Okada Y. ClC-3-independent, PKC-dependent activity of volume-sensitive Cl channel in mouse ventricular cardiomyocytes. Cell Physiol Biochem. 2004;14:213–224. doi: 10.1159/000080330. [DOI] [PubMed] [Google Scholar]
- Graves AR, Curran PK, Smith CL. Mindell JA. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–792. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- Gründer S, Thiemann A, Pusch M. Jentsch TJ. Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature. 1992;360:759–762. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- Günther W, Luchow A, Cluzeaud F, Vandewalle A. Jentsch TJ. ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc Natl Acad Sci U S A. 1998;95:8075–8080. doi: 10.1073/pnas.95.14.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther W, Piwon N. Jentsch TJ. The ClC-5 chloride channel knock-out mouse – an animal model for Dent’s disease. Pflügers Arch. 2003;445:456–462. doi: 10.1007/s00424-002-0950-6. [DOI] [PubMed] [Google Scholar]
- Guzman RE, Alekov AK, Filippov M, Hegermann J. Fahlke C. Involvement of ClC-3 chloride/proton exchangers in controlling glutamatergic synaptic strength in cultured hippocampal neurons. Front Cell Neurosci. 2014;8:143. doi: 10.3389/fncel.2014.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman RE, Grieschat M, Fahlke C. Alekov AK. ClC-3 is an intracellular chloride/proton exchanger with large voltage-dependent nonlinear capacitance. ACS Chem Neurosci. 2013;4:994–1003. doi: 10.1021/cn400032z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara-Chikuma M, Wang Y, Guggino SE, Guggino WB. Verkman AS. Impaired acidification in early endosomes of ClC-5 deficient proximal tubule. Biochem Biophys Res Commun. 2005;329:941–946. doi: 10.1016/j.bbrc.2005.02.060. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S. Verkman AS. ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J Biol Chem. 2005;280:1241–1247. doi: 10.1074/jbc.M407030200. [DOI] [PubMed] [Google Scholar]
- Haug K, Warnstedt M, Alekov AK, Sander T, Ramirez A, Poser B, Maljevic S, Hebeisen S, Kubisch C, Rebstock J, Horvath S, Hallmann K, Dullinger JS, Rau B, Haverkamp F, Beyenburg S, Schulz H, Janz D, Giese B, Muller-Newen G, Propping P, Elger CE, Fahlke C. Lerche H. Retraction: mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2009;41:1043. doi: 10.1038/ng0909-1043. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL. Horowicz P. The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J Physiol. 1959;148:127–160. doi: 10.1113/jphysiol.1959.sp006278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegg-Beiler MB, Sirisi S, Orozco IJ, Ferrer I, Hohensee S, Auberson M, Gödde K, Vilches C, de Heredia ML, Nunes V, Estévez R. Jentsch TJ. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun. 2014;5:3475. doi: 10.1038/ncomms4475. [DOI] [PubMed] [Google Scholar]
- Hryciw DH, Ekberg J, Lee A, Lensink IL, Kumar S, Guggino WB, Cook DI, Pollock CA. Poronnik P. Nedd4-2 functionally interacts with ClC-5: involvement in constitutive albumin endocytosis in proximal tubule cells. J Biol Chem. 2004;279:54996–55007. doi: 10.1074/jbc.M411491200. [DOI] [PubMed] [Google Scholar]
- Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P. Guggino WB. Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines. J Biol Chem. 2003;278:40169–40176. doi: 10.1074/jbc.M307890200. [DOI] [PubMed] [Google Scholar]
