

Abstract
Keywords: -synuclein, calcium channels, lipid rafts, plasma membrane
Alpha-synuclein (Syn) is a cytosolic, soluble, and unstructured protein, which may also associate with membranes by acquiring an α-helical conformation. High Syn concentrations are present in genetic Parkinson disease (PD) due to duplication/triplication of SNCA gene, and are also associated with polymorphisms found in idiopathic PD cases.1 Syn has been shown to act on several intracellular targets, and to alter synaptic function. The discovery of the presence of Syn in cerebrospinal fluid and of its release from neurons, as demonstrated by numerous studies,2 suggests that Syn-driven pathology may spread in the brain, although the mechanism of Syn release is still unclear. There is evidence that Syn overexpression modifies the structure of the cellular membrane, however a possible effect of excess Syn on the pre- or post-synaptiC-terminal from the extracellular milieu has yet to be fully analyzed.
In our recent paper,3 we show that treatment of isolated rat cortical neurons with recombinant, highly purified, monomeric Syn leads to an increase in KCl−stimulated calcium influx through Cav2.2 (N-type) calcium channels. The increase in calcium entry is evident at the presynaptic compartment, as shown by the use of the synaptic vesicles-targeted calcium dye SyGCaMP3, suggesting that Syn may profoundly affect synaptic neurotransmitter release.
The activating effect on calcium channels has been confirmed in primary cultures of superior cervical ganglion neurons (SCGN), a model used to study the biophysical properties of Cav2.2 in a physiological context. Syn pre-treatment significantly increases both calcium conductance and voltage sensitivity of the channels. Accordingly, augmented cholinergic transmission has been observed in long-term cultured SCGN after acute Syn application. Cav2.2 channels play a fundamental role in the release of dopamine in the striatum, where dopaminergic innervations target and control function. Our study shows that Syn perfusion of striatal brain slices induces a significant increase of dopamine release, an effect confirmed in vivo by microdialysis of Syn in rat striatum.
Cav2.2 channels are multimeric protein complexes that allow functional coupling between action potentials and calcium entry into nerve terminals, thus controlling neurotransmitter release. Cav2.2 regulation occurs by the integration of different pathways involving G protein subunits, synaptic proteins such as syntaxin, and kinases such as PKC. However, we show that Syn does not affect any of these targets; rather, analysis of channel biophysical parameters favors that Syn facilitates Cav2.2 transition from a closed to an open state by allowing movement of channel gating charge. This effect can be related to changes occurring in the stiffness of the plasma membrane where the channel is inserted, as dictated by lipid composition and organization of the membrane. In this regard, the level of cholesterol in the membrane affects its structure, as demonstrated by the use of reagents that either decrease or increase cellular cholesterol content. Our study shows that, although Syn treatment leaves the total cellular cholesterol content unchanged, it decreases the level of the lipid in the plasma membrane. Interestingly, this effect is accompanied by a translocation of Cav2.2 from high to low cholesterol compartments. Lipid rafts are patches in the membrane with high cholesterol content that constitute ordered lipid domains, compared to the disordered bilayer. We propose that the demonstrated shift of Cav2.2 channels into non-raft domains, where lower energy is required for membrane deformation, favors channel transition to the open state. The net activity of the channel will then derive from a balance between its localization in the membrane and the relative localization of its regulatory proteins.
Reports in the literature show that cholesterol depletion by methyl-β-cyclodextrin induces an increase in calcium current, as we observe with Syn treatment. Moreover cholesterol loading of neurons causes a clear decrease in calcium current.4 We show that methyl-β-cyclodextrin causes an increase in calcium entry, but it also decreases synaptic vesicles release by altering membrane curvature;5 we further show that extracellular Syn, acting only at the plasma membrane level, is able to couple the increase in Cav2.2 activity and intracellular calcium levels with an increase in neurotransmitter release. Conversely, cholesterol loading causes a concentration-dependent decrease in excitatory postsynaptic potential amplitude in SCGN synapses, consistent with a reduction in calcium current (Fig. 1).
Figure 1.
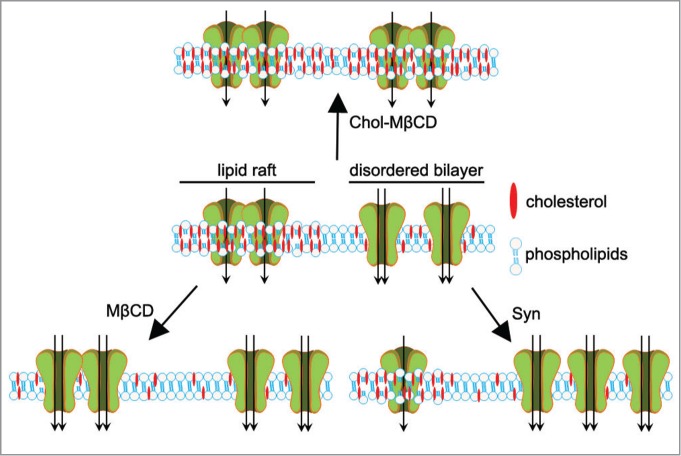
Cartoon showing the effect of cholesterol-loaded methyl-β-cyclodextrin (Chol-MβCD), methyl-β-cyclodextrin (MβCD), and Syn on the plasma membrane cholesterol content and on the activity of Cav2.2.
The effect of Syn on cholesterol is still unknown, whether inducing an internalization of cholesterol-rich domains, or a change in size/number of lipid rafts; however, the effect of Syn appears selective for Cav2.2 over the other major presynaptic calcium channel, Cav2.1. This may suggest that insertion and re-localization pathways differ for these channels. Evidence suggests that Cav2.2 and Cav2.1 insert into different ‘slots’ in central synapses,6 this difference may extend to re-localization by Syn.
Syn-mediated enhancement of Cav2.2 activity may explain the increase in DA release that precedes degeneration of dopaminergiC-terminals observed in animal models of PD,7 and recently it was shown that mutant Syn increases the firing rate of substantia nigra neurons, a subtype of dopaminergic neurons selectively vulnerable to degeneration.8 The increased concentration of DA may therefore account for the toxicity observed in dopaminergic neurons at later stages of disease.
References
- 1.Venda LL, et al. Trends Neurosci 2010; 33:559-68; PMID:20961626; http://dx.doi.org/ 10.1016/j.tins.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marques O, Outeiro TF. Cell death & disease 2012; 3: e350; PMID:22825468; http://dx.doi.org/ 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ronzitti G, et al.. J Neurosci 2014; 34: 10603-15; PMID:25100594; http://dx.doi.org/ 10.1523/JNEUROSCI.0608-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toselli M, et al. Biophys J 2005; 89: 2443-57; PMID:16040758; http://dx.doi.org/ 10.1529/biophysj.105.065623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rituper B, et al. Biochimica et biophysica acta 2013; 1831: 1228-38; PMID:24046863; http://dx.doi.org/ 10.1016/j.bbalip.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Cao YQ, Tsien RW. J Neurosci 2010; 30:4536-46; PMID:20357104; http://dx.doi.org/ 10.1523/JNEUROSCI.5161-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lam HA, et al.. J Neurosci Res 2011; 89:1091-102; PMID:21488084; http://dx.doi.org/ 10.1002/jnr.22611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subramaniam M, et al.. J Neurosci 2014; 34: 13586-99; PMID:25297088; http://dx.doi.org/ 10.1523/JNEUROSCI.5069-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]