

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) display anti-inflammatory, antipyretic and analgesic properties by inhibiting cyclooxygenases and blocking prostaglandin production. Previous studies, however, suggested that some NSAIDs also modulate peroxisome proliferator activated receptors (PPARs), raising the possibility that such off target effects contribute to the spectrum of clinically relevant NSAID actions. In this study, we set out to understand how peroxisome proliferator activated receptor-γ (PPARγ/PPARG) interacts with NSAIDs using X-ray crystallography and to relate ligand binding modes to effects on receptor activity. We find that several NSAIDs (sulindac sulfide, diclofenac, indomethacin and ibuprofen) bind PPARγ and modulate PPARγ activity at pharmacologically relevant concentrations. Diclofenac acts as a partial agonist and binds to the PPARγ ligand binding pocket (LBP) in typical partial agonist mode, near the β-sheets and helix 3. By contrast, two copies of indomethacin and sulindac sulfide bind the LBP and, in aggregate, these ligands engage in LBP contacts that resemble agonists. Accordingly, both compounds, and ibuprofen, act as strong partial agonists. Assessment of NSAID activities in PPARγ-dependent 3T3-L1 cells reveals that NSAIDs display adipogenic activities and exclusively regulate PPARγ-dependent target genes in a manner that is consistent with their observed binding modes. Further, PPARγ knockdown eliminates indomethacin activities at selected endogenous genes, confirming receptor-dependence of observed effects. We propose that it is important to consider how individual NSAIDs interact with PPARγ to understand their activities, and that it will be interesting to determine whether high dose NSAID therapies result in PPAR activation.
Keywords: non-steroidal anti-inflammatory drugs, peroxisome proliferator activated receptors, X-ray structure, gene expression, 3T3-L1, partial agonist
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) possess anti-inflammatory, antipyretic and analgesic activity. Therapeutic actions are attributed to inhibition of cyclooxygenases (COX-1 and COX-2) [Smith et al., 1994], enzymes that catalyze the first steps in conversion of arachidonic acid (AA) to prostaglandins (PGs). NSAIDs attach to a helical motif that protects the COX active site [Pountos et al., 2011]; preventing passage of AA towards the active site and blocking AA to PG conversion [2-5]. While the COX enzymes constitute major NSAID targets [Smith et al., 1994], they may not be the only pharmacologically relevant site of action [Brune et al., 1991; Gilroy and Colville-Nash, 2000a; Mukherjee et al., 1996]; there are also suggestions that some NSAID effects are mediated by peroxisome proliferator-activated receptors (PPARs) [Gilroy and Colville-Nash, 2000b; Jaradat et al., 2001; Lehmann et al., 1997a; Wick et al., 2002], which are ligand-activated transcription factors that belong to the nuclear receptor family [Berger and Moller, 2002; Schoonjans et al., 1997]. Higher NSAID concentrations are needed for inhibition of COX-2 than for COX-1 [Jaradat et al., 2001]. Given that these concentrations may approach levels required for PPAR activation in vitro, it is conceivable that high doses of certain NSAIDs could modulate PPARs [Gilroy and Colville-Nash, 2000a; Lehmann et al., 1997b].
There are three PPAR subtypes (α, β and γ) that each bind to a variety of synthetic and natural ligands, sometimes in greater than 1:1 stoichiometry [Forman et al., 1996; Kliewer et al., 1994]. PPARγ is expressed mainly in adipose tissue, macrophages and inflammatory cells [Braissant et al., 1996; Ricote et al., 1998a; Tontonoz et al., 1994a; Tontonoz et al., 1994b] and mediates anti-inflammatory, adipogenic and insulin sensitizing effects of a well-known anti-diabetic drug class, thiazolidinediones (TZDs) [Jiang et al., 1998; Lehrke and Lazar, 2005; Ricote et al., 1998b; Ricote et al., 2000]. PPARα ligands also act through their cognate receptor to inhibit pro-inflammatory effects and reduce production of inflammatory interleukin-6 and PGs in smooth muscle cells, possibly reducing the risk for atherosclerosis [Li and Yang, 2011; Staels et al., 1998].
Precise position of PPARγ ligands in the ligand binding pocket (LBP) influences receptor transcriptional outputs by modulation of activation function 2 (AF-2), a coactivator binding surface comprised of residues from C-terminal helix (H) 12, H3 and H5. Full PPARγ agonists, such as TZDs, occupy two sub-pockets of the Y-shaped buried ligand binding pocket (LBP) in the PPAR ligand binding domain (LBD) [Nolte et al., 1998]. Here, the thiazolidine group of the ligand forms strong contacts with a tyrosine residue (Tyr473) on the inner surface of H12, thereby docking H12 into a rigid active position and promoting AF-2 activity. By contrast, H12 appears more mobile in the presence of partial agonists and the degree of H12 mobility inversely correlates with magnitude of AF-2 activity and agonist response. Partial agonists often occupy locations in the LBP that are close to H3 and the β-sheet region [Bruning et al., 2007]. Such compounds partly stabilize H12 indirectly via effects upon H3, which forms part of the H12 docking site. Solution nuclear magnetic resonance studies has also revealed that some partial agonists fluctuate between binding modes and that this phenomenon is associated with slow equilibrium between different receptor conformations and incomplete H12 stabilization [27,28]. Finally, we showed that a very weak partial agonist/antagonist, the natural product luteolin, binds preferentially to a crystallographic PPARγ conformer with H12 in an inactive position [Puhl et al., 2012]. Accordingly, PPARγ ligands display a continuum of graded agonist/partial agonist responses that are linked to binding position and capacity to stabilize H12.
