

Abstract
Cardiac arrhythmias are often associated with mutations in SCN5A the gene that encodes the cardiac paralogue of the voltage-gated sodium channel, NaV1.5. The NaV1.5 mutants R1193Q and E1784K give rise to both long QT and Brugada syndromes. Various environmental factors, including temperature, may unmask arrhythmia. We sought to determine whether temperature might be an arrhythmogenic trigger in these two mixed syndrome mutants. Whole-cell patch clamp was used to measure the biophysical properties of NaV1.5 WT, E1784K and R1193Q mutants. Recordings were performed using Chinese hamster ovary (CHOk1) cells transiently transfected with the NaV1.5 α subunit (WT, E1784K, or R1193Q), β1 subunit, and eGFP. The channels’ voltage-dependent and kinetic properties were measured at three different temperatures: 10ºC, 22ºC, and 34ºC. The E1784K mutant is more thermosensitive than either WT or R1193Q channels. When temperature is elevated from 22°C to 34°C, there is a greater increase in late INa and use-dependent inactivation in E1784K than in WT or R1193Q. However, when temperature is lowered to 10°C, the two mutants show a decrease in channel availability. Action potential modelling using Q10 fit values, extrapolated to physiological and febrile temperatures, show a larger transmural voltage gradient in E1784K compared to R1193Q and WT with hyperthermia. The E1784K mutant is more thermosensitive than WT or R1193Q channels. This enhanced thermosensitivity may be a mechanism for arrhythmogenesis in patients with E1784K sodium channels.
Key points
The E1784K mixed syndrome mutant of the cardiac voltage-gated sodium channel, NaV1.5, responds differently to temperature changes compared to the R1193Q mutant and wild-type (WT) NaV1.5.
In E1784K, elevated temperature causes a larger increase in persistent current; there is also an increase in use-dependent inactivation at 1 Hz, which is not apparent at 3 Hz.
WT NaV1.5 and R1193Q channels respond similarly to temperature changes.
Action potential modelling (from extrapolated temperature coefficient (Q10)values) predicts the effects of differential temperature sensitivity on the cardiac action potential: greater attenuation of the epicardial action potential occurs in E1784K as temperature shifts from hypothermic to hyperthermic conditions, and when transient outward potassium currents are increased.
The results from the action potential model predict that, at febrile temperatures, E1784K channels results in a larger transmural voltage gradient.
Hyperthermia exacerbates the Brugada syndrome 1 (BrS1) phenotype, which may be arrhythmogenic in E1784K mutants.
Introduction
Voltage-gated sodium channels are responsible for the upstroke of action potentials in most cardiac, skeletal, and neuronal tissue. In this study we focus on NaV1.5, the sodium channel most prevalent in the cardiac conduction system and cardiomyocytes (Remme et al. 2009). Mutations in SCN5A, the gene encoding NaV1.5, give rise to a large spectrum of potentially arrhythmogenic disorders including Brugada and long QT-3 syndromes.
Long QT syndrome 3 (LQT3) is due to gain-of-function mutants of NaV1.5, in which the S4 voltage sensor, domain III–IV linker segment, and the C-terminus are most commonly affected (Glaaser et al. 2012). These mutants hinder fast inactivation and lead to an increase in late INa which disrupts the normal balance between outward and inward currents that maintain the plateau phase of the cardiac action potential (AP). An increase in late INa delays cardiomyocyte repolarization and prolongs the cardiac action potential. In contrast, loss-of-function NaV1.5 mutants that diminish peak INa can cause Brugada syndrome 1 (BrS1). Many of the mechanisms underlying BrS1 involve stabilizing inactivated states and/or destabilizing the activated state (Grant et al. 2002). Due to heterogeneous channel expression in the heart tissue, BrS1 preferentially affects the right epicardial AP. Relatively high levels of IK,To (transient outward potassium current), found in the right epicardium, can cause the loss of the AP plateau in myocytes with decreased INa. This loss generates a transmural voltage gradient causing ST segment elevation, visible in the right precordial leads. This phenomenon is also known as ‘phase 2 reentry’ (Chen et al. 1998; Antzelevitch et al. 2005). There are three different manifestations of this phenomenon in the ECG: Type I is characterized by a J-point elevation, a coved ST-segment, and an inverted T wave in V1-V3; Type II shows a saddleback-type ST-segment elevation; Type III shows an ST-segment elevation accompanied by a J-point elevation. Types II and III are not diagnostic of BrS (Antzelevitch et al. 2005).
BrS1 and LQT3 have been reported to precipitate into ventricular tachycardia and ventricular fibrillation leading to sudden death in men and women, and sudden infant death syndrome in young children (Priori et al. 2000; Garcia-Borbolla et al. 2007; Makaryus et al. 2009). BrS1 may be unmasked by decreases in plasma pH, changes in body core temperatures, and pharmacological agents that block sodium channels (Mok et al. 2003; Makaryus et al. 2009; Doetzer et al. 2011). Elevated temperatures exacerbate the decrease in channel function in the BrS1 mutant T1620M by causing a larger destabilization of the activated state (Dumaine et al. 1999). Additionally, there is an enhanced onset into inactivation at elevated temperatures. In another thermosensitive-mutant, F1344S, the conductance is further destabilized at elevated temperatures (Keller et al. 2006).
Interestingly, some NaV1.5 mutants, including R1193Q and E1784K, can cause both LQT3 and BrS1 and are referred to as mixed syndromes (Bezzina et al. 1999; Veldkamp et al. 2000; Grant et al. 2002). Molecular genetic screening revealed that a heterozygous mutation, in which a guanine was replaced with an adenine in the 3578th position in exon 20 of SCN5A, results in the R1193Q mutant (Huang et al. 2006). R1193Q is located in the intracellular domain II–III linker region. This mutant stabilizes the fast-inactivated state (Wang et al. 2004; Huang et al. 2006). Enhanced inactivation may lead to a decrease in sodium currents, consistent with the phenotype of BrS1. However, the mutant increases late INa, associated with LQT3 (Sun et al. 2008). A guanine to an adenine mutation in the 5349th position of SCN5A results in the E1784K mutant in the C-terminus of NaV1.5 (Splawski et al. 2000; Tester et al. 2005). E1784K tends to stabilize steady-state inactivation but also increases late INa (Wei et al. 1999; Wang et al. 2004; Makita et al. 2008).
Our study focused on the effects of temperature on wild-type (WT), R1193Q, and E1784K channels. Our results suggest differential thermosensitivity in the E1784K and R1193Q mutants. E1784K is most strongly affected by temperature changes. Action potential simulations suggest that increasing temperature may attenuate the epicardial AP dome in cardiomyocytes expressing the E1784K mutant. Temperature increases may therefore be arrhythmogenic in E1784K.
Methods
Ethical approval
The research was approved by Biohazards review 251–2012 issued by the office of the Environmental Health and Safety at Simon Fraser University, Burnaby, BC, Canada.
Cell culture
Chinese hamster ovary (CHOk1) cells (Sigma-Aldrich, St. Louis, MO, USA) were grown at pH 7.4 in filter sterile F12 (Ham) nutrient medium (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 5% FBS and maintained in a humidified environment at 37°C with 5% CO2. Twenty-four hours prior to electrophysiology experiments, cells were transfected with cDNA for the sodium channel α and β subunits, as well as green fluorescent protein (eGFP). Eight hours after transfection, cells were dissociated with 0.25% trypsin–EDTA (Life Technologies) and then plated on sterile cover slips. These time intervals were used to control for channel expression.
Transfection
Transfection followed the procedures suggested by Qiagen. Briefly, 1 μg of the NaV1.5 α subunit, 0.5 μg of the sodium channel β1 subunit, and 1 μg of eGFP were allowed to incubate with 15 μl of polyfect transfection reagent (Qiagen, Toronto, ON, Canada) and 147 μl of unsupplemented medium for 10 min. The cDNA mixture was then allowed to incubate with the CHOk1 cells for 8 h before plating on coverslips. NaV1.5 mutations were generously provided by Dr Charles Antzelevitch (E1784K) and Dr Mohamed Chahine (R1193Q).
Electrophysiology
Whole-cell recordings were performed in extracellular solution containing (mm): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10 Hepes (pH 7.4). Solutions were titrated with CsOH to pH 7.4. Pipettes were fabricated with a P-1000 puller using borosilicate glass (Sutter Instruments, Novato, CA, USA), dipped in dental wax to reduce capacitance, then thermally polished to a resistance of 1.0-1.5 MΩ. Low resistance electrodes were used to minimize series resistance between pipette and intracellular solution resulting in typical access resistances of 3.5 MΩ or less, thereby minimizing voltage measurement error. Pipettes were filled with intracellular solution, containing (mm): 130 CsF, 10 NaCl, 10 Hepes, and 10 EGTA titrated to pH 7.4.
All recordings were made using an EPC-9 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany) digitized at 20 kHz using an ITC-16 interface (HEKA Elektronik). Data was acquired and low-pass-filtered (5 kHz) using PatchMaster/FitMaster software (HEKA Elektronik) running on an Apple iMac (Apple Computer, Cupertino, CA, USA). Leak subtraction was performed online using a P/4 procedure. Bath solution temperature was controlled using a Peltier device driven by a TC-10 Temperature Controller (Dagan, Minneapolis, MN, USA). Bath temperature was maintained at 10°C, 22°C or 34°C. Experiments were not performed at physiological temperatures because of the inherent instability of cells at temperatures above 34°C. Using a Q10 relationship we extrapolated data to physiological temperatures (described below). After a giga-ohm seal resistance was achieved, the whole-cell configuration was attained. Currents were then allowed to stabilize such that currents measured by successive trains of five 10 ms depolarizations at 1 Hz to 0 mV were similar. Run-down was assessed by comparing peak current amplitudes before and after each protocol. With the exception of use-dependent inactivation protocols, only protocols with less than 5% run-down were used. Use-dependent protocols showing more than 5% run-down were corrected post hoc. The holding potential between protocols was −110 mV. We recorded INa from cells that expressed currents no greater than –5 nA. Cells with larger currents were not used since they gave rise to voltage-error issues. The average voltage error calculated for all cells used in this study is 6.15 mV obtained from a total of 102 cells (Table1). There are no differences between the voltage errors in the different conditions (P > 0.05).
