

Abstract
We recently reported that knocking down the enzyme phosphatidylserine decarboxylase, which synthesizes the phospholipid phosphatidylethanolamine (PE) in mitochondria, perturbs the homeostasis of the human Parkinson disease (PD) protein α-synuclein (expressed in yeast or worms). In yeast, low PE in the psd1Δ deletion mutant induces α-synuclein to enter cytoplasmic foci, the level of this protein increases 3-fold compared to wild-type cells, and the mutant cells are severely sick. The metabolite ethanolamine protects both yeast and worms from the deleterious synergistic effects of low mitochondrial PE and α-synuclein. Here we highlight a Drosophila mutant called easily shocked—thought to be a model of epilepsy—that cannot use ethanolamine to synthesize PE. We also highlight recently identified mutated genes associated with defective lipid metabolism in PD and epilepsy patients. We propose that disruptions in lipid homeostasis (synthesis and degradation) may be responsible for some cases of PD and epilepsy.
Keywords: α-synuclein, epilepsy, Kennedy pathway, lipid disequilibrium, Parkinson disease, phosphatidylethanolamine, phosphatidylserine decarboxylase
PD is a devastating disease that affects 1–2% of the population over 65 y of age. Because the costs associated with caring for PD patients will skyrocket as our population ages, we need early biomarkers that can identify at-risk individuals, who can then be treated with novel therapeutics that halt neurodegeneration before symptoms emerge. The main culprit in this disease is thought to be the protein α-synuclein.1 Aging—for unknown reasons—triggers α-synuclein to transform from a non-toxic conformation into a toxic one. The toxic conformation kills the host neurons and then spreads to neighboring healthy neurons.2 How α-synuclein changes conformations, kills, and spreads are the subjects of intense investigations. We believe that changes in the cellular level of PE may be the triggering factor that converts α-synuclein into a toxic protein.
PE is Synthesized in Mitochondria and in the Endoplasmic Reticulum
The enzymes that synthesize PE in yeast, worms, and flies have mammalian counterparts.3,4 First, lodged in the inner membrane, the enzyme phosphatidylserine decarboxylase converts phosphatidylserine to PE (Fig. 1). PE synthesized in mitochondria can spread via mitochondrial associated membranes to other cellular compartments.5 Second, the Kennedy pathway consists of 3 enzymes that convert the metabolite ethanolamine into PE; the last enzyme in this pathway is embedded in the membranes of the endoplasmic reticulum (ER) (Fig. 1). In some cells phosphatidylserine decarboxylase may synthesize most of the PE, whereas in other cells the Kennedy pathway may synthesize the most of the PE.
Figure 1.
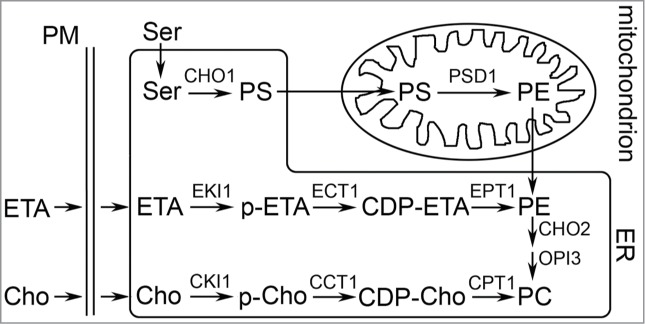
Scheme for phosphatidylethanolamine (PE) synthesis in mitochondria and the endoplasmic reticulum (ER) in yeast. CDP, cytidine diphosphate; Cho, choline; ETA, ethanolamine; p-ETA/p-Cho, phosphorylated ETA/choline; PM, plasma membrane. EKI1, ETA kinase; ECT1, ETA-phosphate cytidylytransferase; EPT1, ETA/Cho phosphotransferase. This figure was taken from Wang et al.6
Low Mitochondrial PE Induces ER Stress and α-synuclein Enters Cytoplasmic Foci; Ethanolamine Rescues
We found that the psd1Δ deletion mutant has low PE, intense ER stress, and a slight growth defect compared to wild-type cells.6 When α-synuclein is expressed in these PE-deficient cells, α-synuclein forms intracytoplasmic foci, the level of α-synuclein is 3-fold higher than identically treated wild-type cells, and growth is severely inhibited. Treating such cells with exogenous ethanolamine boosted the level of PE through the Kennedy pathway, which eliminated these various defects due to initially low PE. These findings were replicated in transgenic C. elegans worms in which α-synuclein was expressed in each of the worm's 6 dopaminergic neurons. Such worms exhibit a gradual