

Abstract
Neuronal activity results in release of K+ into the extracellular space of the central nervous system. If the excess K+ is allowed to accumulate, neuronal firing will be compromised by the ensuing neuronal membrane depolarization. The surrounding glial cells are involved in clearing K+ from the extracellular space by molecular mechanism(s), the identity of which have been a matter of controversy for over half a century. Kir4.1-mediated spatial buffering of K+ has been promoted as a major contributor to K+ removal although its quantitative and temporal contribution has remained undefined. We discuss the biophysical and experimental challenges regarding determination of the contribution of Kir4.1 to extracellular K+ management during neuronal activity. It is concluded that 1) the geometry of the experimental preparation is crucial for detection of Kir4.1-mediated spatial buffering and 2) Kir4.1 enacts spatial buffering of K+ during but not after neuronal activity.
Keywords: glia, ion transport, Kir4.1, Na+/K+-ATPase, NKCC1, potassium clearance, spatial buffering
Abbreviations
- Cx30
Connexin 30
- Cx43
Connexin 43
- Kir
Inward rectifier K+ channel
- NKCC1
Na+/K+/2Cl− cotransporter 1
- Veq
Equilibrium potential
- VK
Equilibrium potential for K+
- Vm
membrane potential
Introduction
Nearly 60 years ago Frankenhaeuser and Hodgkin demonstrated that action potentials in the squid giant axon caused a significant release of K+ into the surrounding fluid.1 At the time, it was established that the extracellular K+ concentration had to be restored to basal levels to prevent a global membrane depolarization which would severely compromise neuronal firing capability. The extracellular K+ level, [K+]o, in the mammalian brain has indeed proven to be under strict control with a baseline ∼3 mM K+, typical increases of less than 1 mM K+ during physiological neuronal activity,2,3 and a ceiling level of ∼12 mM K+, at which point no further rise in [K+]o will occur despite more intense electrical stimulation.4,5 The high K+ permeability of the surrounding glia cells caused researchers to propose a role for glia cells in shielding the neurons from high loads of extracellular K+ via their ability to redistribute K+ to sites where [K+]o remained undisturbed.6 It was later demonstrated that K+ was indeed able to move through a network of gap junction-coupled astrocytes during clearance from the extracellular space and not solely by diffusion in the extracellular space.7,8
During, but not after, neuronal activity, K+ exits from the neurons while it accumulates in the surrounding astrocytes9-12 (Fig. 1). At the termination of the neuronal activity, K+ is lost from the astrocytes and returns to the neurons, indicating that the astrocytic compartment acts as a K+ sink only during the time of K+ efflux from the neuronal structure.9-12 The role of the different astrocytic K+ transporting mechanisms in the absorption of K+ from the extracellular space following neuronal activity has been debated for decades.6,13-16 In a recent publication, we therefore set out to determine the quantitative and temporal contribution of the different transport mechanisms to K+ clearance from the extracellular space17: The Na+/K+/2Cl− cotransporter (NKCC1) appeared to serve no role in clearance of K+ transients from the extracellular space whereas the Na+/K+-ATPase acted as the key mechanism for restoration of extracellular K+ concentrations following neuronal activity. The K+ channel Kir4.1 enacted its role in K+ clearance exclusively during continuous delivery of K+ into the extracellular space. In this accompanying paper, we aim to further discuss and clarify some issues and challenges associated with experimental determination of a quantitative physiological role for Kir4.1-mediated spatial buffering.
Figure 1.

Astrocytic K+ accumulation during neuronal activity. Electrical stimulation of the guinea-pig lateral olfactory tract (l.o.t.), marked by black bar at the bottom. The extracellular K+ concentration (upper panel) and the intracellular K+ concentration (middle panel) were measured as K+ activities (aK) with ion-sensitive microelectrodes and Em is the glial membrane potential. The intracellular K+ concentration increased slower than that of the extracellular space, probably due to the kinetics of the K+ uptake mechanism. Adapted from9 with permission.