While some NSAIDs bind PPARs at high doses [Jaradat et al., 2001; Lehmann et al., 1997a; Wick et al., 2002], the nature of PPAR/NSAID interactions and consequences for gene expression are poorly understood. In this study, we used X-ray crystallography to define how NSAIDs interact with the PPARγ LBP and to relate these binding events to effects of these compounds on PPARγ activity. We find that different NSAIDs display different PPARγ binding modes with features that resemble agonists or partial agonists. NSAID-dependent changes in gene expression correlate well with predictions about varying degrees of PPARγ partial agonism deduced from their binding modes. We propose that it is important to consider how individual NSAIDs interact with PPARγ to fully understand their pharmacologic activities.
Materials and Methods
Reagents
NSAIDs ibuprofen, sodium diclofenac, indomethacin (Cayman Chemical); sulindac sulfide and rosiglitazone (Sigma Aldrich); PolarScreen™ PPARγ Competitor Assay, Green (Life Technologies); EnVision® Multilabel Reader (Perkin Elmer); Mouse whole genome expression arrays (Illumina BeadChip Array MouseWG-6v2).
Fluorescence polarization assay
Competition data was generated in a 384-well Optiplate (Perkin Elmer, Cat. No. 6007270). Initial serial dilutions of the ligands were made in DMSO and added to the sample buffer. The assay plate was protected from light and incubated at room temperature for two hours before measurement. Fluorescence polarization was measured in the EnVision® Multilabel Reader. Curve fitting was performed using Prism software from GraphPad Software, Inc. Error bars represent one standard deviation from the mean of triplicate reaction wells.
Transactivation assays
Cellular transactivation assays were performed in HeLa cells as described [Puhl et al., 2012]. We used 100 ng of plasmid PPRE-LUC (Firefly luciferase reporter vector), 10 ng of a CMV-driven PPARγ expression vector (Promega), and 2.5 ng of pRL-TK, which contains Renilla luciferase (Dual-Luciferase Report Assay system Promega, Madison, WI). NSAIDs (±) were tested for PPARγ activation. EC50 value was calculated from plots of the relationship between luminescence and ligand concentrations (10-9 to 10-3 M).
Protein expression and purification
The plasmid pET28a(+) (Novagen) encoding a human PPARγ LBD, fused in frame to the C-terminus of a polyhistidine (His) tag was used for expression of PPARγ in Escherichia coli strain BL21 (DE3). The expression and purification was conducted as described previously [Puhl et al., 2012].
Crystallization, data collection and structure determination
PPARγ LBD at 10-15 mg/mL was mixed with 2 mM ligands on ice and allowed to stand at 4°C overnight. The crystallization screens were performed under conditions similar to those described previously [Nolte et al., 1998] and also with several crystallization kits by sitting drop method using the robot Mosquito (TTP LABTech) and 0.5 µl of protein complex solution mixed with 0.5 µl precipitant solution and equilibrated against a 100 mL reservoir solution. Suitable crystals of PPARγ in complex with sodium diclofenac were obtained in the condition containing 1 M sodium citrate, 0.1 M HEPES pH 7.5, and 10 mM MgCl2, whereas crystals in complex with indomethacin were grown in 0.95 M sodium citrate and 0.1 M HEPES pH 8.0. Crystals of PPARγ in complex with sulindac sulfide were grown in 25% (w/v) PEG 6000 and 0.1 M Tris-HCl pH 8.5. Prior to data collection, crystals were soaked in a cryoprotectant containing the same reservoir solution complemented with 15% (v/v) ethylene glycol and rapidly cooled in a gaseous nitrogen stream at 100 K. X-ray diffraction data were collected in the protein crystallography MX2 beamline at the Laboratório Nacional de Luz Síncrotron (LNLS, Campinas, Brazil) [Guimarães et al., 2009] and 5.0.1 beamline of Advanced Light Source (ALS) - Lawrence Berkeley National Laboratory (Berkeley, CA, EUA). Diffraction data were processed using MOSLFM [Leslie, 1999] and scaled with SCALA from the CCP4 program suite [Collaborative Computational Project, 1994]. The structures were determined by molecular replacement using the program PHASER from CCP4 Packages and the PPARγ LBD (PDB code: 3SZ1[Puhl et al., 2012]) structure as a model. The programs PHENIX and COOT were used to alternately run cycles of refinement and model building [Adams et al., 2010; Emsley and Cowtan, 2004].
Adipocyte differentiation
3T3-L1 preadipocytes were cultured as previously described [29]. Two days post-confluency, cells were induced to differentiate using DMEM/F12 medium supplemented with 167 nM insulin, 1 μM dexamethasone and 0.5 mM IBMX with or without Rosiglitazone or NSAIDs for three days [Klemm et al., 2001]. Cells were then maintained in Zen Bio AM-1-L1 medium (Zen-Bio, Inc., Research Triangle Park, NC). On day 8, lipid accumulation in the adipocytes was assessed using Oil Red O staining method as per manufacturer protocol [Klemm et al., 2001]. Test ligands were: control (DMSO); 1 μM rosiglitazone; 10 μM indomethacin; 75 μM ibuprofen; 25 μM sodium diclofenac. For image quantification, 4 random field 10x images of the 3T3-L1 cells were taken using a conventional light microscope and camera and exported to ImageJ software (NIH) and converted to grayscale. Posterior segmentation (isolation) was performed to obtain the red-stained area using thresholding and measured. Posterior adjustment to number of cells per image was obtained by multiplying % of stained area by count in a Neubauer chamber-like grid, calculated with statistical analysis and graphed with Graph Pad Prism.