age-dependent loss (t1/2 = 3.6 d) of the dopaminergic neurons due to the expression of α-synuclein. RNAi depletion of psd-1 in neurons that express α-synuclein accelerated the age-dependent cell loss (t1/2 = 2.3 d), whereas RNAi depletion of psd-1 in neurons lacking a-syn expression did not affect cell loss over time. Ethanolamine rescued cell loss due to the expression of α-synuclein in the neurons with psd-1 depleted (t1/2 = 7.9 d). Ethanolamine even rescued the age-dependent degeneration that occurs in worms that express α-synuclein but without RNAi depletion of psd-1. Such a result suggests that aging worms have a deficit of neuronal PE. Given that stimulating PE synthesis through the Kennedy pathway ameliorates α-synuclein toxicity in worm neurons, we asked whether the Kennedy pathway might be associated with other neurological diseases.
Failure to Synthesize PE from Ethanolamine Causes Seizures in Flies
A class of Drosophila mutants that exhibit a “transient paralysis following a brief mechanical shock” was identified in a behavioral screen and reported in 1971.7 One of these mutants was called easily shocked (eas). Subjecting eas flies to a brief mechanical shock (bang or vortex) triggers a transitory seizure that persists for several seconds, but then the flies recover. Tanouye and colleagues later characterized the eas mutant and showed that the paralytic phenotype was due to a mutation in the gene for ethanolamine kinase,8 which phosphorylates ethanolamine to yield phosphoethanolamine (Fig. 1). Biochemical analysis of whole fly lipid extracts showed that the eas flies lacked ethanolamine kinase activity and had a deficit of PE compared to wild type flies. Low PE in the eas mutant might cause membrane leakiness or decrease ion channel activity, which in either case leads to the transient paralytic phenotype. We suggest that chronically low PE in the membranes of the endoplasmic reticulum will likely lead to chronic stress in this compartment, which, over time, can lead to neuronal failure.Table 1 shows the effects of low PE on different organisms.
Table 1.
Low PE harms membranes
| Gene | Comments | Reference |
|---|---|---|
| Phosphatidylserine decarboxylase |
Yeast: Deletion of PSD1 causes intense ER stress, α-synuclein foci, protein trafficking defects, PE/PC ratio decreases by 70% (vs wild-type). ETA rescues. Worms: Knocking down psd-1 with RNAi enhances the toxicity of α-synuclein expressed in worm neurons; ETA rescues. ETA also protects from α-synuclein-induced neurodegeneration in the absence of psd-1 knockdown. |
Ref. 6 |
| Ethanolamine kinase |
Flies: i) null-activity mutant of eas: a brief mechanical shock induces transient paralysis. PE decreases, and the PE/PC ratio decreases by 18% (vs wild-type). ii) null-activity mutant of eas: increases triglycerides and causes tachycardia, cardiac relaxation problems, and SREBP activation. |
i) Ref. 8 ii) Ref. 14 |
Mutated Lipid Metabolism Genes Associated with PD and Epilepsy
If lipid disequilibrium is indeed a problem for PD and/or epilepsy, then one would expect that lipid-related genes would be identified from sequencing studies. Mutations in FASN (fatty acid synthase) and PLCB1 (phospholipase C isoform β1) were recently identified from a small number of individuals with a severe form of childhood epilepsy.9 Mutations in GBA (codes for enzyme that cleaves glucose from glucocerebroside) have been linked to a higher risk of PD,10 and single nucleotide polymorphisms in PLA2G6 (calcium-independent phospholipase A2)11 and in SREBP-1 (sterol-regulatory binding protein)12 have been identified in large genome-wide association studies of individuals with PD. Curiously, the enzymes coded by PLCB1 and PLA2G6 degrade phospholipids to yield important lipid second messengers. While these sequencing results are tantalizing, clearly more sequencing needs to be done to establish whether mutations in lipid metabolism genes are significant risk factors for epilepsy and PD.