- Hu H, Haas SA, Chelly J, vanEsch H, Raynaud M, de Brouwer APM, Weinert S, Froyen G, Frints SGM, Laumonnier F, Zemojtel T, Love MI, Richarrd H, Emde AK, Bienek M, Jensen C, Hambrok M, Fischer U, Langnick C, Feldkamp M, Wissink-Lindhout E, Lebrun N, Castelnau L, Rucci J, Montjean R, Dorseuil O, Billuart P, Stuhlmann T, Shaw M, Corbett MA, Gardner A, Willis-Owen S, Tan C, Friend KL, Belet S, van Roozendaal KE, Jimenez-Pocquet M, Moizard MP, Ronce N, Sun R, O’Keeffe S, Chenna R, van Bömmel A, Göke J, Hackett A, Field M, Christie L, Boyle J, Haan E, Nelson J, Turner G, Baynam G, Gillessen-Kaesbach G, Müller U, Steinberger D, Budny B, Badura-Stronka M, Latos-Bielenska A, Ousager LB, Wieacker P, Rodríguez Criado G, Bondeson ML, Annerén G, Dufke A, Cohen M, Van Maldergem L, Vincent-Delorme C, Echenne B, Simon-Bouy B, Kleefstra T, Willemsen M, Fryns JP, Devriendt K, Ullmann R, Vingron M, Wrogemann K, Wienker TF, Tzschach A, van Bokhoven H, Gecz J, Jentsch TJ, Chen W, Ropers HH, Kalscheuer VM. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry. 2015 doi: 10.1038/mp.2014.193. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes., doi: 10.1038/mp.2014.193 in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Jeong HJ, Park J, Jeon S. Chloride channel 4 is required for nerve growth factor-induced TrkA signaling and neurite outgrowth in PC12 cells and cortical neurons. Neuroscience. 2013;253:389–397. doi: 10.1016/j.neuroscience.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Ignoul S, Simaels J, Hermans D, Annaert W. Eggermont J. Human ClC-6 Is a late endosomal glycoprotein that associates with detergent-resistant lipid domains. PLoS ONE. 2007;2:e474. doi: 10.1371/journal.pone.0000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, García AM. Lodish HF. Primary structure of a novel 4-acetamido-4-isothiocyanostilbene-2,2-disulphonic acid (SITS)-binding membrane protein highly expressed in Torpedo californica electroplax. Biochem J. 1989;261:155–166. doi: 10.1042/bj2610155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Günther W, Pusch M. Schwappach B. Properties of voltage-gated chloride channels of the ClC gene family. J Physiol (Lond) 1995;482:19S–25S. doi: 10.1113/jphysiol.1995.sp020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Keller SK, Koch M. Wiederholt M. Evidence for coupled transport of bicarbonate and sodium in cultured bovine corneal endothelial cells. J Membr Biol. 1984;81:189–204. doi: 10.1007/BF01868713. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Maritzen T, Keating DJ, Zdebik AA. Thevenod F. ClC-3 – a granular anion transporter involved in insulin secretion? Cell Metab. 2010;12:307–308. doi: 10.1016/j.cmet.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Steinmeyer K. Schwarz G. Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature. 1990;348:510–514. doi: 10.1038/348510a0. [DOI] [PubMed] [Google Scholar]
- Jeworutzki E, Lagostena L, Elorza-Vidal X, Lopez-Hernandez T, Estevez R. Pusch M. GlialCAM, a CLC-2 Cl− channel subunit, activates the slow gate of CLC chloride channels. Biophys J. 2014;107:1105–1116. doi: 10.1016/j.bpj.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeworutzki E, López-Hernández T, Capdevila-Nortes X, Sirisi S, Bengtsson L, Montolio M, Zifarelli G, Arnedo T, Müller CS, Schulte U, Nunes V, Martínez A, Jentsch TJ, Gasull X, Pusch M. Estévez R. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl− channel auxiliary subunit. Neuron. 2012;73:951–961. doi: 10.1016/j.neuron.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE. Jentsch TJ. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997;16:1582–1592. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poët M, Steinfeld R, Schweizer M, Kornak U. Jentsch TJ. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079–1091. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieferle S, Fong P, Bens M, Vandewalle A. Jentsch TJ. Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc Natl Acad Sci U S A. 1994;91:6943–6947. doi: 10.1073/pnas.91.15.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Uchida S, Mizutani S, Sasaki S. Marumo F. Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol. 2001;12:1327–1334. doi: 10.1681/ASN.V1271327. [DOI] [PubMed] [Google Scholar]
- Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann-Horn F, Grzeschik KH. Jentsch TJ. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Lodish HF. Primary structure and transmembrane orientation of the murine anion exchange protein. Nature. 1985;316:234–238. doi: 10.1038/316234a0. [DOI] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G. Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–215. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ. Kubisch C. Mutations in the a3 subunit of the vacuolar H+-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059–2063. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]
- L’Hoste S, Diakov A, Andrini O, Genete M, Pinelli L, Grand T, Keck M, Paulais M, Beck L, Korbmacher C, Teulon J. Lourdel S. Characterization of the mouse ClC-K1/barttin chloride channel. Biochim Biophys Acta. 2013;1828:2399–2409. doi: 10.1016/j.bbamem.2013.06.012. [DOI] [PubMed] [Google Scholar]
- Lange PF, Wartosch L, Jentsch TJ. Fuhrmann JC. ClC-7 requires Ostm1 as a β-subunit to support bone resorption and lysosomal function. Nature. 2006;440:220–223. doi: 10.1038/nature04535. [DOI] [PubMed] [Google Scholar]
- Leegwater PA, Yuan BQ, van der Steen J, Mulders J, Konst AA, Boor PK, Mejaski-Bosnjak V, van der Maarel SM, Frants RR, Oudejans CB, Schutgens RB, Pronk JC. van der Knaap MS. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68:831–838. doi: 10.1086/319519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisle L, Ludwig CF, Wagner FA, Jentsch TJ. Stauber T. ClC-7 is a slowly voltage-gated 2Cl−/1H+-exchanger and requires Ostm1 for transport activity. EMBO J. 2011;30:2140–2152. doi: 10.1038/emboj.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang T, Zhao Z. Weinman SA. The ClC-3 chloride channel promotes acidification of lysosomes in CHO-K1 and Huh-7 cells. Am J Physiol Cell Physiol. 2002;282:C1483–C1491. doi: 10.1152/ajpcell.00504.2001. [DOI] [PubMed] [Google Scholar]
- Lipicky RJ. Bryant SH. Sodium, potassium, and chloride fluxes in intercostal muscle from normal goats and goats with hereditary myotonia. J Gen Physiol. 1966;50:89–111. doi: 10.1085/jgp.50.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipicky RJ, Bryant SH. Salmon JH. Cable parameters, sodium, potassium, chloride, and water content, and potassium efflux in isolated external intercostal muscle of normal volunteers and patients with myotonia congenita. J Clin Invest. 1971;50:2091–2103. doi: 10.1172/JCI106703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lísal J. Maduke M. The ClC-0 chloride channel is a ‘broken’ Cl−/H+ antiporter. Nat Struct Mol Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Hernández T, Ridder MC, Montolio M, Capdevila-Nortes X, Polder E, Sirisi S, Duarri A, Schulte U, Fakler B, Nunes V, Scheper GC, Martínez A, Estévez R. van der Knaap MS. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422–432. doi: 10.1016/j.ajhg.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Pusch M. Jentsch TJ. Heteromultimeric CLC chloride channels with novel properties. Proc Natl Acad Sci U S A. 1996;93:13362–13366. doi: 10.1073/pnas.93.23.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig U, Pusch M. Jentsch TJ. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 1996;383:340–343. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]
- Ludwig CF, Ullrich F, Leisle L, Stauber T. Jentsch TJ. Common gating of both CLC transporter subunits underlies voltage-dependent activation of the 2Cl−/1H+ exchanger ClC-7/Ostm1. J Biol Chem. 2013;288:28611–28619. doi: 10.1074/jbc.M113.509364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Rychkov GY, Bykova EA, Zheng J. Bretag AH. Movement of hClC-1 carboxyl termini during common gating and limits on their cytoplasmic location. Biochem J. 2011;436:415–428. doi: 10.1042/BJ20102153. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC. Thornton CA. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- Maritzen T, Keating DJ, Neagoe I, Zdebik AA. Jentsch TJ. Role of the vesicular chloride transporter ClC-3 in neuroendocrine tissue. J Neurosci. 2008;28:10587–10598. doi: 10.1523/JNEUROSCI.3750-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic S. Dutzler R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure. 2007;15:715–725. doi: 10.1016/j.str.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Volk KA, Collins MM, Moreland JG. Lamb FS. Overexpression of ClC-3 in HEK293T cells yields novel currents that are pH-dependent. Am J Physiol Cell Physiol. 2008;294:C251–C262. doi: 10.1152/ajpcell.00338.2007. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Uchida S, Kondo Y, Miyazaki H, Ko SB, Hayama A, Morimoto T, Liu W, Arisawa M, Sasaki S. Marumo F. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat Genet. 1999;21:95–98. doi: 10.1038/5036. [DOI] [PubMed] [Google Scholar]
- Meindl A, Hosenfeld D, Bruckl W, Schuffenhauer S, Jenderny J, Bacskulin A, Oppermann HC, Swensson O, Bouloux P. Meitinger T. Analysis of a terminal Xp22.3 deletion in a patient with six monogenic disorders: implications for the mapping of X linked ocular albinism. J Med Genet. 1993;30:838–842. doi: 10.1136/jmg.30.10.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S. Dutzler R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure. 2006;14:299–307. doi: 10.1016/j.str.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Meyer S, Savaresi S, Forster IC. Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat Struct Mol Biol. 2007;14:60–67. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- Middleton RE, Pheasant DJ. Miller C. Purification, reconstitution, and subunit composition of a voltage- gated chloride channel from Torpedo electroplax. Biochemistry. 1994;33:13189–13198. doi: 10.1021/bi00249a005. [DOI] [PubMed] [Google Scholar]
- Middleton RE, Pheasant DJ. Miller C. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- Miller C. Open-state substructure of single chloride channels from Torpedo electroplax. Phil Trans R Soc Lond. 1982;299:401–411. doi: 10.1098/rstb.1982.0140. [DOI] [PubMed] [Google Scholar]
- Miller C. White MM. Dimeric structure of single chloride channels from Torpedo electroplax. Proc Natl Acad Sci U S A. 1984;81:2772–2775. doi: 10.1073/pnas.81.9.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad-Panah R, Harrison R, Dhani S, Ackerley C, Huan LJ, Wang Y. Bear CE. The chloride channel ClC-4 contributes to endosomal acidification and trafficking. J Biol Chem. 2003;278:29267–29277. doi: 10.1074/jbc.M304357200. [DOI] [PubMed] [Google Scholar]
- Neagoe I, Stauber T, Fidzinski P, Bergsdorf EY. Jentsch TJ. The late endosomal CLC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J Biol Chem. 2010;285:21689–21697. doi: 10.1074/jbc.M110.125971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, Baggs R, Shull GE. Melvin JE. Loss of hyperpolarization-activated Cl− current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem. 2002;26:23604–23611. doi: 10.1074/jbc.M202900200. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Cid LP, Sepúlveda FV, Blanz J, Auberson M. Jentsch TJ. No evidence for a role of CLCN2 variants in idiopathic generalized epilepsy. Nat Genet. 2010;42:3. doi: 10.1038/ng0110-3. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Yusef YR, Cornejo I, Flores CA, Sepúlveda FV. Cid LP. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol Genomics. 2004;19:74–83. doi: 10.1152/physiolgenomics.00070.2004. [DOI] [PubMed] [Google Scholar]
- Noda M, Ikeda T, Suzuki H, Takeshima H, Takahashi T, Kuno M. Numa S. Expression of functional sodium channels from cloned cDNA. Nature. 1986;322:826–828. doi: 10.1038/322826a0. [DOI] [PubMed] [Google Scholar]