Kir4.1-Mediated Spatial Buffering of K+
The spatial buffering concept relies on the high conductance (permeability) of glial cells to K+: As the membrane potential (Vm) of a cell is determined by the weighted average of the permeable ions’ equilibrium potentials (Veq), the astrocytic membrane potential, in essence, follows that of the equilibrium potential for K+ (VK).18,19 Under basal conditions, the astrocytic membrane potential is generally slightly less negative than the equilibrium potential for K+, promoting an outwardly directed driving force for K+. An increase in [K+]o will generate a less negative VK which is thus mirrored by an (almost) equal shift in the membrane potential. For K+ to move inward through a channel, the membrane potential must be rendered more negative than the equilibrium potential for K+, which experimentally is readily achievable with an intracellular electrode providing a negative voltage clamp20-23 while more biophysically challenging to obtain in vivo. Channel-mediated spatial buffering of K+ thus requires that only part of the collective cell membrane of the electrically coupled syncytium is exposed to a change in [K+]o and that the combined astrocyte membrane potential therefore does not change with the same magnitude as the local change in VK. Consequently, in the area of local activity VK would be rendered less negative than Vm of the astrocytic syncytium and the requirement for an inward channel-mediated K+ current would thus be met6,15 (Fig. 2).
Figure 2.

Principle of channel-mediated spatial buffering. Neuronal activity causes a release of K+ into the extracellular space. The elevation of [K+]o may occur in a local area and only part of the astrocytic membrane will thus be exposed to this increase. Consequently the equilibrium potential for K+ is changed in this particular region, illustrated in the figure by a VK of around -60 mV. In the more distant regions, where [K+]o is undisturbed, VK remains at around -90 mV. The membrane potential is decided by the weighted average of the equilibrium potentials for K+ along the astrocytic membrane (or astrocyte syncytium). As such it can be considered “internally clamped” at an intermediate value, represented by a Vm of −75 mV in this figure. With a Vm more negative than VK, an inward driving force is generated across the perisynaptic astrocytic membrane (left side of the figure). At distal parts of the astrocyte (or astrocytic syncytium) an outward driving force for K+ promotes the exit of K+ from the astrocytic compartment (right side of the figure).
The K+ entering the glia cell in this manner spreads electrotonically through the astrocytic syncytium (via the gap junctions Cx30/Cx4324) or the Müller cell and exits at distal endfeet; ‘one ion in, one ion out’.16,25,26 This manner of ionic movement is faster than diffusion along the extracellular space27 and precludes net accumulation of cytoplasmic K+ (and thus osmolytes).6,28 Channel-mediated intracellular accumulation of K+ could take place with parallel transport of its counter-ion (like Cl−). In this scenario, however, no net charge would enter the cell and therefore electrotonic movement of positive charges, and thus spatial buffering in the original sense, would be non-existent. As a consequence, and due to no such Cl− channel having been identified, this possibility will not be considered further in the present context. Independently of the molecular machinery involved in K+ uptake from the extracellular space, K+ subsequently gets released from astrocytes along K+'s electrochemical gradient by means of open K+ channels, among them Kir4.1. We are, nonetheless, reluctant to consider this perisynaptic K+ release ‘Kir4.1-mediated spatial buffering’ since the prerequisite for reverse spatial buffering (significantly increased local [K+]o) is not present at the endfeet and Kir4.1-mediated K+ uptake at the endfeet has not been demonstrated.