Microarray analysis
3T3-L1 adipocytes were maintained and differentiated in the absence of Rosiglitazone and NSAIDs. On day 8, cells were treated with either vehicle 1 μM Rosiglitazone, 25 μM sodium diclofenac, 25 μM sodium diclofenac or 75 μM ibuprofen for 24hrs. Total RNA was extracted using Aurum Total RNA Fatty and Fibrous kit (BioRad). cRNA synthesis and labeling were performed using Illumina® TotalPrep™-96 RNA Amplification Kit (Ambion). Biotin labeling in vitro transcription reaction was performed at 37°C for 14 h. Biotinylated cRNA samples were hybridized to MouseWG-6v2 arrays at 58°C for 18 h according to manufacturer's protocol, and scanned with an iScan reader. Raw data was obtained from GenomeStudio and subsequently background-subtracted and quantile-normalized using the lumi package [Du et al., 2008] and analyzed with the limma package [Smyth, 2004] within R [Team, 2011]. All analysis was corrected for multiple hypothesis testing [Benjamini and Hochberg, 1995], and effects determined to be significant with a ≥2-fold change an adjusted p-value ≤0.05 when compared to the vehicle (DMSO) control. To facilitate comparisons among the various treatments, all data were uploaded into a SQLite3 database (http://www.sqlite.org/). Data are publicly deposited as GEO dataset GSE64075.
RT-qPCR
One μg of isolated total RNA was reverse transcribed using the iScript cDNA Synthesis kit (Bio-Rad) according to the manufacturer’s recommendations. Quantitative PCR (RT-qPCR) was performed using Roche LightCycler 480 II Instrument with SYBR Green Mastermix (Roche). Primers were designed to span exon-exon boundaries to eliminate amplification of genomic DNA (primer sequences are listed in Table 1). Normalized Cp values (Critical point; CpTarget - Cp18S) of at least 3 biological replicates were analyzed by ANOVA followed by t-tests of DMSO (vehicle) vs. treatment, with treatment effect determined to be significant with a Bonferroni Corrected p-value ≤ 0.05 (GraphPad Software, version 5.01; GraphPad).
Table 1. Primer Sequences used for RT-qPCR.
| Gene | Forward primer (5’- 3’) | Reverse primer (3’ – 5’) |
|---|---|---|
|
Acaa1b |
CAG GAC GTG AAG CTA AAG CCT |
CTC CGA AGT TAT CCC CAT AGG AA |
|
Fgf21 |
GTG TCA AAG CCT CTA GGT TTC TT |
GGT ACA CAT TGT AAC CGT CCT C |
|
Gyk |
GTC ACA ATG GAG CGG TTT GAA |
TTA TGG GAT ACC ACT TTC TGG AG |
|
Gys2 |
CGC TCC TTG TCG GTG ACA TC |
CAT CGG CTG TCG TTT TGG C |
|
Fabp5 |
TGA AAG AGC TAG GAG TAG GAC TG |
CTC TCG GTT TTG ACC GTG ATG |
|
Irs2 |
GCA CCT ATG CAA GCA TCG AC |
GCG CTT CAC TCT TTC ACG AC |
|
Acsm3 |
CTT TGG CCC CAG CAG TAG ATG |
GGC TGT CAC TGG CAT ATT TCA T |
|
Orm2 |
ATT GGT GCG GCT GTC CTA AA |
ACA CAG TGG TCA TCT ATG GTG T |
|
Vcam1 |
ATG TCA ACG TTG CCC CCA A |
GCT GTC TGC TCC ACA GGA TT |
|
S100a8b |
TTC AAG ACA TCG TTT GAA AGG AAA |
AGG TTG CTC AAG GCC TTC TC |
|
Ptgis |
ATG CAG TGT CAA AAA CCG CC |
TGG GAC CCA TAT TCC CCT GT |
|
Lgals9 |
TAC CAA CAC CGC GTA CCC TA |
GCT GCA GAG TTC TGG AAG GTG |
| 18s | CGG ACA GGA TTG ACA GAT TG | CAA ATC GCT CCA CCA ACT AA |
Results
PPARγ agonist and partial agonist activities of NSAIDs
We performed ligand binding and cellular transactivation assays to evaluate the NSAIDs as PPARγ ligands. We first determined relative affinity of each compound for the PPARγ LBD in a fluorescence polarization ligand displacement assay with the labeled PPARγ ligand, Fluormone™ PPARγ Green (Figure 1A). All four NSAIDs competed with the bound PPARγ ligand. IC50 values of 1.87 μM, 3.70 μM, 21.3 μM, and 80.6 μM were obtained for sulindac sulfide, sodium diclofenac, indomethacin, and ibuprofen, respectively.
Figure 1. NSAIDs bind PPARγ and modulate PPARγ activity.

(A) Results of displacement assay using fluorescently labeled ligand. Y-axis represents fluorescence polarization units, X-axis represents concentration of the PPARγ competitor ligands rosiglitazone, sulindac, diclofenac, indomethacin, and ibuprofen. Error bars represent one standard deviation from the mean of triplicate reaction wells. (B) Results of HeLa cell transactivation assays. Transfections used a PPARγ expression vector and a PPAR response element driven luciferase promoter. Cells were treated with varying concentrations (10-9 to 10-3 M) of rosiglitazone, sodium diclofenac, indomethacin and ibuprofen. Assays were performed in three replicates and normalized for differences in efficiency by measuring internal control Renilla luciferase activity in the same lysate. Error bars represent standard deviation.