- Noda M, Takahashi H, Tanabe T, Toyosato M, Furutani Y, Hirose T, Asai M, Inayama S, Miyata T. Numa S. Primary structure of alpha-subunit precursor of Torpedo californica acetylcholine receptor deduced from cDNA sequence. Nature. 1982;299:793–797. doi: 10.1038/299793a0. [DOI] [PubMed] [Google Scholar]
- Nomura N, Tajima M, Sugawara N, Morimoto T, Kondo Y, Ohno M, Uchida K, Mutig K, Bachmann S, Soleimani M, Ohta E, Ohta A, Sohara E, Okado T, Rai T, Jentsch TJ, Sasaki S. Uchida S. Generation and analyses of R8L barttin knockin mouse. Am J Physiol Renal Physiol. 2011;301:F297–F307. doi: 10.1152/ajprenal.00604.2010. [DOI] [PubMed] [Google Scholar]
- Novarino G, Weinert S, Rickheit G. Jentsch TJ. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science. 2010;328:1398–1401. doi: 10.1126/science.1188070. [DOI] [PubMed] [Google Scholar]
- Nozu K, Inagaki T, Fu XJ, Nozu Y, Kaito H, Kanda K, Sekine T, Igarashi T, Nakanishi K, Yoshikawa N, Iijima K. Matsuo M. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet. 2008;45:182–186. doi: 10.1136/jmg.2007.052944. [DOI] [PubMed] [Google Scholar]
- Ogura T, Furukawa T, Toyozaki T, Yamada K, Zheng YJ, Katayama Y, Nakaya H. Inagaki N. ClC-3B, a novel ClC-3 splicing variant that interacts with EBP50 and facilitates expression of CFTR-regulated ORCC. FASEB J. 2002;16:S63–S65. doi: 10.1096/fj.01-0845fje. [DOI] [PubMed] [Google Scholar]
- Okkenhaug H, Weylandt KH, Carmena D, Wells DJ, Higgins CF. Sardini A. The human ClC-4 protein, a member of the CLC chloride channel/transporter family, is localized to the endoplasmic reticulum by its N-terminus. FASEB J. 2006;20:2390–2392. doi: 10.1096/fj.05-5588fje. [DOI] [PubMed] [Google Scholar]
- Picollo A. Pusch M. Chloride / proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- Piwon N, Günther W, Schwake M, Bösl MR. Jentsch TJ. ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408:369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- Poët M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S, Wurst W, Schmitt A, Fuhrmann JC, Planells-Cases R, Mole SE, Hübner CA. Jentsch TJ. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci U S A. 2006;103:13854–13859. doi: 10.1073/pnas.0606137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preobraschenski J, Zander JF, Suzuki T, Ahnert-Hilger G. Jahn R. Vesicular glutamate transporters use flexible anion and cation binding sites for efficient accumulation of neurotransmitter. Neuron. 2014;84:1287–1301. doi: 10.1016/j.neuron.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Pressey SN, O’Donnell KJ, Stauber T, Fuhrmann JC, Tyynelä J, Jentsch TJ. Cooper JD. Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J Neuropathol Exp Neurol. 2010;69:1228–1246. doi: 10.1097/NEN.0b013e3181ffe742. [DOI] [PubMed] [Google Scholar]
- Pusch M, Jordt SE, Stein V. Jentsch TJ. Chloride dependence of hyperpolarization-activated chloride channel gates. J Physiol (Lond) 1999;515:341–353. doi: 10.1111/j.1469-7793.1999.341ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Rehfeldt A. Jentsch TJ. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- Pusch M, Steinmeyer K. Jentsch TJ. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys J. 1994;66:149–152. doi: 10.1016/S0006-3495(94)80753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Steinmeyer K, Koch MC. Jentsch TJ. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the ClC-1 chloride channel. Neuron. 1995;15:1455–1463. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP. Patapoutian A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 2014;157:447–458. doi: 10.1016/j.cell.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratté S. Prescott SA. ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci. 2011;31:15838–15843. doi: 10.1523/JNEUROSCI.2748-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed AA, Loh NY, Terryn S, Lippiat JD, Partridge C, Galvanovskis J, Williams SE, Jouret F, Wu FT, Courtoy PJ, Nesbit MA, Rorsman P, Devuyst O, Ashcroft FM. Thakker RV. CLC-5 and KIF3B interact to facilitate CLC-5 plasma membrane expression, endocytosis, and microtubular transport: relevance to pathophysiology of Dent’s disease. Am J Physiol Renal Physiol. 