The generation of spatial buffer currents has been convincingly demonstrated in response to increased [K+]o.26,29 Inhibition of the spatial buffer currents by BaCl2 assigned the inward rectifier Kir4.1 as the molecular identity of the involved K+ channel.26,30 Ba2+ concentrations up to 100 μM predominantly inhibit K+ channels of the Kir subfamily whereas higher concentrations (used in certain studies21,31,32) affect the Na+/K+-ATPase33 as well as other ion channels such as the IH current34,35 and the Ca2+-activated K+ channels.36,37 Inwardly rectifying channels favor inward current during hyperpolarization of the membrane over outward current during depolarization.38 Inward rectification thus describes the shape of the I/V relation obtained with these channels and not the direction of the K+ flow, which is exclusively determined by the electrochemical driving force for K+ in relation to the membrane potential. Astrocytes and glial Müller cells of the retina present with currents that are consistent with a prevalent presence of Ba2+-sensitive inwardly rectifying K+ channels of the Kir4.1 subtype.30,39-41 It should be noted, though, that astrocytic expression of other Kir isoforms (Kir2.1 in particular) has been reported42-44 in addition to K+ channels of the TREK subfamily.41 Kir4.1 has received the majority of the experimental attention regarding spatial buffering due to its distinct localization at synaptic regions and at the perivascular endfeet of astrocyte and Müller glia processes.25,45-47 Genetic ablation of Kir4.1 illustrates the importance of this channel in setting the membrane potential in astrocytes and Müller cells: The highly negative membrane potential of these glial cells (around −85 mV) is markedly depolarized in Kir4.1−/− mice20,21,42,48 as also observed following Ba2+-mediated block of Kir4.1 in the WT mice.20,49 Both the global knock-out mice50 and the conditional knockout mice20 die in their fourth post-natal week with compromised motor functions, small brains, enlarged lateral ventricles, general dysmyelination, axonal degeneration, vacuolization and gliosis.20,42,50 It is therefore evident that Kir4.1 serves a crucial function in the brain. It could be advocated, though, that care should be taken when attributing data obtained with these knock-out mice solely to a physiological role for Kir4.1. The general pathological state of these animals, up- or down regulation of other proteins, putative changes in size of the extracellular space, astrocytic membrane depolarization, and large animal/slice variations in stimulus-induced ΔK+ (since each brain slice can no longer serve as its own control) may possibly affect the K+ homeostasis and its quantification in an indirect, and yet undefined, manner. However, the discovery of variant forms of Kir4.1, and the altered localized expression pattern of Kir4.1, in patients with temporal lope epilepsy point to its physiological importance.51,52
Taken together, retinal Müller glia and the astrocytic syncytium express Kir4.1 in an appropriately localized manner and are indeed able to release K+ at sites distal from the local increase in extracellular [K+] and carry spatial buffer currents in the process. Nevertheless, the quantitative contribution of Kir4.1-mediated spatial buffering to removal of K+ from the extracellular space following neuronal activity has remained under debate for decades. A finite determination of its contribution has been complicated by experimental and biophysical challenges as well as by issues regarding temporal quantification.
Requirements for Channel-Mediated Spatial Buffering of K+
Evaluation of Kir4.1-mediated spatial buffering in brain slices has been determined with electrical stimulation, often at high-frequency, to provide the means to achieve K+ release from activated neurons.17,48,49,53-55 With this experimental approach, one may envision a scenario of global K+ release in which no nearby local region surrounded by basal [K+]o remains (Fig. 3A). Given the biophysical requirements for Kir4.1-mediated inward flux of K+ (see above), this experimental approach might actually limit or even preclude K+ removal from the extracellular space by Kir4.1-mediated spatial buffering. Accordingly, the majority of slice studies relying on a stimulation approach have reported no, or a minor, delay in post-stimulus clearance of K+ from the extracellular space upon inhibition or knockout of Kir4.117,21,48,49,54-56 (Fig. 4A).
Figure 3.

High frequency stimulation may prevent Kir4.1-mediated spatial buffering. (A) During high-frequency stimulation, global synaptic activity may elevate [K+]o in a large region of the brain slice and thus leave no nearby region with basal [K+]o to which K+ can be redistributed. (B) Focal K+ application can be obtained with iontophoresis by which K+ is released to a local area by a current-pulse through a micropipette (illustrated in upper right corner). This mode of K+ elevation fulfills the requirements for channel-mediated spatial buffering, which can thereby be experimentally evaluated.
Figure 4.

Inhibition of Kir4.1 increases the peak [K+]o but does not affect the rate of K+ recovery. (A) Representative stimulus-evoked [K+]o changes in a rat hippocampal slice before (black trace) and during Ba2+ application (gray trace), stimulus protocol indicated by black bar. Normalized and summarized K+ decay rates (obtained between the dashed lines) indicated in the insert. (B) Focal K+ application in rat hippocampal slices was obtained with iontophoresis in the presence of neurotransmitter receptor blockers before (black trace) and during Ba2+ application (gray trace), iontophoretic pulse indicated with black bar. Consecutive control traces with identical amplitude are indicated in the insert. Normalized and summarized peak [K+]o is illustrated as an insert and the K+ decay rate (obtained between the dashed lines) indicated in Panel A, insert. Adapted from17 with permission.