Table 1.
Voltage error between the channel variants at different temperatures
| Temperature | Voltage error (mV) | n | |
|---|---|---|---|
| Wild type | 10°C | 6.22 ± 0.84 | 14 |
| 22°C | 5.15 ± 0.95 | 11 | |
| 34°C | 6.49 ± 1.08 | 10 | |
| R1193Q | 10°C | 5.89 ± 1.01 | 12 |
| 22°C | 6.58 ± 1.37 | 5 | |
| 34°C | 5.95 ± 0.74 | 12 | |
| E1784K | 10°C | 6.22 ± 1.47 | 10 |
| 22°C | 5.58 ± 0.85 | 12 | |
| 34°C | 7.23 ± 0.70 | 16 |
Analysis
Analysis and graphing were done using FitMaster software (HEKA Elektronik) and Igor Pro (Wavemetrics, Lake Oswego, OR, USA) with statistical information derived using JMP statistical software. All data acquisition and analysis programs were run on an Apple iMac (Apple Computer). Statistical significance was accepted at P < 0.05 using a two-factor completely randomized design (CRD) ANOVA test followed by a post hoc Tukey test. We used a post hoc Student’s t test to analyse temperature effect on persistent current (22°C and 34°C) and steady-state slow inactivation (10°C and 22°C). Statistical analysis was performed on the averages between three variables: channel variant (WT, R1193Q, E1784K), temperature (10°C, 22°C, 34°C), and channel variant × temperature. The latter represented the statistical significance of a change in channel variants as a result of temperature changes. All values reported are given as means ± standard error of means for n cells.
Voltage protocols
Activation
To determine the voltage dependence of activation, we measured the peak current amplitude at test pulse potentials ranging from −100 mV to +80 mV in increments of +10 mV for 19 ms. Prior to the test pulse, channels were allowed to recover from fast inactivation at −130 mV for 197 ms. Channel conductance was calculated from peak INa.
where GNa is sodium channel conductance, INa is peak sodium current in response to the command potential V, and Erev is the reversal potential. Calculated values for conductance were fitted with the Boltzmann function:
where G/G max is the normalized conductance amplitude, Vm is the command potential, z is the apparent valency, e0 is the elementary charge, V½ is the midpoint voltage, k is the Boltzmann constant, and T is temperature in °K.
Steady-state fast inactivation (SSFI)
The voltage dependence of SSFI was measured by preconditioning the channels to a hyperpolarizing potential of −130 mV and then eliciting prepulse potentials that range from −130 or −150 to +10 mV in increments of 10 mV for 200 ms. Channel availability was assessed by a test pulse to 0 mV. Different hyperpolarizing prepulse potentials (−130 mV or −150 mV) were used to obtain a plateau for the SSFI curve. Normalized current amplitude as a function of voltage was fitted using the Boltzmann function:
where I/Imax is the normalized current amplitude, z is apparent valency, e0 is the elementary charge, Vm is the prepulse potential, V½ is the midpoint voltage of SSFI, k is the Boltzmann constant, and T is temperature in oK.
Fast inactivation onset
Time constants for open-state fast inactivation were derived by fitting a single exponential function to the decay of current obtained from the activation protocol. To measure onset into fast inactivation, channels were preconditioned at −130 mV prior to a prepulse at −50 mV, −70 mV, or −90 mV for 0–0.256 s. Current amplitude was measured during a test pulse to 0 mV for 20 ms. Normalized current amplitudes as a function of time were fitted using a single exponential equation:
where I is current amplitude, Iss is the plateau amplitude, α is the amplitude at time 0 for time constant τ, and t0 is initial time. Rates of recovery from and onset into fast inactivation were calculated from the reciprocal of the time constants (τ) obtained from the single-exponential fits.
Fast inactivation recovery
Channels were fast-inactivated during a 200 ms depolarizing step to 0 mV. Recovery was measured during a 19 ms test pulse to 0 mV following 0–1.024 s conditioning pulses at −130 mV, −110 mV, or −90 mV. Time constants of fast inactivation recovery as a function of time were fitted using a single exponential equation, as above.
Persistent current
Persistent current was measured between 45 and 50 ms during a 50 ms depolarizing pulse to 0 mV from a holding potential of –130 mV. An average of 60 pulses was used to increase the signal-to-noise ratio.
Use-dependent inactivation (1 Hz and 3 Hz)
Channels accumulated into a use-dependent inactivated state during either a series of 300 × 380 ms depolarizing pulses to 0 mV followed by a 615 ms –90 mV recovery pulse at a frequency 1 Hz, or 500 × 220 ms depolarizing pulses to 0 mV followed by a 110 ms –90 mV recovery pulse at a frequency 3 Hz. Normalized current amplitude as a function of time was fitted with a double exponential.
where I is current amplitude, Iss is the plateau amplitude, α1 and α2 are the amplitudes at time 0 for time constants τ1 and τ2, and t is time.
Slow inactivation onset
To measure onset into slow inactivation, channels were preconditioned at −130 mV for 30 s prior to a prepulse at 0 mV for 0–64 s. A test pulse to 0 mV followed a −130 mV recovery pulse from fast inactivation for 20 ms. Normalized current amplitudes as a function of time were fitted using a double exponential equation.
Steady-state slow inactivation (SSSI)
The voltage dependence of SSSI was measured by preconditioning the channels to a hyperpolarizing potential of −150 mV for 30 s and then eliciting prepulse potentials that ranged from −150 to −10 mV in increments of 20 mV for 60 s. Channel availability was assessed by a test pulse to 0 mV following a −130 mV recovery pulse from fast inactivation at 20 ms. Normalized current amplitude as a function of voltage was fitted using a modified Boltzmann function:
where I1 and I2 are maximum and minimum values of fit. The other symbols are as previously stated.
Slow inactivation recovery
To measure recovery from slow inactivation, channels were preconditioned at −130 mV for 30 s prior to a prepulse at 0 mV for 60 s, followed by series of test pulses to 0 mV for 20 ms between increasing incremental recovery durations at −130 mV for 0–32 s. Normalized current amplitudes as a function of time were fitted using a double exponential equation with the plateau equal to 1.00.
Q10 coefficients
To determine an appropriate fit for the kinetic or thermodynamic parameters plotted as a function of temperature, we used the Q10 formula:
where R is the rate and T is temperature (1 and 2 refer to initial and secondary, respectively). Rate was calculated by the inverse of the τ value. Q10 fits for steady-state midpoints and slopes were calculated by replacing the R values with V½ and z values. Fits for y0 were calculated based of the 1/y0 to yield optimal Q10 values. The best fit was used with three points obtained at 10°C, 22°C and 34°C to increase the accuracy of the Q10 fit. The fit was extrapolated to physiological (37°C) and febrile (41°C) temperatures.
Action potential modelling
Cardiac action potential modelling was based on a modified Ten Tusscher 2006 model, previously described (Ten Tusscher et al. 2004; Ten Tusscher & Panfilov, 2006; Jones et al. 2011). All action potentials were programmed and run in the Python language. The Ito formulation and maximal conductance were modified as suggested by Dumaine et al. 1999. The epicardial L-type calcium current was decreased by 50% to obtain a more realistic action potential length (Dumaine et al. 1999; Xia et al. 2006). Simulations were run at 1 Hz. The 34°C data was incorporated into the model to account for hypothermic conditions. Predicted channel kinetics and steady-state properties at 37°C (normothermia) and 41°C (hyperthermia) temperatures were obtained from the Q10 extrapolations. Our model only accounted for current density, activation, steady-state fast inactivation, fast inactivation kinetics, and persistent INa. The slow inactivation parameters were excluded from the model since we did not have a full slow inactivation kinetics profile. As described earlier, it is challenging to patch at elevated temperatures because of a loss of patch stability. Temperature shifts affect the kinetics of other channels that contribute to the maintenance of the cardiac action potential, thus changes in temperature will have other effects on cardiac action potential morphology, for which we are unable to account. We used the Ten Tusscher model to determine the effects of temperature on cardiac action potential based only on shifts occurring in NaV1.5. We modelled epicardial action potentials with increasing IK,To to simulate changes across the heart wall and between the right and left ventricles.
Results
Activation
We measured current density from the ratio of peak current amplitude to the cell membrane capacitance (μA μF–1). Representative raw current traces are shown for the different channel variants at the three temperatures studied (Fig. 1Aa–Cc). Current densities were different in the channel variants (WT, E1784K, and R1193Q; Table2). The R1193Q mutant significantly decreased current density compared to E1784K and WT (P < 0.01, Fig. 1D). Current density was also significantly affected by temperature (P < 0.01; Table2). Peak INa was lower at 10°C and 22°C compared to 34°C (Fig. 1D). However, temperature increases from 22°C to 34°C significantly increased the current densities in WT and E1784K compared to R1193Q (Fig. 1D, P < 0.01). The R1193Q mutant did not respond to any temperature changes. The Q10 values and extrapolated 37°C and 41°C values (shown in bold) are reported in Table2 and are consistent with temperature insensitivity in R1193Q compared to WT and E1784K.
Figure 1.