2010;298:F365–F380. doi: 10.1152/ajprenal.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazanski V, Deriy LV, Shevchenko PD, Le B, Gomez EA. Nelson DJ. Presynaptic CLC-3 determines quantal size of inhibitory transmission in the hippocampus. Nat Neurosci. 2011;14:487–494. doi: 10.1038/nn.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Anwar S, Fischer M, Ahmed ZM, Khan SY, Janssen AG, Zafar AU, Scholl U, Husnain T, Belyantseva IA, Friedman PL, Riazuddin S, Friedman TB. Fahlke C. Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am J Hum Genet. 2009;85:273–280. doi: 10.1016/j.ajhg.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard EA. Miller C. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 1990;247:1208–1210. doi: 10.1126/science.2156338. [DOI] [PubMed] [Google Scholar]
- Rickheit G, Maier H, Strenzke N, Andreescu CE, De Zeeuw CI, Muenscher A, Zdebik AA. Jentsch TJ. Endocochlear potential depends on Cl− channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2008;27:2907–2917. doi: 10.1038/emboj.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickheit G, Wartosch L, Schaffer S, Stobrawa SM, Novarino G, Weinert S. Jentsch TJ. Role of ClC-5 in renal endocytosis is unique among CLC exchangers and does not require PY-motif-dependent ubiquitylation. J Biol Chem. 2010;285:17595–17603. doi: 10.1074/jbc.M110.115600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke I, Artmann J. Stein V. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci. 2010;30:4776–4786. doi: 10.1523/JNEUROSCI.6299-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JL, Kolmakova-Partensky L. Miller C. Design, function and structure of a monomeric ClC transporter. Nature. 2010;468:844–847. doi: 10.1038/nature09556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Adler DA, Borsani G, Tsuchiya K, Franco B, Hauge X, Disteche C, Chapman V. Ballabio A. Different chromosomal localization of the Clcn4 gene in Mus spretus and C57BL/6 J mice. Nat Genet. 1995;10:466–471. doi: 10.1038/ng0895-466. [DOI] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Astill DS, Roberts ML, Jentsch TJ. Bretag AH. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J Physiol. 1996;497:423–435. doi: 10.1113/jphysiol.1996.sp021778. (London) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov GY, Pusch M, Roberts ML, Jentsch TJ. Bretag AH. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. J Gen Physiol. 1998;111:653–665. doi: 10.1085/jgp.111.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Love R, Styers ML, Werner E, Peden A, Rodriguez S, Gearing M, Wainer BH. Faundez V. AP-3-dependent mechanisms control the targeting of a chloride channel (ClC-3) in neuronal and non-neuronal cells. J Biol Chem. 2004;279:25430–25439. doi: 10.1074/jbc.M402331200. [DOI] [PubMed] [Google Scholar]
- Saviane C, Conti F. Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J Gen Physiol. 1999;113:457–468. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel O, Zdebik A, Lourdel S. Jentsch TJ. Voltage-dependent electrogenic chloride proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Schenck S, Wojcik SM, Brose N. Takamori S. A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosci. 2009;12:156–162. doi: 10.1038/nn.2248. [DOI] [PubMed] [Google Scholar]
- Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, Seyberth HW. Waldegger S. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350:1314–1319. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- Schmidt-Rose T. Jentsch TJ. Reconstitution of functional voltage-gated chloride channels from complementary fragments of CLC-1. J Biol Chem. 1997;272:20515–20521. doi: 10.1074/jbc.272.33.20515. [DOI] [PubMed] [Google Scholar]