To circumvent the stimulus-evoked global release of K+, local increases in [K+]o can be obtained by focal application of K+ delivered by iontophoresis17,32,57,58 (Fig. 3B). To prevent spontaneous neuronal spiking activity, iontophoretically-induced increase in [K+]o is routinely carried out in the presence of neurotransmitter receptor blockers.32,57 It must thus be kept in mind that any putative modulatory effect of these receptors on K+ clearance will be lost in this type of experiment. Inhibition of Kir4.1 with BaCl2 did not alter the rate of K+ removal following termination of K+ delivery into the extracellular space but increased the peak [K+]o during the iontophoretical pulse17,32,57,58 (Fig. 4B). This temporal effect of BaCl2 on iontophoretically applied K+ indicates that Kir4.1 would enact its spatial buffering of K+ exclusively during neuronal activity. Subsequently, when neuronal activity terminates, the K+ flow through Kir4.1 gradually reverts to a solely outward direction as typical at resting conditions. In that sense Kir4.1 would be involved in post-stimulus K+ homeostasis though most likely not due to reverted Kir4.1-mediated spatial buffering currents originating from inwardly-directed Kir4.1 mediated current at the endfeet processes. Rather, Kir4.1 would facilitate the release of K+ accumulated by the Na+/K+-ATPase.17
The geometry of the retina provides an advantageous experimental preparation for detection of spatial buffering with a physiologically relevant stimulus giving rise to a localized [K+]o increase: light stimulus causes neuronal release of K+ in the local region of the inner plexiform layer. Subsequently the rise in [K+]o generates a current through the glial Müller cells, which are specialized retinal glial cells located with their nuclear part in close approximation to the neuronal K+ release site and attached via a stalk region to the endfoot facing the vitreous humor25 (Fig. 5A). With a terminal Kir4.1-mediated K+ efflux from the Müller cell vitreal endfeet, the vitreous humor then serves as a reservoir/sink for K+26. Due to the unicellular movement of K+ through the individual Müller cells, as opposed to the astrocytic syncytium, this particular flow of K+ current has been termed K+ siphoning instead of spatial buffering.27
Figure 5.
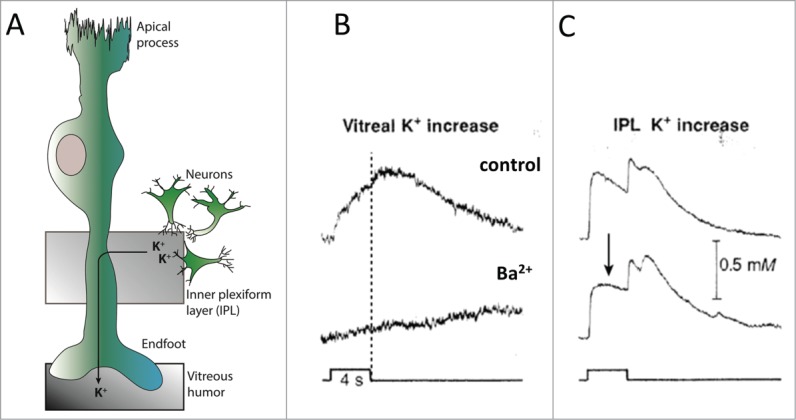
Ba2+ blocks Kir4.1-mediated vitreal release of K+ by the Müller cell. (A) A retinal Müller glia cell consists of an apical process, a nuclear part localized in the vicinity of the K+-releasing neurons in the inner plexiform layer, and a vitreal endfoot from which K+ is released into the vitreous humor during spatial buffering/K+ siphoning. (B) Light-evoked [K+]o changes in the vitreous humor of the frog eyecup before (control, upper panel) and during Ba2+ application (Ba2+, lower panel), light stimulus indicated at the bottom. Dashed line indicates the termination of the light stimulus. (C) Light-evoked [K+]o changes in the inner plexiform layer (IPL) of the frog eyecup before (control, upper panel) and during Ba2+ application (Ba2+, lower panel), light stimulus indicated at the bottom. The arrow points to the Ba2+-induced reduction in K+ clearance during the stimulus. The secondary peak in the IPL at light ‘off’ is the delayed off response. Panel B and C adapted from26 with permission according to agreement.