Na+ currents
Current recordings of the channel variants are shown at three temperatures (A1-A3 (WT), B1-B3 (R1193Q), C1-C3 (E1784K)). Panel D shows the current density for the different channel variants as a function of temperature. Panel E shows the time to half peak of maximal Na current measured at 0 mV.
Table 2.
Current density and time to half-peak INa
| Current density | Time to half- | ||||
|---|---|---|---|---|---|
| Temperature | (μA μF–1) | n | peak (μs) | n | |
| Wild type | Q10 = 1.20 | Q10 = 1.75 | |||
| 10°C | 313 ± 30 | 11 | 514 ± 50 | 11 | |
| 22°C | 458 ± 65 | 9 | 277 ± 26 | 9 | |
| 34°C | 1009 ± 104 | 8 | 292 ± 37 | 8 | |
| 37°C | 1189 | — | 256 | — | |
| 41°C | 1487 | — | 237 | — | |
| R1193Q | Q10 = 1.22 | Q10 = 1.27 | |||
| 10°C | 328 ± 51 | 8 | 785 ± 32 | 8 | |
| 22°C | 361 ± 48 | 8 | 283 ± 50 | 8 | |
| 34°C | 508 ± 69 | 9 | 331 ± 13 | 9 | |
| 37°C | 528 | — | 274 | — | |
| 41°C | 572 | — | 249 | — | |
| E1784K | Q10 = 1.94 | Q10 = 1.87 | |||
| 10°C | 264 ± 22 | 8 | 829 ± 48 | 8 | |
| 22°C | 407 ± 41 | 9 | 290 ± 15 | 9 | |
| 34°C | 1030 ± 80 | 10 | 150 ± 13 | 10 | |
| 37°C | 1258 | — | 122 | — | |
| 41°C | 1643 | — | 95 | — |
Values in bold were determined by extrapolation of Q10 fits to measured values.
We measured activation rate by fitting the rise phase of INa at 0 mV, from the beginning of the test pulse to the half-peak INa. Time to half-peak was significantly different between the channel variants (Fig. 1E and Table2). Both mutants, E1784K and R1193Q, were slower to reach half-peak than WT channels at 10°C (P < 0.01). Increasing the temperature from 10°C to 22°C resulted in a larger decrease in time to half-peak in E1784K (539 ± 51.1 μs) and R1193Q (501 ± 52.6 μs) compared to WT (237 ± 47 μs, Fig. 1E). Furthermore, elevating temperature from 22°C to 34°C resulted in a large decrease in time to half-peak of E1784K (140 ± 48.3 μs) as opposed to a smaller change in WT and R1193Q (Fig. 1E). Table2 includes Q10 fit values which are consistent with overall trends in which E1784K exhibits higher thermosensitivity compared to WT and R1193Q. The Q10 extrapolation predicts that a febrile state markedly decreases the time taken to reach half-peak.
We show conductance as a function of membrane potential in Fig. 2 for the channel variant effect and the temperature effect. Figure 4A, and B shows the interaction effect for both the conductance midpoint and conductance slope presented as Q10 fits. The midpoint of activation (V½) was significantly different between the channel variants (P < 0.01): V½ for E1784K was depolarized relative to WT and R1193Q, which were not significantly different from one another (Figs 2A–C, 4A, and Table3). This mutant effect on V½ was only evident at 10°C (P < 0.01; Fig. 2A). Temperature affected V½ in a non-linear fashion: 10°C was positively shifted compared to both 22°C and 34°C (Figs 2D–F and 4A). The effects of temperature on V½ are significantly different between the channel variants (P < 0.05). Both E1784K and R1193Q had a significant shift of ∼−10 mV in V½ as temperature was increased from 10°C to 22°C compared to WT channels (P > 0.05). The V½ for WT and R1193Q was not significantly different between 22°C and 34°C, as opposed to a slight hyperpolarization of –3.88 ± 2.36 mV shift in E1784K. This shift is too small to allow meaningful interpretation. The Q10 fit values are reported in Table3 (and shown in Fig. 4A) and are consistent with a slightly higher Q10 value for E1784K compared to R1193Q and WT. The conductance slope (z) was significantly different between the channel variants in which E1784K had a lower slope value compared to WT and R1193Q (P < 0.05; Fig. 4B). Temperature significantly shifted the slope factor to higher values as temperature increased from 10°C to 22°C to 34°C (P < 0.01; Table3). Nevertheless, the changes in slope due to temperature were not significantly different between the channel variants (P > 0.05). The Q10 fit values reported in Table3 are relatively equal between the channel variants.
Figure 2.

Activation
Panels A–F show the voltage-dependence of activation as normalized conductance plotted against membrane potential plotted versus membrane potential. The insets show pulse protocols used to measure INa at different voltages. Panels A–C show the channel variant effect at 10°C, 22°C and 34°C. Panels D–F show the temperature effect for WT, R1193Q and E1784K.
Figure 4.
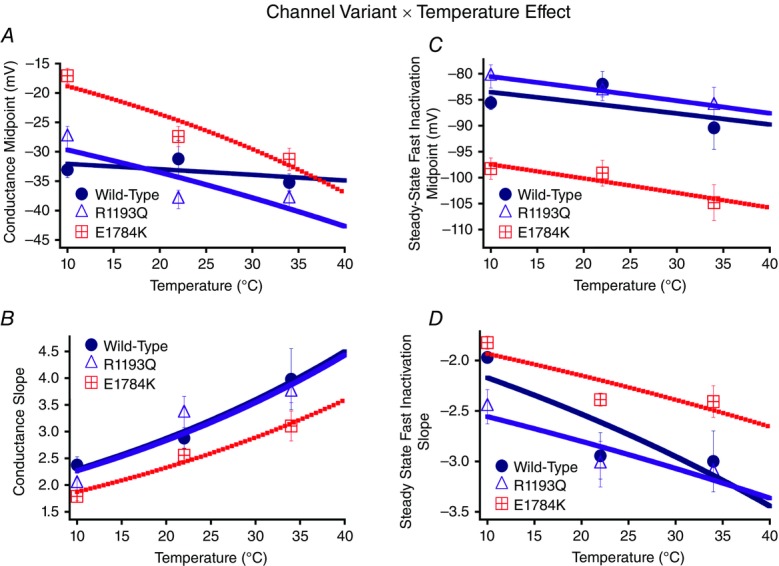
Activation and steady-state fast inactivation Q10 curves
Panels A and B show the conductance midpoint and conductance slope for the channel variants as a function of temperature fit with Q10 curves. Panels C and D show the steady-state fast inactivation midpoint and slope Q10 curves.
Table 3.
Conductance and steady-state fast inactivation
| Activation | Steady-state fast inactivation | ||||||
|---|---|---|---|---|---|---|---|
| Temperature | GV-V½ (mV) | GV-z | n | SSFI-V½ (mV) | SSFI-z | n | |
| Wild type | Q10 = 1.03 | Q10 = 1.25 | Q10 = 1.02 | Q10 = 1.17 | |||
| 10°C | −33.06 ± 1.32 | 2.38 ± 0.15 | 7 | −85.58 ± 1.32 | −1.97 ± 0.05 | 5 | |
| 22°C | −31.21 ± 3.12 | 2.88 ± 0.20 | 9 | −82.01 ± 2.45 | −2.95 ± 0.23 | 5 | |
| 34°C | −35.24 ± 1.49 | 3.98 ± 0.57 | 5 | −90.41 ± 4.14 | −3.00 ± 0.30 | 5 | |
| 37°C | −34.63 | 4.22 | — | −89.26 | −3.30 | — | |
| 41°C | −35.05 | 4.62 | — | −90.00 | −3.51 | — | |
| R1193Q | Q10 = 1.13 | Q10 = 1.25 | Q10 = 1.03 | Q10 = 1.10 | |||
| 10°C | −27.51 ± 1.83 | 2.02 ± 0.11 | 11 | −80.51 ± 2.22 | −2.46 ± 0.17 | 6 | |
| 22°C | −38.12 ± 1.58 | 3.34 ± 0.32 | 7 | −83.34 ± 1.81 | −3.03 ± 0.23 | 5 | |
| 34°C | −38.05 ± 1.45 | 3.74 ± 0.19 | 10 | −86.14 ± 3.52 | −3.11 ± 0.09 | 5 | |
| 37°C | −41.32 | 4.16 | — | −86.92 | −3.28 | — | |
| 41°C | −43.38 | 4.56 | — | −87.91 | −3.40 | — | |
| E1784K | Q10 = 1.25 | Q10 = 1.24 | — | Q10 = 1.03 | Q10 = 1.11 | ||
| 10°C | −17.07 ± 1.28 | 1.79 ± 0.08 | 8 | −98.27 ± 2.07 | −1.82 ± 0.06 | 5 | |
| 22°C | −27.40 ± 1.71 | 2.56 ± 0.13 | 11 | −99.13 ± 2.50 | −2.39 ± 0.06 | 5 | |
| 34°C | −31.29 ± 1.87 | 3.10 ± 0.28 | 15 | −104.83 ± 3.46 | −2.41 ± 0.16 | 6 | |
| 37°C | −34.79 | 3.38 | — | −105.45 | −2.58 | — | |
| 41°C | −38.01 | 3.69 | — | −106.23 | −2.69 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
Fast inactivation
Normalized currents obtained from the SSFI protocols are plotted as a function of membrane potential (Fig. 3). Figure 4C, and D shows the SSFI midpoint and slope Q10 fits for the channel variants as a function of temperature. The midpoint of steady-state fast inactivation (SSFI-V½) was significantly different between the channel variants (P < 0.01; Fig. 4C and Table3): E1784K-V½ was hyperpolarized compared to WT and R1193Q at all temperatures (Fig. 3A–C and Fig. 4C). Temperature also significantly affected SSFI-V½ (P < 0.05). At 34°C, V½ was negatively shifted compared to 22°C and 10°C (Fig. 3D–F). The differences in V½ due to temperature were not significantly different between the channel variants (P > 0.05, Fig. 4C). The Q10 values reported in Table3 are almost identical between the different channel variants. The E1784K mutant had a significantly lower slope (z) value than R1193Q and WT (P < 0.01; Fig. 4D and Table3). The slope was significantly lower at 10°C compared to 22°C and 34°C (P < 0.01, Fig. 4D). We found the changes in slope as a function of temperature were not significantly different between the channel variants (P > 0.05; Fig. 4D). The Q10 fit values are relatively consistent between the different channel variants reported in Table3.