- Scholl U, Hebeisen S, Janssen AG, Müller-Newen G, Alekov A. Fahlke C. Barttin modulates trafficking and function of ClC-K channels. Proc Natl Acad Sci U S A. 2006;103:11411–11416. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M, Friedrich T. Jentsch TJ. An internalization signal in ClC-5, an endosomal Cl−-channel mutated in Dent’s disease. J Biol Chem. 2001;276:12049–12054. doi: 10.1074/jbc.M010642200. [DOI] [PubMed] [Google Scholar]
- Scimeca JC, Franchi A, Trojani C, Parrinello H, Grosgeorge J, Robert C, Jaillon O, Poirier C, Gaudray P. Carle GF. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (ococ) mutants. Bone. 2000;26:207–213. doi: 10.1016/s8756-3282(99)00278-1. [DOI] [PubMed] [Google Scholar]
- Sekine T, Komoda F, Miura K, Takita J, Shimadzu M, Matsuyama T, Ashida A. Igarashi T. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant. 2014;29:376–384. doi: 10.1093/ndt/gft394. [DOI] [PubMed] [Google Scholar]
- Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E. Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- Staley K, Smith R, Schaack J, Wilcox C. Jentsch TJ. Alteration of GABAA receptor function following gene transfer of the CLC-2 chloride channel. Neuron. 1996;17:543–551. doi: 10.1016/s0896-6273(00)80186-5. [DOI] [PubMed] [Google Scholar]
- Stauber T. Jentsch TJ. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J Biol Chem. 2010;285:34537–34548. doi: 10.1074/jbc.M110.162545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ. Grinstein S. A cation counterflux supports lysosomal acidification. J Cell Biol. 2010;189:1171–1186. doi: 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S. Jentsch TJ. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. 1991;354:304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Lorenz C, Pusch M, Koch MC. Jentsch TJ. Multimeric structure of ClC-1 chloride channel revealed by mutations in dominant myotonia congenita (Thomsen) EMBO J. 1994;13:737–743. doi: 10.1002/j.1460-2075.1994.tb06315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer K, Ortland C. Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 1991;354:301–304. doi: 10.1038/354301a0. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Schwappach B, Bens M, Vandewalle A. Jentsch TJ. Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J Biol Chem. 1995;270:31172–31177. doi: 10.1074/jbc.270.52.31172. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H, Draguhn A, Jahn R. Jentsch TJ. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/s0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Rai T, Hayama A, Sohara E, Suda S, Itoh T, Sasaki S. Uchida S. Intracellular localization of ClC chloride channels and their ability to form hetero-oligomers. J Cell Physiol. 2006;206:792–798. doi: 10.1002/jcp.20516. [DOI] [PubMed] [Google Scholar]
- Thiemann A, Gründer S, Pusch M. Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- Thomsen J. Tonische Krämpfe in willkürlich beweglichen Muskeln in Folge von ererbter psychischer Disposition. Arch Psychiatr Nervenkrankh. 1876;6:702–718. [Google Scholar]
- Uchida S, Sasaki S, Nitta K, Uchida K, Horita S, Nihei H. Marumo F. Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J Clin Invest. 1995;95:104–113. doi: 10.1172/JCI117626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst MA, Bassi MT, Borsani G, Wapenaar MC, Ferrero GB, de Conciliis L, Rugarli EI, Grillo A, Franco B, Zoghbi HY, et al. A gene from the Xp22.3 region shares homology with voltage-gated chloride channels. Hum Mol Genet. 1994;3:547–552. doi: 10.1093/hmg/3.4.547. [DOI] [PubMed] [Google Scholar]
- Vandewalle A, Cluzeaud F, Bens M, Kieferle S, Steinmeyer K. Jentsch TJ. Localization and induction by dehydration of ClC-K chloride channels in the rat kidney. Am J Physiol. 1997;272:F678–F688. doi: 10.1152/ajprenal.1997.272.5.F678. Renal Physiol. [DOI] [PubMed] [Google Scholar]