With this preparation, Newman and colleagues elegantly illustrated that the Kir4.1-mediated release of K+ into the vitreous humor was apparent during the time of the light pulse and not in the K+ recovery phase following termination of the light pulse, despite the prolonged elevated level of [K+]o immediately following the stimulus26 (Fig. 5B). This temporal vitreal K+ release pattern supports the notion that Kir4.1-mediated spatial buffering occurs exclusively during neuronal activity and that no electrotonic redistribution of K+ takes place in the post-stimulus phase of K+ recovery. Simultaneous recording of light-induced K+ transients in the inner plexiform layer demonstrated a Ba2+-dependent reduction in the rate of K+ clearance during the pulse of light26,59 (Fig. 5C). Contrary, the rate of post-stimulus K+ recovery appeared unaltered by inhibition of Kir4.1 in the retina preparation26,59 (Fig 5C), in line with data obtained in hippocampus and optic nerve with either iontophoretically-applied K+ or electric stimulation of the slice.17,21,48,49,54-56
Quantification of Post-Stimulus Clearance
Given the indications of the temporal contribution from Kir4.1 in the retinal preparations and iontophoretic K+ applications, one may propose future focus on the role of Kir4.1-mediated spatial buffering in K+ clearance from the extracellular space be directed toward the phase during the neuronal activity. Although some studies did quantify the effect of Ba2+ on the peak [K+]o using the stimulation paradigm,21,24,55,56 the majority of the studies reported on the K+ decay phase17,21,24,48,49,53,54,56 in which Kir4.1-mediated spatial buffering may actually not be in effect.
Quantification of the decay phase of the K+ transients and the effect of inhibiting Kir4.1 with either BaCl2 or genetic deletion is complicated by the dual action of Kir4.1. Determination of the time constant, τ, or the half-time recovery, t1/2, is based on the recovery from the achieved peak back to baseline. Consequently, if either the peak or, more prominently, the baseline is changed following inactivation of Kir4.1 (pharmacologically or genetically), the decay rate comparison with the control trace can become skewered despite a possibly unchanged rate of K+ removal (as in mM/sec post-stimulus). Pharmacological block of Kir4.1 increases the iontophoretically delivered peak [K+]o17,32 and in some studies also the stimulus-induced peak [K+]o,55,59 whereas a post-clearance undershoot in [K+]o (below the baseline level) arises upon disruption of Kir4.1 in several studies.21,48,53,55,56 The Ba2+/Kir4.1−/−-evoked [K+]o undershoot is thought to represent Kir4.1-mediated K+ efflux from the glia cells following the neuronal activity. Such an efflux would facilitate the return of K+ to the neuronal compartment.48,54-56 The temporally-dictated oppositely-directed flow of K+ through Kir4.1 would clearly interfere with a quantitative measurement involving the entire decay phase including altered peak and baseline.
Conclusion
Glial cells, whether retinal Müller cells or astrocytes, act as K+ sinks during neuronal K+ release after which K+ returns to the neuronal structure.9-12 Kir4.1-mediated spatial buffering contributes to the K+ management in the extracellular space during the neuronal activity whereas post-stimulus recovery of [K+]o back to baseline does not require Kir4.1-mediated spatial buffering. Instead, Kir4.1 appears to serve its dual role in the latter part of the recovery phase as an exit route for accumulated glial K+. The glial K+ accumulation observed during neuronal activity9-12 is not compatible with spatial buffering being the sole K+-removing candidate as no net K+ accumulation occurs via this clearance mechanism. In addition, complete disruption of the astrocytic coupling (Cx30−/−/Cx43−/−), which is required for K+ redistribution, conferred only a slight delay in hippocampal K+ clearance, indicating that gap junction-mediated spatial buffering of K+ represents a limited fraction of the combined K+ clearance capacity.24 We therefore propose, at least in hippocampal slices, glial and neuronal Na+/K+-ATPase as key contributors to K+ management in the extracellular space in association with neuronal activity.17
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This project was supported by the Lundbeck foundation.