Figure 3.

Steady-state fast inactivation
Panels A–F show the voltage-dependence of steady-state fast inactivation as normalized current plotted against membrane potential. The insets show pulse protocols used to measure INa at different voltages. Panels A–C show the channel variant effect at 10°C, 22°C and 34°C. Panels D–F show the temperature effect for WT, R1193Q and E1784K.
Time constants measured from single exponential fits to the recovery from fast inactivation at −130 mV are shown in Fig. 5. Insets in Fig. 5 show time constants plotted against the membrane potential. The values of the time constants (ms) are reported in Tables4 and 5, for time constants between −130 mV to −70 mV, and −50 mV to +10 mV, respectively. E1784K had enhanced kinetics of fast inactivation between −90 mV to +10 mV compared to R1193Q and WT (P < 0.01; Fig. 6). Fast inactivation kinetics of all channel variants between −130 mV to −90 mV, and −30 mV to −10 mV had a significantly larger (P < 0.01) time constants (decelerated kinetics) at 10°C compared to 22°C and 34°C, which were not significantly different (P > 0.05). At −70 mV, both WT and R1193Q fast inactivation time constants decrease significantly from 10°C to 34°C (P < 0.01) compared to an insignificant change in E1784K (P > 0.05). At −50 mV WT and E1784K channel had reduced time constants when temperature increased from 10°C to 34°C compared to R1193Q, which decreased from 10°C to 22°C (P < 0.01). The Q10 fit values for the different channel variants are shown in bold in Tables4 and 5. The Q10 curves are shown in Fig. 6, with the E1784K exhibiting relatively faster kinetics than both WT and R1193Q at most voltages.
Figure 5.

Fast inactivation (FI) time constants
Panels A–F show the recovery from fast inactivation at −130 mV as normalized currents versus recovery time duration. The insets show the FI time constants plotted against the membrane potential. Panels A–C show the channel variant effects at 10°C, 22°C and 34°C. Panels D–F show the temperature effect for WT, R1193Q and E1784K. Pulse protocols of recovery and onset were not shown for clarity. Please refer to Methods.
Table 4.
Fast inactivation time constants between −130 mV and −70 mV
| Fast inactivation time constants (ms) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Temperature | −130 mV | n | −110 mV | n | −90 mV | n | −70 mV | n | |
| Wild type | Q10 = 1.86 | Q10 = 1.92 | Q10 = 2.18 | Q10 = 4.03 | |||||
| 10°C | 18.0 ± 1.8 | 5 | 36.4 ± 5.2 | 5 | 176.0 ± 52.5 | 4 | 55.0 ± 10.0 | 4 | |
| 22°C | 5.5 ± 1.3 | 6 | 12.4 ± 2.6 | 5 | 46.5 ± 8.3 | 4 | 57.2 ± 3.1 | 5 | |
| 34°C | 3.0 ± 0.5 | 5 | 6.2 ± 0.8 | 5 | 19.9 ± 2.5 | 5 | 8.2 ± 2.1 | 5 | |
| 37°C | 2.4 | — | 5.0 | — | 15.5 | — | 5.3 | — | |
| 41°C | 1.9 | — | 3.9 | — | 11.3 | — | 3.0 | — | |
| R1193Q | Q10 | Q10 = 1.56 | Q10 = 1.71 | Q10 = 1.96 | Q10 = 2.19 | ||||
| 10°C | 15.4 ± 1.8 | 7 | 54.4 ± 6.8 | 4 | 103.2 ± 19.2 | 4 | 55.5 ± 6.2 | 4 | |
| 22°C | 4.4 ± 0.2 | 5 | 14.7 ± 4.4 | 5 | 57.2 ± 6.2 | 5 | 38.5 ± 10.5 | 5 | |
| 34°C | 3.4 ± 0.6 | 5 | 9.6 ± 2.2 | 5 | 23.7 ± 4.2 | 4 | 12.6 ± 3.0 | 4 | |
| 37°C | 2.9 | — | 7.8 | — | 19.2 | — | 9.9 | — | |
| 41°C | 2.4 | — | 6.3 | — | 14.7 | — | 7.2 | — | |
| E1784K | Q10 | Q10 = 2.27 | Q10 = 1.82 | Q10 = 1.74 | Q10 = 1.83 | ||||
| 10°C | 16.0 ± 1.8 | 5 | 25.7 ± 1.9 | 6 | 47.8 ± 7.5 | 6 | 23.8 ± 2.8 | 6 | |
| 22°C | 5.5 ± 1.5 | 5 | 8.9 ± 1.1 | 5 | 16.6 ± 2.3 | 5 | 8.3 ± 1.8 | 5 | |
| 34°C | 2.1 ± 0.3 | 5 | 4.8 ± 0.9 | 4 | 9.8 ± 2.2 | 5 | 4.5 ± 1.4 | 4 | |
| 37°C | 1.6 | — | 3.9 | — | 8.1 | — | 3.6 | — | |
| 41°C | 1.2 | — | 3.1 | — | 6.4 | — | 2.9 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
Table 5.
Fast inactivation time constants between −50 mV and +10 mV
| Fast inactivation time constants (ms) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Temperature | −50 mV | n | −30 mV | n | −10 mV | n | +10 mV | n | |
| Wild type | Q10 = 5.80 | Q10 = 2.13 | Q10 = 1.80 | Q10 = 1.79 | |||||
| 10°C | 24.40 ± 4.86 | 4 | 7.39 ± 0.48 | 9 | 4.96 ± 0.29 | 9 | 4.34 ± 0.29 | 9 | |
| 22°C | 20.10 ± 5.11 | 5 | 1.98 ± 0.29 | 8 | 0.96 ± 0.10 | 8 | 0.67 ± 0.09 | 8 | |
| 34°C | 2.09 ± 0.59 | 5 | 0.88 ± 0.16 | 8 | 0.61 ± 0.13 | 8 | 0.45 ± 0.09 | 8 | |
| 37°C | 1.19 | — | 0.69 | — | 0.49 | — | 0.36 | — | |
| 41°C | 0.59 | — | 0.51 | — | 0.39 | — | 0.29 | — | |
| R1193Q | Q10 = 2.02 | Q10 = 1.89 | Q10 = 1.90 | Q10 = 1.82 | |||||
| 10°C | 23.60 ± 3.69 | 4 | 7.08 ± 0.42 | 10 | 4.49 ± 0.35 | 10 | 3.76 ± 0.24 | 10 | |
| 22°C | 3.34 ± 0.54 | 6 | 1.29 ± 0.10 | 6 | 0.73 ± 0.03 | 6 | 0.53 ± 0.04 | 6 | |
| 34°C | 1.80 ± 0.27 | 11 | 0.75 ± 0.12 | 11 | 0.43 ± 0.07 | 11 | 0.35 ± 0.04 | 11 | |
| 37°C | 1.41 | — | 0.60 | — | 0.34 | — | 0.28 | — | |
| 41°C | 1.06 | — | 0.46 | — | 0.27 | — | 0.22 | — | |
| E1784K | Q10 = 1.99 | Q10 = 2.03 | Q10 = 1.84 | Q10 = .84 | |||||
| 10°C | 8.59 ± 0.71 | 6 | 4.42 ± 0.36 | 8 | 3.47 ± 0.26 | 8 | 3.20 ± 0.21 | 8 | |
| 22°C | 1.73 ± 0.23 | 10 | 0.90 ± 0.12 | 10 | 0.58 ± 0.06 | 10 | 0.49 ± 0.05 | 10 | |
| 34°C | 0.91 ± 0.18 | 10 | 0.45 ± 0.07 | 10 | 0.36 ± 0.05 | 10 | 0.31 ± 0.05 | 10 | |
| 37°C | 0.72 | — | 0.36 | — | 0.29 | — | 0.25 | — | |
| 41°C | 0.54 | — | 0.27 | — | 0.23 | — | 0.19 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
Figure 6.