- Vandewalle A, Cluzeaud F, Peng KC, Bens M, Lüchow A, Günther W. Jentsch TJ. Tissue distribution and subcellular localization of the ClC-5 chloride channel in rat intestinal cells. Am J Physiol Cell Physiol. 2001;280:C373–C381. doi: 10.1152/ajpcell.2001.280.2.C373. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, Barth-Maron A, Greenberg ME, Stuhlmann T, Weinert S, Jentsch TJ, Pazzi M, Restifo LL, Talwar D, Erickson RP. Hammer MF. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–1281. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T. Jentsch TJ. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 2014;344:634–638. doi: 10.1126/science.1252826. [DOI] [PubMed] [Google Scholar]
- Waldegger S, Jeck N, Barth P, Peters M, Vitzthum H, Wolf K, Kurtz A, Konrad M. Seyberth HW. Barttin increases surface expression and changes current properties of ClC-K channels. Pflügers Arch. 2002;444:411–418. doi: 10.1007/s00424-002-0819-8. [DOI] [PubMed] [Google Scholar]
- Waldegger S. Jentsch TJ. Functional and structural analysis of ClC-K chloride channels involved in renal disease. J Biol Chem. 2000;275:24527–24533. doi: 10.1074/jbc.M001987200. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K. Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, Thakker RV, Guggino S. Guggino WB. Mice lacking renal chloride channel, CLC-5, are a model for Dent’s disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet. 2000;9:2937–2945. doi: 10.1093/hmg/9.20.2937. [DOI] [PubMed] [Google Scholar]
- Wartosch L, Fuhrmann JC, Schweizer M, Stauber T. Jentsch TJ. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009;23:4056–4068. doi: 10.1096/fj.09-130880. [DOI] [PubMed] [Google Scholar]
- Waters CW, Varuzhanyan G, Talmadge RJ. Voss AA. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci U S A. 2013;110:9160–9165. doi: 10.1073/pnas.1220068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Hohensee S, Chan WL, Kornak U. Jentsch TJ. Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 2014;15:784–791. doi: 10.15252/embr.201438553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U. Jentsch TJ. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science. 2010;328:1401–1403. doi: 10.1126/science.1188072. [DOI] [PubMed] [Google Scholar]
- Weinreich F. Jentsch TJ. Pores formed by single subunits in mixed dimers of different CLC chloride channels. J Biol Chem. 2001;276:2347–2353. doi: 10.1074/jbc.M005733200. [DOI] [PubMed] [Google Scholar]
- White MM. Miller C. A voltage-gated anion channel from the electric organ of Torpedo californica. J Biol Chem. 1979;254:10161–10166. [PubMed] [Google Scholar]
- Wrong OM, Norden AG. Feest TG. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87:473–493. [PubMed] [Google Scholar]
- Yoshikawa M, Uchida S, Ezaki J, Rai T, Hayama A, Kobayashi K, Kida Y, Noda M, Koike M, Uchiyama Y, Marumo F, Kominami E. Sasaki S. CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes Cells. 2002;7:597–605. doi: 10.1046/j.1365-2443.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- Zdebik AA, Cuffe J, Bertog M, Korbmacher C. Jentsch TJ. Additional disruption of the ClC-2 Cl− channel does not exacerbate the cystic fibrosis phenotype of CFTR mouse models. J Biol Chem. 2004;279:22276–2226783. doi: 10.1074/jbc.M309899200. [DOI] [PubMed] [Google Scholar]
- Zdebik AA, Zifarelli G, Bergsdorf E-Y, Soliani P, Scheel O, Jentsch TJ. Pusch M. Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J Biol Chem. 2008;283:4219–4227. doi: 10.1074/jbc.M708368200. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Tseng PY. Chen TY. ATP inhibition of CLC-1 is controlled by oxidation and reduction. J Gen Physiol. 2008;132:421–428. doi: 10.1085/jgp.200810023. [DOI] [PMC free article] [PubMed] [Google Scholar]