References
- 1. Frankenhaeuser B, Hodgkin AL. The after-effects of impulses in the giant nerve fibres of Loligo. J Physiol 1956; 131:341-76; PMID:13320339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singer W, Lux HD. Extracellular potassium gradients and visual receptive fields in the cat striate cortex. Brain Res 1975; 96:378-83; PMID:1175021; http://dx.doi.org/ 10.1016/0006-8993(75)90751-9 [DOI] [PubMed] [Google Scholar]
- 3. Syková E, Rothenberg S, Krekule I. Changes of extracellular potassium concentration during spontaneous activity in the mesencephalic reticular formation of the rat. Brain Res 1974; 79:333-7; http://dx.doi.org/ 10.1016/0006-8993(74)90428-4 [DOI] [PubMed] [Google Scholar]
- 4. Heinemann U, Lux HD. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res 1977; 120:231-49; PMID:832122; http://dx.doi.org/ 10.1016/0006-8993(77)90903-9 [DOI] [PubMed] [Google Scholar]
- 5. Prince DA, Lux HD, Neher E. Measurement of extracellular potassium activity in cat cortex. Brain Res 1973; 50:489-95; PMID:4705519; http://dx.doi.org/ 10.1016/0006-8993(73)90758-0 [DOI] [PubMed] [Google Scholar]
- 6. Orkand R, Nicholls J, SW K. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol 1966; 29:788-806; PMID:5966435 [DOI] [PubMed] [Google Scholar]
- 7. Cordingley GE, Somjen GG. The clearing of excess potassium from extracellular space in spinal cord and cerebral cortex. Brain Res 1978; 151:291-306; PMID:209864; http://dx.doi.org/ 10.1016/0006-8993(78)90886-7 [DOI] [PubMed] [Google Scholar]
- 8. Gardner-Medwin AR. A study of the mechanisms by which potassium moves through brain tissue in the rat. J Physiol 1983; 335:353-74; PMID:6875883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ballanyi K, Grafe P, Bruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol 1987; 382:159-74; PMID:2442359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grafe P, Ballanyi K. Cellular mechanisms of potassium homeostasis in the mammalian nervous system. Can J Physiol Pharmacol 1987; 65:1038-42; PMID:3621030; http://dx.doi.org/ 10.1139/y87-164 [DOI] [PubMed] [Google Scholar]
- 11. Coles JA, Schneider-Picard G. Increase in glial intracellular K+ in drone retina caused by photostimulation but not mediated by an increase in extracellular K+. Glia 1989; 2:213-22; PMID:2527820; http://dx.doi.org/ 10.1002/glia.440020402 [DOI] [PubMed] [Google Scholar]
- 12. Coles JA, Orkand RK. Modification of potassium movement through the retina of the drone (Apis mellifera male) by glial uptake. J Physiol 1983; 340:157-74; PMID:6887045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hertz L. Possible role of neuroglia: a potassium-mediated neuronal–neuroglial–neuronal impulse transmission system. Nature 1965; 206:1091-4; PMID:5325441; http://dx.doi.org/ 10.1038/2061091a0 [DOI] [PubMed] [Google Scholar]
- 14. Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 2000; 36:291-300; PMID:10732996; http://dx.doi.org/ 10.1016/S0197-0186(99)00137-0 [DOI] [PubMed] [Google Scholar]
- 15. MacAulay N, Zeuthen T. Glial K+ clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res 2012; 37:2299-309; PMID:22367475; http://dx.doi.org/ 10.1007/s11064-012-0731-3 [DOI] [PubMed] [Google Scholar]
- 16. Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience 2004; 129:1043-54; http://dx.doi.org/ 10.1016/j.neuroscience.2004.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N. Contributions of the Na+K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 2014; 62:608-22; PMID:24482245; http://dx.doi.org/ 10.1002/glia.22629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophysiol 1966; 29:768-87; PMID:5966434 [DOI] [PubMed] [Google Scholar]
- 19. Ransom B, Goldring S. Slow depolarization in cells presumed to be glia in cerebral cortex of cat. J Neurophysiol 1973; 36:869-78; PMID:4805016 [DOI] [PubMed] [Google Scholar]
- 20. Djukic B, Casper KB, Philpot BD, Chin L-S, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 2007; 27:11354-65; PMID:17942730; http://dx.doi.org/ 10.1523/JNEUROSCI.0723-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, et al. Lack of the Kir4.1 channel subunit abolishes K +buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol 2006; 95:1843-52; PMID:16306174; http://dx.doi.org/ 10.1152/jn.00996.2005 [DOI] [PubMed] [Google Scholar]