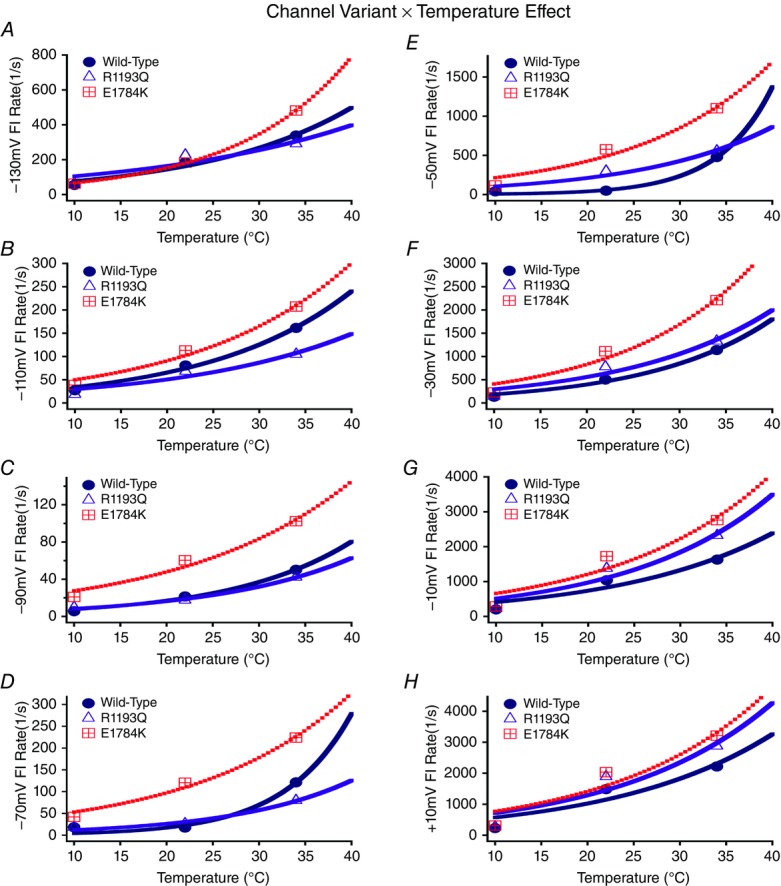
Fast inactivation (FI) kinetics Q10 fits
Panels A–H show the rates of fast inactivation for WT, R1193Q and E1784K at different voltages plotted as a function of temperature.
Late sodium current
We show representative normalized current traces of late INa for the channel variants only at 22°C and 34°C in Fig. 7. Figure 7 includes the channel variant and temperature effects (panels A–E) along with the Q10 fit in panel F. We eliminated 10°C persistent current traces since currents at 10°C took a longer time to reach plateau. Channel variants and temperature significantly affect normalized late INa (P < 0.01; Fig. 7A–E). The percentage of late INa was significantly larger in E1784K compared to WT and R1193Q (P < 0.01; Table6). This effect is only present at 34°C compared to 22°C (P < 0.01; Fig. 7F). Increasing temperature from 22°C to 34°C increases late INa by 3.10% ± 0.39% in E1784K compared to a minor change in WT and R1193Q (Fig. 7F). The Q10 fit values shown in bold in Table6 are consistent with a higher thermosensitivity in E1784K compared to WT and R1193Q. At febrile temperatures (41°C) E1784K has 9.24% late current.
Figure 7.

Persistent sodium currents
Panels A–F show normalized current plotted as a function of time with insets that focus on a narrower current window to show persistent INa. Panels A and B show the channel variant effect at both 22°C and 34°C. Panels C and D show the temperature effect for WT, R1193Q and E1784K. The pulse protocol inset is not shown in Panel E for clarity purposes. Panel F shows the Q10 fit for persistent current of all the channel variants plotted as a function of temperature.
Table 6.
Persistent sodium current
| Temperature | Persistent current | n | |
|---|---|---|---|
| Wild type | Q10 = 1.99 | ||
| 22°C | 0.52 ± 0.07 | 6 | |
| 34°C | 1.19 ± 0.39 | 4 | |
| 37°C | 1.49 | — | |
| 41°C | 1.96 | — | |
| R1193Q | Q10 = 1.78 | ||
| 22°C | 0.64 ± 0.14 | 6 | |
| 34°C | 1.28 ± 0.29 | 5 | |
| 37°C | 1.54 | — | |
| 41°C | 1.94 | — | |
| E1784K | Q10 = 2.87 | ||
| 22°C | 1.22 ± 0.21 | 5 | |
| 34°C | 4.32 ± 0.44 | 6 | |
| 37°C | 6.04 | — | |
| 41°C | 9.24 | — |
Values in bold were determined by extrapolation of Q10 fits to measured values.
Use-dependent inactivation
Normalized currents from 1 Hz use-dependent protocols are plotted against time in Fig. 8. Both the channel variant effect and temperature effect are shown in Fig. 8. Figure 0A–C show the Q10 fits for the channel variants as a function of temperature of use-dependent inactivation parameters. The decay of current was best fitted by a double exponential equation. The τ1 was unaffected by the channel variant or temperature, separately (Fig. 8 and Table7); however, there was an interaction effect on τ1 (P < 0.05). When temperature increased from 10°C to 34°C, τ1 decreased significantly in E1784K by 8.20 s ± 2.39 s compared to a non-significant change in WT and R1193Q (Fig. 10A and Table7). The τ2 value significantly decreased in E1784K (P < 0.05) by 82.8 s ± 21.5 s when temperature increased from 10°C to 34°C (Fig. 10B and Table7). The y0 was not significantly affected by the channel variant or temperature (Fig. 8 and Table7). Temperature increases from 10°C to 34°C caused the y0 plateau to decrease significantly in E1784K by 0.21 ± 0.07 (P < 0.05) as opposed to minor shifts in WT and R1193Q (Fig. 10C and Table7). The reported Q10 fit values in Table7 are consistent with a markedly heightened thermosensitivity in E1784K τ1, τ2 and y0 compared to R1193Q and WT.
Figure 8.

Use-dependent inactivation (1 Hz)
Panels A–F show normalized current plotted as a function of time. Panels A–C show the channel variant effect for 10°C, 22°C and 34°C. Panels D–F show the temperature effect for WT, R1193Q and E1784K. Insets show the pulse protocols used to measure use-dependent inactivation (UDI) at 1 Hz.
Figure 10.
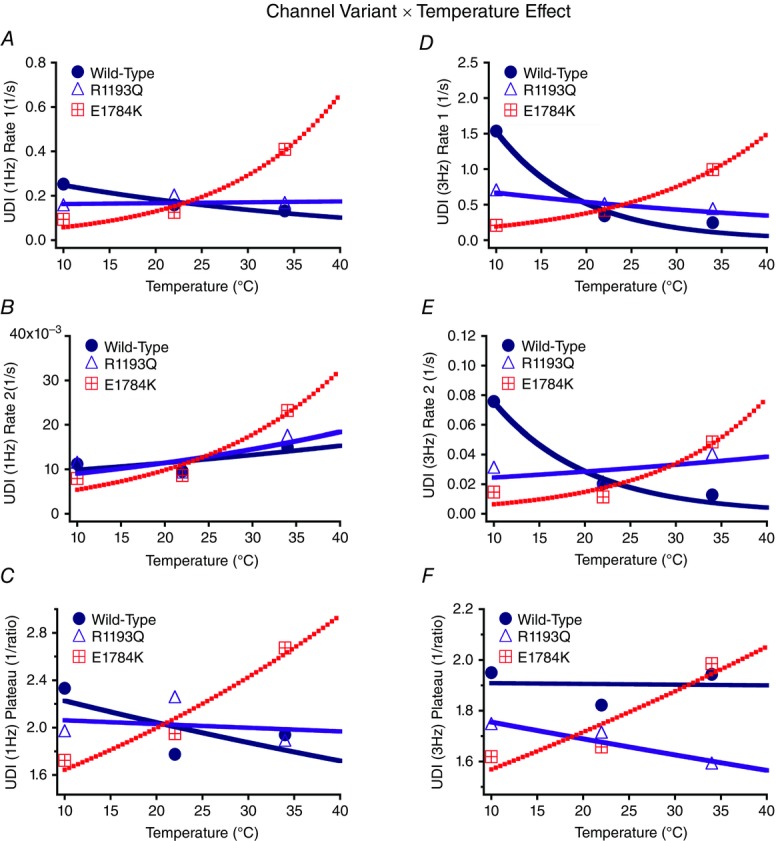
Use-dependent inactivation (UDI; 1 Hz and 3 Hz) parameters Q10 fits
Panels A and D show the initial rate of use-dependent inactivation for 1 Hz and 3 Hz, respectively. Panels B and E show the secondary rate of use-dependent inactivation for 1 Hz and 3 Hz, respectively. Panels C and F show the inverse of plateau of use-dependent inactivation for 1 Hz and 3 Hz, respectively.
Table 7.