To assess NSAID activities, HeLa cells were transfected with PPARγ and a PPAR responsive luciferase reporter and exposed to ligands. Rosiglitazone activated PPARγ with EC50 <100 nM (Figure 1B). Sodium diclofenac only elicited modest PPARγ activation, reaching about 15% of the levels of activation obtained with rosiglitazone at maximal doses (Figure 1B). However, diclofenac displayed high potency, with an EC50 value just over 1 µM. By contrast, indomethacin and ibuprofen activated PPARγ efficiently, but with low potency. Both ligands reached near-maximal activation at the highest doses used, with indomethacin yielding close to 100% of the activity of rosiglitazone and ibuprofen around 50%. Assuming maximal responses at highest doses used, EC50 values were 21 µM and 56.8 µM, respectively. Sulindac sulfide also elicited PPARγ activation which approached 50-100% of that obtained with rosiglitazone in the 3-100 µM concentration range (not shown). This ligand, however, also appeared toxic, as judged by inhibitory influences on renilla luciferase control and changes in cell appearance (not shown), making it difficult to assess true levels of agonism/partial agonism.
We conclude that assessments of rank order of ligand potency (Figure 1B) agree with deduced affinities (Figure 1A). Further, overall NSAID behavior is consistent with the idea that PPARγ ligands display graded agonist/partial agonist responses, with diclofenac weakest, ibuprofen intermediate and indomethacin strongest. Sulindac sulfide may also fit into the latter category. Throughout the remainder of the paper, we refer to diclofenac as a weak partial agonist (<20% of activity of full agonists such as TZDs) and other NSAIDs as strong partial agonists (>50% of TZD activity).
NSAIDs exhibit diverse PPARγ binding modes
To understand how NSAIDs bind to PPARγ, we performed crystallization trials with bacterially expressed PPARγ LBD and sodium diclofenac, indomethacin, sulindac sulfide and ibuprofen. We were not able to obtain crystals of PPARγ with ibuprofen. However, three-dimensional co-crystal structures of the PPARγ LBD with diclofenac, indomethacin and sulindac sulfide were solved in the monoclinic C2 space group and showed good geometric and crystallographic parameters, summarized in Table 2. The asymmetric unit contains a homodimer of the PPARγ LBD, with one monomer (Chain A) in an active form and the other (Chain B) in an inactive conformation with C-terminal helix 12 (H12) displaced, probably as a consequence of crystallographic contacts reported earlier [Nolte et al., 1998]. Structures are shown in Figure 2A-C and chemical structures and binding modes of the three NSAIDs are compared in Figure 3. During the course of our study, Waku and co-workers described PPARγ structures with several indole derivatives [Waku et al., 2010], including indomethacin (PDB: 3ADS). The ligand binding mode is essentially identical to our structure described below, but we have included our own structure because ligand position comparisons were performed with our own datasets.
Table 2. Data collection and structure refinement statistics.
| Diclofenac | Indomethacin | Sulindac sulfide | |
|---|---|---|---|
| Data Collection | |||
| Synchrotron | LNLS | LNLS | ALS |
| Wavelength (λ) | 1.4586 Å | 1.4586 Å | 0.999 Å |
| Space group | C2 | C2 | C2 |
| Unit cell parameters | |||
| a (Å) | 92.40 | 93.04 | 91.77 |
| b (Å) | 62.12 | 62.14 | 63.63 |
| c (Å) | 117.18 | 118.54 | 119.65 |
| β ° | 101.02 | 102.13 | 102.38 |
| Molecules in asymetric unit | 2 | 2 | 2 |
| Resolution range (Å) | 45.04 – 2.50 (2.64 – 2.50) |
38.63 – 2.40 (2.53 – 2.40) |
51.91 – 2.22 (2.34 – 2.22) |
| Unique reflections | 21965 (3111) | 26080 (3799) | 33524 (4898) |
| Multiplicity | 3.0 (3.0) | 4.5 (4.4) | 6.9 (7.1) |
| Completeness (%) | 96.8 (94.8) | 99.8 (100) | 100 (100) |
| Rsyma | 0.06 (0.496) | 0.069 (0.502) | 0.064 (0.471) |
| <I>/<σ(I)> | 11 (2.0) | 12.1 (2.8) | 16.7 (4.2) |
|
B-factor Wilson plot (Å2) |
61.2 |
51.4 |
39.4 |
|
Refinement |
|||
| Rfactorb (%) | 21.21 | 18.82 | 17.24 |
| Rfreeb (%) | 27.40 | 23.96 | 22.26 |
| B-factors | |||
| Protein (2 molecules) | 89.32 | 53.02 | 74.55 |
| Ligands | 95.53 | 64.85 | 96.98 |
| Water | 69.65 | 51.75 | 58.31 |
| rmsd bond lengths (Å) | 0.007 | 0.007 | 0.006 |
| rmsd bond angles (°) | 0.993 | 1.076 | 1.159 |
| Ramachandran outliers | 0 | 0 | 0 |
| PDB | 4XTA | 4XUM | 4XUH |
a values in parameters refer to the last resolution shell
b Rsym: ∑hkl,j (|Ihkl - <Ihkl>|)/∑hkl,j Ihkl, where <Ihkl> is the average intensity for a set of j symmetry-related reflections, and Ihkl is the value of the intensity for a single reflection within a set of symmetry-related reflections.
cRfactor: ∑hkl (||Fo| - |Fc||)/∑hkl |Fo|, where Fo is the observed structure factor amplitude, and Fc is the calculated structure factor amplitude.
d Rfree: ∑hkl,T(||Fo| - |Fc||)/∑hkl |Fo|, where a test set, T (5% of data), is omitted from the refinement.