- 22. Skatchkov SN, Krušek J, Reichenbach A, Orkand RK. Potassium buffering by Müller cells isolated from the center and periphery of the frog retina. Glia 1999; 27:171-80; PMID:10417816; http://dx.doi.org/ 10.1002/(SICI)1098-1136(199908)27:2%3c171::AID-GLIA7%3e3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- 23. Sibille J, Pannasch U, Rouach N. Astroglial potassium clearance contributes to short-term plasticity of synaptically evoked currents at the tripartite synapse. J Physiol 2014; 592:87-102; PMID:24081156; http://dx.doi.org/ 10.1113/jphysiol.2013.261735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 2006; 26:5438-47; PMID:16707796; http://dx.doi.org/ 10.1523/JNEUROSCI.0037-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newman EA. Regional specialization of retinal glial cell membrane. Nature 1984; 309:155-7; PMID:6717594; http://dx.doi.org/ 10.1038/309155a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karwoski C, Lu H-K, Newman E. Spatial buffering of light-evoked potassium increases by retinal Müller (Glial) cells. Science 1989; 244:578-80; PMID:2785716; http://dx.doi.org/ 10.1126/science.2785716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newman E, Frambach D, Odette L. Control of extracellular potassium levels by retinal glial cell K +siphoning. Science 1984; 225:1174-5; PMID:6474173; http://dx.doi.org/ 10.1126/science.6474173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gardner-Medwin AR. Analysis of potassium dynamics in mammalian brain tissue. J Physiol 1983; 335:393-426; PMID:6875885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strohschein S, Hüttmann K, Gabriel S, Binder DK, Heinemann U, Steinhäuser C. Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 2011; 59:973-80; PMID:21446052; http://dx.doi.org/ 10.1002/glia.21169 [DOI] [PubMed] [Google Scholar]
- 30. Newman EA. Potassium conductance block by barium in amphibian Müller cells. Brain Res 1989; 498:308-14; PMID:2790485; http://dx.doi.org/ 10.1016/0006-8993(89)91109-8 [DOI] [PubMed] [Google Scholar]
- 31. Kivi A, Lehmann TN, Kovács R, Eilers A, Jauch R, Meencke HJ, Von Deimling A, Heinemann U, Gabriel S. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci 2000; 12:2039-48; PMID:10886343; http://dx.doi.org/ 10.1046/j.1460-9568.2000.00103.x [DOI] [PubMed] [Google Scholar]
- 32. Jauch R, Windmüller O, Lehmann T-N, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res 2002; 925:18-27; PMID:11755897; http://dx.doi.org/ 10.1016/S0006-8993(01)03254-1 [DOI] [PubMed] [Google Scholar]
- 33. Walz W, Shargool M, Hertz L. Barium-induced inhibition of K+ transport mechanisms in cortical astrocytes—its possible contribution to the large Ba2+-evoked extracellular K+ signal in brain. Neuroscience 1984; 13:945-9; PMID:6098861; http://dx.doi.org/ 10.1016/0306-4522(84)90108-8 [DOI] [PubMed] [Google Scholar]
- 34. Bayliss DA, Viana F, Bellingham MC, Berger AJ. Characteristics and postnatal development of a hyperpolarization-activated inward current in rat hypoglossal motoneurons in vitro. J Neurophysiol 1994; 71:119-28; PMID:7512625 [DOI] [PubMed] [Google Scholar]
- 35. van Welie I, Wadman WJ, van Hooft JA. Low affinity block of native and cloned hyperpolarization-activated Ih channels by Ba2+ ions. Eur J Pharmacol 2005; 507:15-20; PMID:15659289; http://dx.doi.org/ 10.1016/j.ejphar.2004.11.051 [DOI] [PubMed] [Google Scholar]
- 36. Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 1995; 268:C799-C822 [DOI] [PubMed] [Google Scholar]
- 37. Skinner LJ, Enée V, Beurg M, Jung HH, Ryan AF, Hafidi A, Aran J-M, Dulon D. Contribution of BK Ca2+-activated K+ channels to auditory neurotransmission in the guinea pig cochlea. J Neurophysiol 2003; 90:320-32; PMID:12611976; http://dx.doi.org/ 10.1152/jn.01155.2002 [DOI] [PubMed] [Google Scholar]
- 38. Hille B. Potassium and Chloride Channels. Ion Channels of Excitable Membranes, third edition. Sunderland, MA: Sinauer Associates, Inc., 2001; pp. 131-67 [Google Scholar]
- 39. Sontheimer H. Voltage-dependent ion channels in glial cells. Glia 1994; 11:156-72; PMID:7523291; http://dx.doi.org/ 10.1002/glia.440110210 [DOI] [PubMed] [Google Scholar]