Use-dependent inactivation (1 Hz)
| Use-dependent inactivation (1 Hz) | |||||
|---|---|---|---|---|---|
| Temperature | 1 Hz–y0 | 1 Hz–τ1 (s) | 1 Hz–τ2 (s) | n | |
| Wild type | Q10 = 0.92 | Q10 = 0.74 | Q10 = 1.16 | ||
| 10°C | 0.43 ± 0.06 | 3.96 ± 1.16 | 89.15 ± 9.89 | 5 | |
| 22°C | 0.56 ± 0.05 | 6.30 ± 0.30 | 104.14 ± 16.62 | 4 | |
| 34°C | 0.52 ± 0.05 | 7.59 ± 2.76 | 66.83 ± 18.32 | 5 | |
| 37°C | 0.57 | 9.06 | 68.03 | — | |
| 41°C | 0.59 | 10.20 | 64.12 | — | |
| R1193Q | Q10 | Q10 = 0.99 | Q10 = 1.02 | Q10 = 1.27 | |
| 10°C | 0.51 ± 0.08 | 6.70 ± 1.92 | 90.04 ± 15.61 | 5 | |
| 22°C | 0.45 ± 0.05 | 5.16 ± 0.87 | 112.23 ± 9.27 | 6 | |
| 34°C | 0.53 ± 0.05 | 6.28 ± 1.63 | 58.30 ± 21.23 | 5 | |
| 37°C | 0.51 | 5.77 | 58.01 | — | |
| 41°C | 0.51 | 5.71 | 52.78 | — | |
| E1784K | Q10 | 1.21 | 2.25 | 1.81 | |
| 10°C | 0.58 ± 0.03 | 10.70 ± 2.82 | 125.85 ± 23.32 | 5 | |
| 22°C | 0.51 ± 0.03 | 7.98 ± 2.17 | 116.01 ± 15.02 | 4 | |
| 34°C | 0.37 ± 0.07 | 2.44 ± 0.70 | 43.05 ± 10.37 | 6 | |
| 37°C | 0.36 | 1.91 | 36.66 | — | |
| 41°C | 0.33 | 1.38 | 28.82 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
To understand the effects of elevated heart rate on channel function, we elicited use-dependent inactivation at 3 Hz. Normalized currents from 3 Hz use-dependent inactivation protocols are plotted against time in Fig. 9 for both the channel variant effect and the temperature effect. Τhe τ1 was not significantly affected by channel variants or temperature, separately. However, as temperature was elevated from 10°C to 34°C, τ1 decreased by 3.81 s ± 1.02 s in E1784K (P < 0.05) versus a non-significant change in WT and R1193Q (Fig. 10D and Table8). When temperature was elevated from 10°C to 34°C, τ1 increased by 3.42 s ± 1.02 s in WT (P < 0.05) compared to non-significant changes in E1784K and R1193Q. The τ2 value was not significantly different between the channel variants (Fig. 9D–F). The τ2 is significantly smaller at 10°C versus 22°C or 34°C (P < 0.05). Importantly, the differences in τ2 due to temperature are significantly different among the channel variants (P < 0.01; Fig. 10E and Table8). When temperature was elevated from 10°C to 34°C, τ2 increased significantly (P < 0.01) by 65.2 s ± 17.3 s in WT versus minimal changes in E1784K and R1193Q (P > 0.05). When temperature was elevated from 22°C to 34°C, τ2 increased by 67.2 s ± 17.3 s for E1784K compared to minimal changes in R1193Q and WT (P > 0.05). The y0 plateau was not significantly affected by channel variant, temperature, and the interaction between both factors (P > 0.05; Fig. 10F). The Q10 fits values for these three parameters are shown in bold in Table8. The trends seen in τ1 and τ2 are consistent with the Q10 values in that E1784K has a higher thermosensitivity than R1193Q and WT. Nevertheless, the Q10 values are relatively consistent for y0 amongst the channel variants.
Figure 9.

Use-dependent inactivation (3 Hz)
Panels A–F show normalized current plotted as a function of time. Panels A–C show the channel variant effect for 10°C, 22°C and 34°C. Panels D–F show the temperature effect for WT, R1193Q and E1784K. Insets show the pulse protocols used to measure use-dependent inactivation (UDI) at 3 Hz.
Table 8.
Use-dependent inactivation (3 Hz)
| Use-dependent inactivation (3 Hz) | |||||
|---|---|---|---|---|---|
| Temperature | 3 Hz–y0 | 3 Hz–τ1 (s) | 3 Hz–τ2 (s) | n | |
| Wild type | Q10 = 1.00 | Q10 = 0.34 | Q10 = 0.38 | ||
| 10°C | 0.51 ± 0.05 | 0.65 ± 0.46 | 13.20 ± 5.48 | 5 | |
| 22°C | 0.55 ± 0.07 | 2.91 ± 0.43 | 49.24 ± 7.04 | 6 | |
| 34°C | 0.52 ± 0.03 | 4.07 ± 0.10 | 78.44 ± 23.15 | 5 | |
| 37°C | 0.53 | 12.51 | 183.02 | — | |
| 41°C | 0.53 | 19.32 | 269.04 | — | |
| R1193Q | Q10 = 0.96 | Q10 = 0.80 | Q10 = 1.16 | ||
| 10°C | 0.58 ± 0.06 | 1.47 ± 0.18 | 33.30 ± 5.37 | 5 | |
| 22°C | 0.59 ± 0.04 | 2.07 ± 0.48 | 49.65 ± 11.49 | 5 | |
| 34°C | 0.63 ± 0.06 | 2.42 ± 0.67 | 25.61 ± 4.63 | 6 | |
| 37°C | 0.63 | 2.72 | 27.14 | — | |
| 41°C | 0.64 | 2.97 | 25.53 | — | |
| E1784K | Q10 = 1.09 | Q10 = 1.98 | Q10 = 2.30 | ||
| 10°C | 0.62 ± 0.04 | 4.82 ± 1.53 | 68.37 ± 17.76 | 5 | |
| 22°C | 0.60 ± 0.03 | 2.38 ± 0.35 | 87.80 ± 15.95 | 5 | |
| 34°C | 0.50 ± 0.06 | 1.01 ± 0.09 | 20.60 ± 4.88 | 5 | |
| 37°C | 0.50 | 0.82 | 16.22 | — | |
| 41°C | 0.48 | 0.62 | 11.61 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
Slow inactivation
Slow inactivation onset was measured with a double-pulse protocol and fitted with a double exponential curve. The τ2 and y0 values were not affected by channel variant, temperature or both interactions (P > 0.05). The Q10 fit values are reported in Table9 which suggest that E1784K has a higher thermosensitivity compared to R1193Q and WT with respect to τ2. The Q10 fit values were comparable for y0. The τ1 value was affected significantly by temperature (P < 0.01). When temperature increased from 10°C to 22°C the WT τ1 increased by 5.47s ± 1.43 s compared to minimal changes in E1784K and R1193Q (Table9). Extrapolations to 37°C and 41°C were not possible in Table9 since the Q10 fit values were markedly skewed from the physiological range.
Table 9.
Slow inactivation onset
| Slow inactivation onset | |||||
|---|---|---|---|---|---|
| Temperature | SIonset – y0 | SIonset – τ1 (s) | SIonset – τ2 (s) | n | |
| Wild type | Q10 = 0.95 | Q10 = 0.05 | Q10 = 0.99 | ||
| 10°C | 0.28 ± 0.03 | 0.15 ± 0.04 | 18.71 ± 4.51 | 6 | |
| 22°C | 0.26 ± 0.04 | 5.62 ± 1.99 | 22.25 ± 6.82 | 5 | |
| 34°C | 0.32 ± 0.05 | 2.49 ± 0.84 | 19.31 ± 6.30 | 5 | |
| 37°C | 0.30 | n/a | 20.40 | — | |
| 41°C | 0.31 | n/a | 20.51 | — | |
| R1193Q | Q10 = 1.13 | Q10 = 0.10 | Q10 = 0.97 | ||
| 10°C | 0.28 ± 0.05 | 0.12 ± 0.06 | 16.35 ± 5.15 | 5 | |
| 22°C | 0.29 ± 0.05 | 2.14 ± 1.00 | 14.97 ± 3.96 | 6 | |
| 34°C | 0.21 ± 0.03 | 1.68 ± 0.35 | 17.61 ± 2.33 | 5 | |
| 37°C | 0.21 | n/a | 17.00 | — | |
| 41°C | 0.20 | n/a | 17.22 | — | |
| E1784K | Q10 = 1.07 | Q10 = 0.12 | Q10 = 1.40 | ||
| 10°C | 0.26 ± 0.07 | 0.26 ± 0.05 | 18.48 ± 1.80 | 4 | |
| 22°C | 0.24 ± 0.05 | 4.02 ± 1.73 | 13.71 ± 2.38 | 5 | |
| 34°C | 0.22 ± 0.05 | 2.89 ± 1.02 | 8.61 ± 4.13 | 5 | |
| 37°C | 0.22 | n/a | 7.82 | — | |
| 41°C | 0.21 | n/a | 6.83 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
SSSI measurement and ANOVA analysis were limited to the channel variant (three levels) and the temperature factor with only two levels (10°C and 22°C). The 34°C data were excluded from the analysis since we were not able to record full steady-state slow inactivation data for E1784K at 34°C. The midpoint of SSSI for E1784K (34°C) was hyperpolarized. To obtain a full plateau the prepulse potential was reduced to potentials like −170 mV. With cellular instability at temperatures of 34°C, cells did not survive this protocol. The SSSI midpoint (V½) of E1784K was significantly hyperpolarized (P < 0.01) compared to WT and R1193Q. The V½ of all channel variants were unaffected by temperature (P > 0.05). The SSSI slope and plateau was unaffected by channel variant, temperature or their interaction (P > 0.05). The Q10 fit values are shown in bold in Table10. We were not able to fit Q10 curves to the E1784K data since our 34°C data was missing.
Table 10.
Steady-state slow inactivation
| Steady-state slow inactivation | |||||
|---|---|---|---|---|---|
| Temperature | SSSI-V½ (mV) | SSSI-z | SSSI-y0 | n | |
| Wild type | Q10 = 0.92 | Q10 = 0.88 | Q10 = 0.88 | ||
| 10°C | −103.81 ± 3.13 | −2.69 ± 0.07 | 0.28 ± 0.03 | 6 | |
| 22°C | −95.27 ± 4.04 | −2.27 ± 0.57 | 0.26 ± 0.04 | 4 | |
| 34°C | −85.76 ± 5.90 | −1.97 ± 0.58 | 0.40 ± 0.06 | 4 | |
| 37°C | −90.41 | −2.12 | 0.37 | — | |
| 41°C | −89.00 | −2.07 | 0.38 | — | |
| R1193Q | Q10 = 0.95 | Q10 = 0.79 | Q10 = 1.03 | ||
| 10°C | −101.66 ± 3.20 | −3.13 ± 0.90 | 0.34 ± 0.03 | 5 | |
| 22°C | −94.76 ± 6.57 | −1.85 ± 0.24 | 0.30 ± 0.02 | 5 | |
| 34°C | −89.03 ± 7.45 | −1.96 ± 0.28 | 0.32 ± 0.06 | 4 | |
| 37°C | −92.03 | −1.96 | 0.31 | — | |
| 41°C | −91.02 | −1.87 | 0.30 | — | |
| E1784K | Q10 n/a | Q10 n/a | Q10 n/a | ||
| 10°C | −117.02 ± 1.90 | −2.27 ± 0.21 | 0.26 ± 0.04 | 6 | |
| 22°C | −114.15 ± 7.09 | −2.31 ± 0.43 | 0.32 ± 0.06 | 5 | |
| 34°C | n/a | n/a | n/a | — | |
| 37°C | n/a | n/a | n/a | — | |
| 41°C | n/a | n/a | n/a | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
The τ1 values of slow inactivation recovery were not affected by channel variant, temperature or both interactions (P > 0.05). The Q10 fit values are shown in bold in Table11 and show that both R1193Q and E1784K have higher thermosensitivities compared to WT. The τ2 value was significantly affected by temperature as 10°C decelerated τ2 kinetics compared to 22°C and 34°C (P < 0.01). The Q10 fit values reported in Table11 show heightened thermosensitivity for τ2 in E1784K compared to R1193Q and WT.