Figure 2. Structures of PPARγ in complex with NSAIDs.
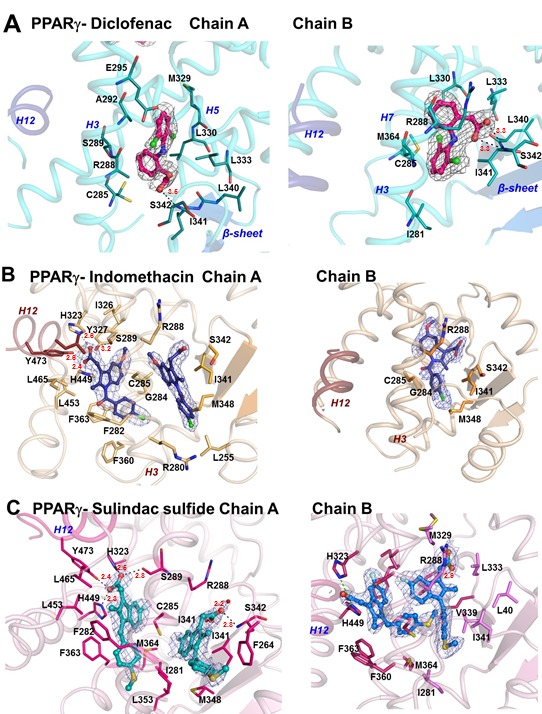
(A) PPARγ/diclofenac structure. Left panel shows overall helical organization of PPARγ Chain B (pale blue) with major structural features labeled, H12 (dark blue) in the inactive position and diclofenac in pink. The right panel represents a close up view of diclofenac (pink stick ball) with LBP amino acids. Interactions are mainly hydrophobic, and the amino acids shown as dots represent Van der Waals radii. (B) PPARγ/Indomethacin structure. The right panel shows ligand-receptor interactions in Chain A, and the left panel shows the same in Chain B. Two molecules of indomethacin (blue stick ball) are present in Chain A and on one in Chain B. (C) PPARγ/sulindac sulfide interactions represented as in Figure 2B. Two molecules of sulindac sulfide are present in Chain A (green stick ball) and B (blue stick ball). A σ-weighted 2 Fo - Fc omit electron-density map is shown contoured at 1.0 σ for the area surrounding all ligands. Hydrogen bond distances are marked in all figures by dotted lines and red figures denoting angstroms (Å).
Figure 3. PPARγ/ NSAID interactions.

(A) Chemical structures and formulae of NSAIDs diclofenac, indomethacin and sulindac sulfide. Oxygen atoms are shaded in red, including those of the carboxylate acidic group that is a common feature of ligands that bind PPARs. (B) Superposition of NSAIDs bound to PPARγ. In Chain A, the indomethacin and sulindac sulfide are bound to site 1, near H12, in a conserved binding mode, making hydrogen bonds with Y473 (H12), H449, H323 and S289, that are canonical interactions found for full agonists. In Chain B, helix 12 is an inactive conformation and sulindac sulfide bound to site 1 adopted a different bound conformation in comparison to Chain A, with interactions with Y473 and S289 not available. NSAIDs adopt different binding modes to site 2 in Chains A and B. Indomethacin and sulindac sulfide display similar binding modes at site 2 in chain A. Although indomethacin adopts the same conformation for site 2 in both chains, sulindac sulfide adopts different positions. Diclofenac is only detected at Chain B site 2 and adopts a distinct position from the other NSAIDs.
The diclofenac binding mode most resembles that of PPARγ partial agonists. The ligand binds both to Chain A and Chain B at a location between H3 and the β-sheets, a common site of PPARγ partial agonist interaction which we term site 2 [Nolte et al., 1998]. While there is significant overlap in ligand:amino acid contacts, diclofenac adopts different binding modes in each subunit (Figure 2A). In both chains, diclofenac engages in hydrophobic contacts with residues L340, L333, L330, M364, C285, I281, I341 and R288. In Chain A, however, the dichloroanilino ring binds at the upper part of the pocket, towards helix H5, making hydrophobic contacts with E295, A292, S289 (H3) and M329 (H5), while in Chain B, the dichloroanilino ring binds in the opposite orientation, along H3 and the β-sheet making hydrophobic interactions with I281 (H3) and M364 (H7) (Figure 2A). Within Chain B, the diclofenac carboxyl group also makes strong hydrogen bond contacts with the side chain of S342, located in the β-sheet (Figure 2A). Interestingly, the Chain B diclofenac exhibits better defined electron density, suggesting that this may be the preferential binding mode. The PPARγ/diclofenac structure also displayed the highest averaged B-factor value (89.32 Å) of the complexes reported here (Table 2) and disordered regions where model building proved unsuccessful. Therefore, the PPARγ-diclofenac complex appears relatively dynamic.