- 40. Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K +buffering to cell differentiation. J Neurochem 2008; 107:589-601; PMID:18691387; http://dx.doi.org/ 10.1111/j.1471-4159.2008.05615.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seifert G, Hüttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhäuser C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 2009; 29:7474-88; PMID:19515915; http://dx.doi.org/ 10.1523/JNEUROSCI.3790-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 2000; 20:5733-40; PMID:10908613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schröder W, Seifert G, Hüttmann K, Hinterkeuser S, Steinhäuser C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol Cell Neurosci 2002; 19:447-58; PMID:11906215; http://dx.doi.org/ 10.1006/mcne.2001.1080 [DOI] [PubMed] [Google Scholar]
- 44. Stonehouse AH, Pringle JH, Norman RI, Stanfield PR, Conley EC, Brammar WJ. Characterisation of Kir2.0 proteins in the rat cerebellum and hippocampus by polyclonal antibodies. Histochem Cell Biol 1999; 112:457-65; PMID:10651097; http://dx.doi.org/ 10.1007/s004180050429 [DOI] [PubMed] [Google Scholar]
- 45. Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K +channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol 2001; 281:C922-C31; PMID:11502569 [DOI] [PubMed] [Google Scholar]
- 46. Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug F-m, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 1999; 26:47-54; PMID:10088671; http://dx.doi.org/ 10.1002/(SICI)1098-1136(199903)26:1%3c47::AID-GLIA5%3e3.0.CO;2-5 [DOI] [PubMed] [Google Scholar]
- 47. Newman EA. High potassium conductance in astrocyte endfeet. Science 1986; 233:453-4; PMID:3726539; http://dx.doi.org/ 10.1126/science.3726539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci 2010; 30:15769-77; PMID:21106816; http://dx.doi.org/ 10.1523/JNEUROSCI.2078-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meeks JP, Mennerick S. Astrocyte membrane responses and potassium accumulation during neuronal activity. Hippocampus 2007; 17:1100-8; PMID:17853441; http://dx.doi.org/ 10.1002/hipo.20344 [DOI] [PubMed] [Google Scholar]
- 50. Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci 2001; 21:5429-38; PMID:11466414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heuser K, Nagelhus EA, Taubøll E, Indahl U, Berg PR, Lien S, Nakken S, Gjerstad L, Ottersen OP. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res 2010; 88:55-64; PMID:19864112; http://dx.doi.org/ 10.1016/j.eplepsyres.2009.09.023 [DOI] [PubMed] [Google Scholar]
- 52. Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 2012; 60:1192-202; http://dx.doi.org/ 10.1002/glia.22313 [DOI] [PubMed] [Google Scholar]
- 53. Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby O, Nagelhus EA. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 2011; 59:1635-42; PMID:21748805; http://dx.doi.org/ 10.1002/glia.21205 [DOI] [PubMed] [Google Scholar]
- 54. Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K +accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol 2000; 522:427-42; PMID:10713967; http://dx.doi.org/ 10.1111/j.1469-7793.2000.00427.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bay V, Butt AM. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia 2012; 60:651-60; PMID:22290828; http://dx.doi.org/ 10.1002/glia.22299 [DOI] [PubMed] [Google Scholar]
- 56. D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na+K+-pump in the regulation of extracellular K+ in rat hippocampus. J Neurophysiol 2002; 87:87-102 [DOI] [PubMed] [Google Scholar]
- 57. Päsler D, Gabriel S, Heinemann U. Two-pore-domain potassium channels contribute to neuronal potassium release and glial potassium buffering in the rat hippocampus. Brain Res 2007; 1173:14-26; http://dx.doi.org/ 10.1016/j.brainres.2007.07.013 [DOI] [PubMed] [Google Scholar]
- 58. Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007; 130:535-47; PMID:17121744; http://dx.doi.org/ 10.1093/brain/awl317 [DOI] [PubMed] [Google Scholar]
- 59. Oakley II B, Katz BJ, Xu Z, Zheng J. Spatial buffering of extracellular potassium by Müller (glial) cells in the toad retina. Exp Eye Res 1992; 55:539-50; PMID:1483500; http://dx.doi.org/ 10.1016/S0014-4835(05)80166-6 [DOI] [PubMed] [Google Scholar]