Table 11.
Slow inactivation recovery
| Slow inactivation recovery | ||||
|---|---|---|---|---|
| Temperature | SIrecovery–τ1 (s) | SIrecovery–τ2 (s) | n | |
| Wild type | Q10 = 1.24 | Q10 = 1.29 | ||
| 10°C | 0.062 ± 0.019 | 3.461 ± 0.861 | 4 | |
| 22°C | 0.036 ± 0.015 | 1.368 ± 0.415 | 5 | |
| 34°C | 0.033 ± 0.017 | 1.444 ± 0.591 | 4 | |
| 37°C | 0.030 | 1.220 | — | |
| 41°C | 0.027 | 1.100 | — | |
| R1193Q | Q10 = 6.21 | Q10 = 4.11 | ||
| 10°C | 0.083 ± 0.034 | 3.264 ± 0.793 | 5 | |
| 22°C | 0.029 ± 0.017 | 1.110 ± 0.188 | 5 | |
| 34°C | 0.040 ± 0.011 | 2.185 ± 0.429 | 5 | |
| 37°C | 0.023 | 1.400 | — | |
| 41°C | 0.011 | 0.792 | — | |
| E1784K | Q10 = 5.36 | Q10 = 8.61 | ||
| 10°C | 0.017 ± 0.005 | 1.655 ± 0.496 | 5 | |
| 22°C | 0.044 ± 0.017 | 1.733 ± 0.434 | 5 | |
| 34°C | 0.041 ± 0.014 | 0.941 ± 0.294 | 6 | |
| 37°C | 0.026 | 0.474 | — | |
| 41°C | 0.013 | 0.200 | — | |
Values in bold were determined by extrapolation of Q10 fits to measured values.
Action potential simulations
We used a modified Ten Tusscher model to simulate epicardial and endocardial action potentials. We focused specific attention on the transmural voltage gradient between both walls of the heart. Figure 1 shows the 1999th and 2000th simulated APs for the different channel variants at three different physiological temperatures (34°C, 37°C, 41°C). The transient outward potassium current conductance was varied in the epicardium of the heart (0.400 pA pF–1 to 0.700 pA pF–1) since its expression is heterogeneous. Across all the temperatures, the E1784K channel had a less negative resting membrane potential (RMP) compared to R1193Q and WT. The E1784K mutant cardiomyoctes showed a decrease in the initial depolarization. The decrease in E1784K AP plateau is not constant across all the temperatures. At 34°C the loss of AP plateau in E1784K alternates with the second AP at 0.700 pA pF–1 of Kto current relative to the lower 0.400 pA pF–1 (Fig. 1). At 37°C there is a loss of AP plateau which is exacerbated at 41°C.
Figure 11.

AP model simulation
Discussion
Temperature is known to unmask Brugada syndrome (Meggiolaro et al. 2013; Salinski & Worrilow, 2014). We characterized the temperature sensitivity of two mixed syndrome mutants, E1784K and R1193Q in the cardiac sodium channel NaV1.5. Long lasting recordings at physiological temperatures are difficult to obtain due to membrane instability. Thus channel behaviour was assessed at three different, albeit non-physiological, temperatures to allow data to be extrapolated to physiologically relevant temperatures. We found the E1784K mutant to be more temperature sensitive than WT NaV1.5 or R1193Q in ways that suggest temperature could be an arrhythmogenic trigger.
Previous studies on the E1784K and R1193Q mutants have shown either no effect or a decrease in channel current density (Wei et al. 1999; Huang et al. 2006; Makita et al. 2008). These inconsistencies may be due to differences in the expression systems used in different studies or to the variability in current amplitudes in transiently transfected heterologous expression systems. One previous study quantifying E1784K expression using fluorescence showed no difference in cell surface expression between E1784K and WT (Makita et al. 2008). Our results using CHOk1 cells also suggest that the E1784K mutant does not affect channel expression compared to WT. Arrhythmogenesis in the E1784K mutant may thus be related to changes in channel gating rather than expression. Current density in E1784K was, however, acutely sensitive to temperature. This result should be interpreted with caution due to the variability in currents as mentioned above. In contrast to E1784K, we show no change in WT conductance midpoints, even with a greater than 10°C change in temperature, consistent with previous studies (Nagatomo et al. 1998).
The greater temperature sensitivity of E1784K is reflected in the AP model, in which E1784K mutant decreases the rise of the initial upstroke of AP and also attenuates the epicardial AP plateau. This effect is exacerbated with elevated temperatures and with larger IK,to. This is not the case with R1193Q as expected from the relatively lower Q10 values for both current density and time to half-peak INa. The increase in late current in E1784K has the greatest temperature sensitivity, suggesting temperature has the potential to be arrhythmogenic in this mixed syndrome mutant. Although the data suggest the AP should be prolonged at greater temperature, consistent with LQT3, the predominant effect in our AP model was a loss-of-function due to decreased epicardial E1784K sodium currents. Decreased sodium current in E1784K could lead to a failure to activate L-type Ca2+ channels and a subsequent loss of the epicardial AP plateau, causing a transmural voltage-gradient between endocardium and epicardium typical of BrS1. In addition, greater use-dependent inactivation in the E1784K mutant, which was not accounted for within the model, is predicted to further exacerbate loss-of-function in INa in vivo. Our in vitro data shows that with higher heart rates the stabilization in use-dependent inactivation in E1784K compared to WT is no longer apparent. Thus, higher frequencies may partially ameliorate the biophysical defects associated with E1784K.
At high stimulation frequencies, we observed a fast rate of entry into use-dependent inactivation in WT below 22°C and above 34°C, yielding a U-shaped temperature dependence. This is consistent with the slow inactivation onset kinetics measured. Previous literature shows that both fast inactivation (with prepulses of at least 500 ms) and slow inactivation are stabilized in Nav1.5 and Nav1.4 channels as temperature decreases, as seen in different heterologous expression systems using different techniques (Murray et al. 1990; Ruff, 1999; Carle et al. 2009). Murray et al. (1990) suggests this effect may be due to disturbances in lipid–channel interactions or metabolic disturbances affecting charge transfer across the membrane. Although biophysically curious, the U-shaped temperature dependence of WT channels does not appear in the range of physiological temperatures extrapolated in this study.
The effects of E1784K on activation, fast inactivation and use-dependent inactivation confirm previous studies that highlight the role of the C-terminus in sodium channel gating (Shah et al. 2006; Sarhan et al. 2012). With a Ca2+ signal, the C-terminus interacts with the domain III–DIV linker through the actions of calmodulin. This interaction was reported to shift the inactivation curve and decrease late currents with increased [Ca2+]i (Shah et al. 2006; Sarhan et al. 2012). We limited our study, however, to the apo-Ca2+ condition, using intracellular EGTA to chelate [Ca2+]i. Other studies reported a direct interaction between the C-terminus and domain III–IV linker under apo-Ca2+ conditions (Cormier et al. 2002; Motoike et al. 2004). In those studies, a series of residues following the IFM motif, PIPR (non-alpha helical structure), was thought to interact with the C-terminus. In this interpretation, the C-terminus does not affect the extent of inactivation but rather ensures that the IFM motif latch is in place upon occluding the pore. The 1885stop mutation (truncation of helix VI of C-terminus) in NaV1.5 causes a large increase in persistent current compared to 1921stop (intact C-terminus including helix VI; Motoike et al. 2004). Helix VI, the IQ motif, plays an essential role in maintaining inactivation, preventing increases in persistent INa. The E1784K mutant is in a region prior to helix I in the C-terminus (EF-hand domain). The E1784K charge reversal mutation may disrupt the interaction between helix VI and helices I–IV, thus altering the mechanism by which helix VI modulates inactivation via III–IV linker, which could explain the large increase in persistent INa. Chimera studies on the domain I–II and domain II–III linkers, as well as the C-terminus, have shown that these regions modulate channel activation and account for many isoform-specific differences (Bennett, 1999, 2001; Choi et al. 2004). Slow inactivation is stabilized when fast inactivation is removed (Featherstone et al. 1996; Richmond et al. 1998). Late current and use-dependent inactivation increases are largest in the E1784K mutant when temperature is elevated to 34°C, consistent with the previously reported inverse relationship between fast and slow inactivation.
In conclusion, we show that the E1784K mutant shows enhanced thermosensitivity compared to WT channels and another mixed syndrome mutant, R1193Q. Heightened thermosensitivity in E1784K may play a role in arrythmogenesis during a fever or intense exercise by prolonging the cardiac action potential or causing loss-of function.
Acknowledgments
The authors thank Dr David Jones and Dr Stanislav Sokolov for their contribution and their support.