Indomethacin and sulindac sulfide adopt binding modes that share features with stronger PPARγ agonists. Both ligands bind to the PPARγ A chain in 2:1 stoichiometry (Figure 2B, C). One ligand binds with its head group adjacent to H12 (site 1) and engages in hydrogen bonds with Tyr473 (H12), Ser289 (H3), His323 (H6) and His449 (H11) and extensive hydrophobic contacts with nearby LBP residues (Figure 2B, 2C and Figure 3B). The second Chain A ligand is located at site 2 [Bruning et al., 2007], between helix 3 and the β-sheet. Both ligands form hydrophobic contacts with site 2 amino acids and the indomethacin carboxylic acid hydrogen bonds with the Ser342 main chain. Indomethacin and sulindac sulfide also bound to Chain B (Figure 2B, 2C and Figure 3B). One indomethacin ligand is present at site 2, where it displays a similar orientation to that seen in Chain A, between the β-sheet and H3 (Figure 2B). There are two sulindac sulfide ligand copies in Chain B. The first binds site 2, but in a different orientation from the equivalent ligand in Chain A site 2 (Figure 2C and Figure 3B). The second occupies site 1, but displays different LBP interactions from its Chain A counterpart because the displaced inactive position of H12 renders this helix unavailable for ligand contact. Here, sulindac sulfide forms hydrogen bonds with H323 and H449 onnly. Together, our data suggest that indomethacin and sulindac sulfide bind the PPARγ active conformer in a mode that allows direct interactions with the inner surface of H12 at site 1, as well as recapitulating partial agonist-like interactions at site 2.
NSAIDs promote adipogenesis in 3T3-L1 cells
To better define the effects of NSAIDs on PPARγ activity, we first determined whether they promote the PPARγ-dependent adipogenic differentiation program in 3T3-L1 cells. A uniform field of fibroblasts wasinduced to differentiate with insulin, dexamethasone and isobutylxanthine in the absence or presence of rosiglitazone, which served as a positive control, or NSAIDs and then subjected to Oil Red O staining (Figure 4A-E). This assay detects lipid staining and therefore reflects both the number of cells that have undergone adipogenic differentiation and amount of lipid in these cells. While the assay is qualitative, we noted a tendency for all NSAIDs to elicit increased lipid staining in the cells (Figure 4C-E), as in the case of rosiglitazone (Figure 4B). Quantitation of Oil Red O (ORO) staining on a per-cell basis supported this notion (Figure 4F), although increases in staining obtained with ibuprofen did not meet statistical significance in this type of analysis. These results indicate that NSAIDs can elicit PPARγ-dependent adipogenic effects in the 3T3-L1 differentiation assay.
Figure 4. NSAIDs display adipogenic activity.
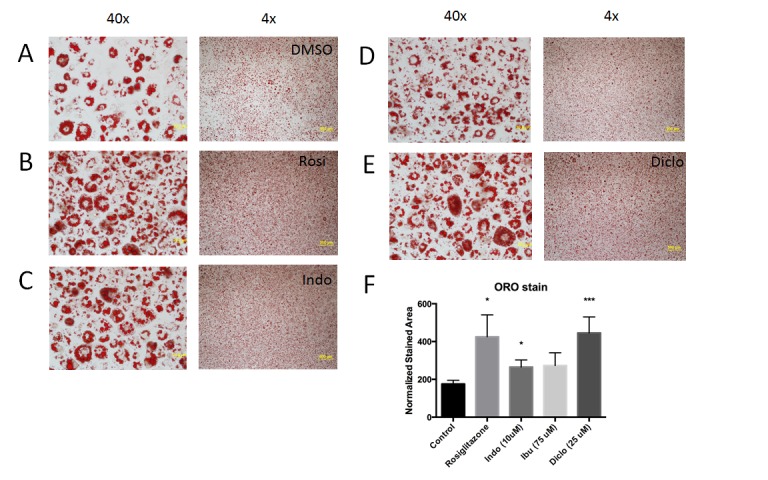
Images of 3T3-L1 cells after ligand treatment. Oil Red O staining of 3T3-L1 cells after adipocyte differentiation in the presence of: (A) DMSO vehicle control, (B) 1 μM rosiglitazone, (C) 10 μM indomethacin, (D) 75 μM ibuprofen or (E) 25 μM sodium diclofenac. Cell images are presented at 4X and 40X magnification. (F) Quantitation of Oil Red O (ORO) staining in each condition. The superscript * = P-value <0.05 and *** = P-value <0.01 in Students t-test.
NSAIDs regulate PPARγ responsive genes in differentiated 3T3-L1 cells
To investigate how NSAIDs influence PPARγ-dependent gene programs, we used an Illumina bead-based microarray system to analyze gene expression patterns in fully differentiated 3T3-L1 cells treated for 24 hrs with rosiglitazone or NSAIDs (Figure 5A). We identified 277 genes regulated ≥2-fold by rosiglitazone and 144, 133 and 18 by indomethacin, ibuprofen and diclofenac, respectively. With the narrow exception of ibuprofen, slightly less than 50% of genes showed increased expression with each treatment, with the remainder showing decreased expression.
Figure 5. NSAIDs regulate similar genes to rosiglitazone.
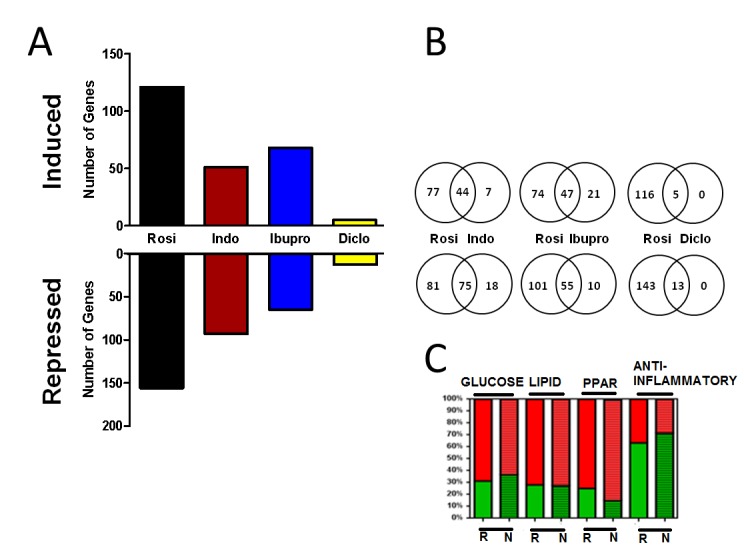
(A) Number of genes regulated ≥2 fold by rosiglitazone and NSAIDs. With the narrow exception of ibuprofen, more than 50% of genes were repressed by the treatments. (B) Shared and unique genes regulated by rosiglitazone and NSAIDs. 82.6% (119) of genes regulated by indomethacin, 76.7% (102) by ibuprofen and 100% (18) by diclofenac were also regulated by rosiglitazone more than ≥2 fold. (C) Percentage of genes with increased (red) and decreased (green) expression in response to rosiglitazone (R) or aggregated NSAIDs (N) in the specified functional categories: glucose metabolism, lipid metabolism, PPAR= PPAR signaling, anti-inflammatory pathway.