In conclusion, how a disruption in phospholipid homeostasis alters the function of the endoplasmic reticulum and how it triggers protein aggregation are relatively unexplored areas. A deeper understanding of the role of phospholipid homeostasis can come about from lipidomic profiling by mass spectrometry of postmortem brain tissue and from magnetic resonance imaging of the brain to detect phospholipid metabolites13 combined with other imaging techniques to detect the proteins such as aggregated α-synuclein.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Auluck PK, Caraveo G, Lindquist S. alpha-Synuclein: membrane interactions and toxicity in Parkinson's disease. Ann Rev Cell Dev Biol 2010; 26:211-33; PMID:20500090; http://dx.doi.org/ 10.1146/annurev.cellbio.042308.113313 [DOI] [PubMed] [Google Scholar]
- 2. Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949-53; PMID:23161999; http://dx.doi.org/ 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vance JE. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: two metabolically related aminophospholipids. J Lipid Res 2008; 49:1377-87; PMID:18204094; http://dx.doi.org/ 10.1194/jlr.R700020-JLR200 [DOI] [PubMed] [Google Scholar]
- 4. Schuiki I, Schnabl M, Czabany T, Hrastnik C, Daum G. Phosphatidylethanolamine synthesized by four different pathways is supplied to the plasma membrane of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 2010; 1801:480-6; PMID:20044027; http://dx.doi.org/ 10.1016/j.bbalip.2009.12.008 [DOI] [PubMed] [Google Scholar]
- 5. Kainu V, Hermansson M, Hanninen S, Hokynar K, Somerharju P. Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim Biophys Acta 2013; 1831:429-37; PMID:23159415; http://dx.doi.org/ 10.1016/j.bbalip.2012.11.003 [DOI] [PubMed] [Google Scholar]
- 6. Wang S, Zhang S, Liou LC, Ren Q, Zhang Z, Caldwell GA, Caldwell KA, Witt SN. Phosphatidylethanolamine deficiency disrupts alpha-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc Natl Acad Sci USA 2014; 111:E3976-85; PMID:25201965; http://dx.doi.org/ 10.1073/pnas.1411694111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benzer S. From the gene to behavior. JAMA 1971; 218:1015-22; PMID:4942064; http://dx.doi.org/ 10.1001/jama.1971.03190200047010 [DOI] [PubMed] [Google Scholar]
- 8. Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell 1994; 79:23-33; PMID:7923374; http://dx.doi.org/ 10.1016/0092-8674(94)90397-2 [DOI] [PubMed] [Google Scholar]
- 9. Euro-EPINOMICS-RES Consortium . De novo mutations in synaptic transmission genes tncluding DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014; 95:360-70; PMID:25262651; http://dx.doi.org/ 10.1016/j.ajhg.2014.08.013; In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004; 351:1972-7; PMID:15525722; http://dx.doi.org/ 10.1056/NEJMoa033277 [DOI] [PubMed] [Google Scholar]
- 11. Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 2006; 38:752-4; PMID:16783378; http://dx.doi.org/ 10.1038/ng1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson's disease. PLoS Genet 2011; 7:e1002141; PMID:21738487; http://dx.doi.org/ 10.1371/journal.pgen.1002141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hattingen E, Magerkurth J, Pilatus U, Mozer A, Seifried C, Steinmetz H, Zanella F, Hilker R. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson's disease. Brain 2009; 132:3285-97; PMID:19952056; http://dx.doi.org/ 10.1093/brain/awp293 [DOI] [PubMed] [Google Scholar]
- 14. Lim HY, Wang W, Wessells RJ, Ocorr K, Bodmer R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev 2011; 25:189-200; PMID:21245170; http://dx.doi.org/ 10.1101/gad.1992411 [DOI] [PMC free article] [PubMed] [Google Scholar]