Glossary
- α1
fast amplitude
- τ1
fast time constant
- α2
slow amplitude
- τ2
slow time constant
- AP
action potential
- BrS1
Brugada syndrome 1
- cDNA
complementary DNA
- CHOK1
Chinese hamster ovary K1
- e0
elementary charge
- ECG
electrocardiogram
- eGFP
green fluorescent protein
- ENa
Na+ Nernst equilibrium potential
- G/Gmax
normalized conductance
- GNa
sodium channel conductance
- GV-V½
conductance midpoint
- GV-z
conductance slope
- I
current amplitude
- I/Imax
normalized current
- IK,To
transient outward potassium current
- INa
Na+ current
- Iss
plateau current amplitude
- LQT3
long QT syndrome 3
- NaV
voltage-gated sodium channel
- NaV1.5
cardiac voltage-gated sodium channel
- RMP
resting membrane potential
- S4
transmembrane segment 4
- SSFI
steady-state fast inactivation
- SSFI-V½
steady-state fast inactivation midpoint
- SSFI-z
steady-state fast inactivation slope
- t
time
- V½
midpoint of voltage dependence
- Vm
command potential
- WT
wild type
- z
apparent valency
Additional information
Competing interests
The authors have no competing interests to disclose.
Author contributions
MA and CHP collected, assembled, analyzed, and interpreted the data, designed the experiments, and drafted the manuscript. PCR conceived the experiments and revised the manuscript critically for important intellectual content. All authors approved the final version of the manuscript and qualify for authorship.
Funding
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada and the Canadian Foundation for Innovation.
References
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, et al. Brugada syndrome: Report of the second consensus conference: Endorsed by the Heart Rhythm Society and the Chinese Heart Rhythm Association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- Bennett ES. Effects of channel cytoplasmic regions on the activation mechanisms of cardiac versus skeletal muscle Na+ channels. Biophys J. 1999;77:2999–3009. doi: 10.1016/S0006-3495(99)77131-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ES. Channel cytoplasmic loops alter voltage-dependent sodium channel activation in an isoform-specific manner. J Physiol. 2001;535:371–381. doi: 10.1111/j.1469-7793.2001.t01-1-00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- Carle T, Fournier E, Sternberg D, Fontaine B. Tabti N. Cold-induced disruption of Na+ channel slow inactivation underlies paralysis in highly thermosensitive paramyotonia. J Physiol. 2009;587:1705–1714. doi: 10.1113/jphysiol.2008.165787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- Choi JS, Tyrrell L, Waxman SG. Dib-Hajj SD. Functional role of the C-terminus of voltage-gated sodium channel Nav1.8. FEBS Lett. 2004;572:256–260. doi: 10.1016/j.febslet.2004.07.047. [DOI] [PubMed] [Google Scholar]
- Cormier JW, Rivolta I, Tateyama M, Yang AS. Kass RS. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- Doetzer AD, Sotomaior VS, Bubna MH. Raskin S. What can be done when asymptomatic patients discover they have Brugada syndrome? A case report of Brugada syndrome. Int J Cardiol. 2011;150:e96–e97. doi: 10.1016/j.ijcard.2010.02.037. [DOI] [PubMed] [Google Scholar]
- Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R. Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE. Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Borbolla M, Garcia-Borbolla R, Valenzuela LF. Trujillo F. Ventricular tachycardia induced by exercise testing in a patient with Brugada syndrome. Rev Esp Cardiol. 2007;60:993–994. doi: 10.1157/13109656. [DOI] [PubMed] [Google Scholar]
- Glaaser IW, Osteen JD, Puckerin A, Sampson KJ, Jin X. Kass RS. Perturbation of sodium channel structure by an inherited long QT syndrome mutation. Nat Commun. 2012;3:706. doi: 10.1038/ncomms1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AO, Carboni MP, Neplioueva V, Starmer CF, Memmi M, Napolitano C. Priori S. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201–1209. doi: 10.1172/JCI15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhao J, Barrane FZ, Champagne J. Chahine M. Nav1.5/R1193Q polymorphism is associated with both long QT and Brugada syndromes. Can J Cardiol. 2006;22:309–313. doi: 10.1016/s0828-282x(06)70915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DK, Peters CH, Tolhurst SA, Claydon TW. Ruben PC. Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys J. 2011;101:2147–2156. doi: 10.1016/j.bpj.2011.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DI, Huang H, Zhao J, Frank R, Suarez V, Delacretaz E, Brink M, Osswald S, Schwick N. Chahine M. A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovasc Res. 2006;70:521–529. doi: 10.1016/j.cardiores.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Makaryus JN, Verbsky J, Schwartz S. Slotwiner D. Fever associated with gastrointestinal shigellosis unmasks probable Brugada syndrome. Case Rep Med. 2009:492031. doi: 10.1155/2009/492031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, Schulze-Bahr E, Fukuhara S, Mochizuki N, Makiyama T, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. doi: 10.1172/JCI34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggiolaro M, Zorzi A, El Maghawry M, Peruzza F, Migliore F. Pittoni GM. Brugada ECG disclosed by acute malaria: Is it all about fever and propofol? J Clin Anesth. 2013;25:483–487. doi: 10.1016/j.jclinane.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Mok NS, Priori SG, Napolitano C, Chan NY, Chahine M. Baroudi G. A newly characterized SCN5A mutation underlying Brugada syndrome unmasked by hyperthermia. J Cardiovasc Electrophysiol. 2003;14:407–411. doi: 10.1046/j.1540-8167.2003.02379.x. [DOI] [PubMed] [Google Scholar]
- Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M. Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KT1, Anno T, Bennett PB. Hondeghem LM. Voltage clamp of the cardiac sodium current at 37 degrees C in physiologic solutions. Biophys J. 1990;57:607–613. doi: 10.1016/S0006-3495(90)82576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatomo T, Fan Z, Ye B, Tonkovich GS, January CT, Kyle JW. Makielski JC. Temperature dependence of early and late currents in human cardiac wild-type and long Q-T DeltaKPQ Na+ channels. Am J Physiol Heart Circ Physiol. 1998;275:H2016–H2024. doi: 10.1152/ajpheart.1998.275.6.H2016. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N. Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Giordano U, Collisani G. Memmi M. Brugada syndrome and sudden cardiac death in children. Lancet. 2000;355:808–809. doi: 10.1016/S0140-6736(99)05277-0. [DOI] [PubMed] [Google Scholar]
- Remme CA, Verkerk AO, Hoogaars WM, Aanhaanen WT, Scicluna BP, Annink C, van den Hoff MJ, Wilde AA, van Veen TA, Veldkamp MW, et al. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol. 2009;104:511–522. doi: 10.1007/s00395-009-0012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Featherstone DE, Hartmann HA. Ruben PC. Slow inactivation in human cardiac sodium channels. Biophys J. 1998;74:2945–2952. doi: 10.1016/S0006-3495(98)78001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben PC, Starkus JG. Rayner MD. Steady-state availability of sodium channels. Interactions between activation and slow inactivation. Biophys J. 1992;61:941–955. doi: 10.1016/S0006-3495(92)81901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff RL. Effects of temperature on slow and fast inactivation of rat skeletal muscle Na+ channels. Am J Physiol Cell Physiol. 1999;277:C937–C947. doi: 10.1152/ajpcell.1999.277.5.C937. [DOI] [PubMed] [Google Scholar]
- Salinski EP. Worrilow CC. ST-segment elevation myocardial infarction vs. hypothermia-induced electrocardiographic changes: A case report and brief review of the literature. J Emerg Med. 2014;46:e107–e111. doi: 10.1016/j.jemermed.2013.08.122. [DOI] [PubMed] [Google Scholar]
- Sarhan MF, Tung CC, Van Petegem F. Ahern CA. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci. 2012;109:3558–3563. doi: 10.1073/pnas.1114748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah VN, Wingo TL, Weiss KL, Williams CK, Balser JR. Chazin WJ. Calcium-dependent regulation of the voltage-gated sodium channel hH1: intrinsic and extrinsic sensors use a common molecular switch. Proc Natl Acad Sci. 2006;103:3592–3597. doi: 10.1073/pnas.0507397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JR. Goldstein SA. Voltage-sensor movements describe slow inactivation of voltage-gated sodium channels II: A periodic paralysis mutation in NaV1.4 (L689I) J Gen Physiol. 2013;141:323–334. doi: 10.1085/jgp.201210910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL. Moss AJ, Schwartz PJ, Towbin JA, Vincent GM. Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Sun A, Xu L, Wang S, Wang K, Huang W, Wang Y, Zou Y. Ge J. SCN5A R1193Q polymorphism associated with progressive cardiac conduction defects and long QT syndrome in a Chinese family. J Med Genet. 2008;45:127–128. doi: 10.1136/jmg.2007.056333. [DOI] [PubMed] [Google Scholar]
- Ten Tusscher KH, Noble D, Noble PJ. Panfilov AV. A model for human ventricular tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1573–H1589. doi: 10.1152/ajpheart.00794.2003. [DOI] [PubMed] [Google Scholar]
- Ten Tusscher KH. Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol. 2006;291:H1088–H1100. doi: 10.1152/ajpheart.00109.2006. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Will ML, Haglund CM. Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AA. Balser JR. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ Res. 2000;86:E91–E97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- Wang Q, Chen S, Chen Q, Wan X, Shen J, Hoeltge GA, Timur AA, Keating MT. Kirsch GE. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. J Med Genet. 2004;41:66. doi: 10.1136/jmg.2003.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wang DW, Alings M, Fish F, Wathen M, Roden DM. George AL Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation. 1999;99:3165–3171. doi: 10.1161/01.cir.99.24.3165. [DOI] [PubMed] [Google Scholar]
- Xia L, Zhang Y, Zhang H, Wei Q, Liu F. Crozier S. Simulation of Brugada syndrome using cellular and three-dimensional whole-heart modelling approaches. Physiol Meas. 2006;27:1125–1142. doi: 10.1088/0967-3334/27/11/006. [DOI] [PubMed] [Google Scholar]