There was striking overlap between genes regulated by rosiglitazone and by NSAIDs. More than 80% of indomethacin responsive genes and 75% of ibuprofen responsive genes were also flagged as rosiglitazone target genes with similar directionality of response (up- or down-regulated) (Figure 5B). All 18 diclofenac responsive genes displayed similar regulation by rosiglitazone. Accordingly, the majority of genes that respond to NSAIDs also respond to rosiglitazone, implying that NSAIDs predominantly regulate PPARγ target genes in this system. We also assessed how the ligands affected genes in particular PPARγ responsive pathways (Figure 5C). To do this, we used GeneCodis [Carmona-Saez et al., 2007; Nogales-Cadenas et al., 2009; Tabas-Madrid et al., 2012] and the online set analysis application Venny [Oliveros, 2007] to group target genes according to function. This method revealed that 5.8% of rosiglitazone regulated genes relate to glucose metabolism, 9.0% to lipid metabolism, 2.9% to the PPAR signaling pathway and 10.8% to inflammatory processes. Importantly, similar proportions of NSAID responsive genes (grouped together) fell into each category and, further, similar proportions of genes in each category were up- or down-regulated by ligand. Cluster analysis confirmed that rosiglitazone and NSAIDs displayed similar effects on individual genes in each functional group (Figure 6), but with rosiglitazone tending to display strongest responses, indomethacin and ibuprofen intermediate responses and diclofenac the weakest responses. Based on these data, rosiglitazone and NSAIDs regulate similar sets of genes and similar pathways.
Figure 6. Effects of individual NSAIDS on gene expression.

The data is presented as a cluster analysis (MultiExperiment Viewer; www.tm4.org/mev/). Genes differentially modulated by rosiglitazone and NSAIDs within the specified functional categories are organized through hierarchical clustering. Red represents gene transcripts with increased, green decreased, and black no changes in expression level. Rosiglitazone and NSAIDs displayed similar pattern of regulation of genes involved in designated biological processes, although rosiglitazone produced a more pronounced response in comparison to NSAIDs.
We validated selected changes in target gene response by RT-qPCR (Figure 7 and not shown). Since we were unable to detect major differences in regulation of PPARγ-dependent pathways in the presence of rosiglitazone and NSAIDs (Figure 6), we focused on genes with interesting response patterns. As predicted by microarray analysis, many positively and negatively regulated genes exhibited strong responses to rosiglitazone, varying levels of intermediate response to indomethacin and ibuprofen and weakest or non-existent responses to diclofenac. Such genes included Acaa1b, which encodes an isoform of Acetyl-Coenzyme A acyltransferase 1 and Fgf21, which encodes a peptide hormone involved in adipogenic differentiation processes and systemic metabolic regulation (see Figure 7A with others not shown).
Figure 7. Validation of microarray analysis.
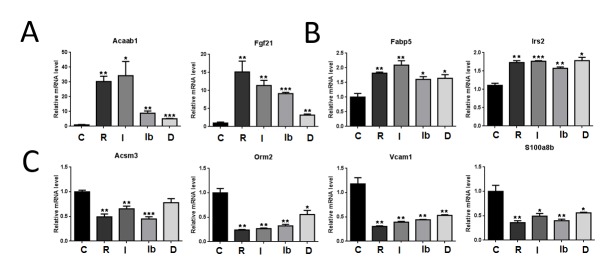
Gene transcripts that displayed a response to rosiglitazone and/or NSAIDs (≥2.0 fold and adjusted p-value ≤ 0.05) were analyzed by RT-qPCR. Normalized Cp values (Critical point; CpTarget - Cp18S) were analyzed by ANOVA followed by t-tests of DMSO (vehicle) vs. treatment (Bonferroni Corrected p-value ≤ 0.05 considered significant) (GraphPad Software, version 5.01; GraphPad). * = p ≤ 0.05, ** = p ≤ 0.01. (A) Genes that show typical NSAID response patterns. C=DMSO control, R= Rosiglitazone, I= Indomethacin, Ib=Ibuprofen, D= Diclofenac. (B) Induced genes with unusual equivalent response to rosiglitazone and NSAIDs. (C) Repressed genes with equivalent response to rosiglitazone and NSAIDs.
We were also able to verify that some genes displayed variations in NSAID efficacy. For example, Fabp2 (which encodes a protein that sequesters fatty acids) and Irs2 (an insulin-signaling pathway component) displayed approximately equivalent activation by rosiglitazone and each of the NSAIDs (Figure 7B). Further, rosiglitazone and NSAIDs displayed equivalent negative regulatory activity at Acsm3, a mitochondrial enzyme involved in fatty acid synthesis, Orm 2, an orosomucoid involved in acute phase response, Vcam1, a cell adhesion molecule involved in inflammatory response and S100a8, a calcium and zinc binding protein, also involved in inflammatory response (Figure 7C).
Remarkably, we were unable to verify any NSAID-specific effect that was not shared with rosiglitazone (not shown). Inspection of microarray datasets and RT-qPCR analysis of 3T3-L1 transcripts that were originally flagged as uniquely NSAID responsive revealed that rosiglitazone responses simply failed to meet the 2-fold cut-off or achieve appropriate statistical significance in these cases. This finding strengthens our conclusion that NSAIDs predominantly regulate PPARγ target genes in this highly PPARγ-dependent system.
Finally, we confirmed that NSAID effects were dependent upon PPARγ using siRNA knockdown and RT-qPCR [Deng et al., 2011]. Transfection of siRNA specific to PPARγ, but not a scrambled control, resulted in a reduction of PPARγ transcript levels by 50-75% (Figure 8). Examination of effects of knockdown on genes that displayed strong responses to rosiglitazone and NSAID revealed that PPARγ siRNA reduced or abolished rosiglitazone and indomethacin induction of Elovl3, part of the adipocyte thermogenic gene expression program, Fgf21, mentioned above, and Gyk, a glycerol kinase involved in lipid biosynthesis. These data indicate that indomethacin acts through PPARγ to induce these genes.
Figure 8. NSAID response requires PPARγ.
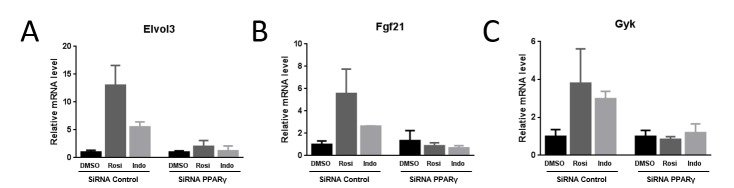
Results of RT-qPCR analysis of cells treated with vehicle, rosiglitazone or indomethacin after transfection with scrambled or PPARγ-specific siRNA. Data is presented as in Figure 7. (A) Elovl3, (B) FGF21, (C) Gyk.
Discussion
NSAIDs display anti-inflammatory, antipyretic and analgesic properties [Smith et al., 1994] and fall into many chemically distinct classes, including oxicams (piroxicam), indole derivatives (indomethacin), acetic acid derivatives (diclofenac), aminoacyl carboxylic acid (flufenamic acid), arylpropionic acid (ibuprofen and fenoprofen) and acid acetylsalicylic (aspirin) [Rao and Knaus, 2008]. While NSAIDs work by inhibiting cyclooxygenases and blocking PG production [Vane et al., 1998], some of these compounds can modulate PPARs [Jaradat et al., 2001; Lehmann et al., 1997b; Wick et al., 2002]. This finding raises the possibility that such off target NSAID effects contribute to the spectrum of actions of these drugs.
In this study, we set out to understand how PPARγ interacts with different NSAIDs and to relate NSAID binding modes to influences on PPARγ activity. An initial screen of NSAIDs (not shown) indicated that PPARγ binds indomethacin, ibuprofen, diclofenac and sulindac sulfide in vitro and we showed that these compounds bind to PPARγ with a range of affinities (sulindac sulfide> diclofenac> indomethacin> ibuprofen). Interestingly, PPARγ agonists are commonly comprised of a lipophilic backbone and an acidic moiety [Nettles, 2008; Pingali et al., 2008], usually a carboxylate, and all four NSAIDs share these characteristics [Jaradat et al., 2001; Lehmann et al., 1997a; Wick et al., 2002]. The four NSAIDs displayed differing degrees of agonism/partial agonism in transfection assays; diclofenac exhibited weak partial agonist activity ibuprofen intermediate levels and indomethacin displayed strong partial agonist activity. General toxicity prevented us from fully assessing activities of sulindac sulfide, but results are most consistent with strong partial agonist activity. These findings agree with previous analyses. Jaradat et al. [Jaradat et al., 2001] found that indomethacin activates PPARγ more effectively than ibuprofen in CV-1 and H4IIEC3 cells. Lehmann and colleagues [Lehmann et al., 1997b] found that high concentrations (100 μM) of ibuprofen activate PPARγ, whereas other NSAIDs do not. Sulindac sulfide, toxic in our assays, has also been reported to activate PPARγ [Wick et al., 2002], although other reports indicate that it does not [Sastre et al., 2006].
X-ray structures suggest possible explanations for observed NSAID activities. The diclofenac binding mode is consistent with partial agonist activity [Bruning et al., 2007]; diclofenac occupies site 2 and does not directly contact H12. Further, diclofenac appears better ordered in the inactive Chain B conformer and, in this regard, its interactions with PPARγ resemble those of the weak partial agonist luteolin, which also binds preferentially to Chain B [29]. Diclofenac adopts different binding modes in the Chain A and Chain B LBPs and analysis of crystallographic parameters is consistent with LBD instability. In light of these findings, it is also interesting to consider the possibility that diclofenac LBP position fluctuates, like MRL20 and MRL24, and whether this could be reflected in fluctuating PPARγ conformations [27, 28]. By contrast to diclofenac, indomethacin and sulindac sulfide display similar binding modes and these share features with stronger PPARγ partial agonists. We detected two copies of each ligand within the active PPARγ conformer (Chain A), one occupies the region near the inner surface of H12 (site 1), and the other occupies a “partial agonist” site 2 position. It seems likely that the combination of direct ligand contacts with the inner surface of H12 in site 1 and partial agonist-like interactions with H3, part of the H12 docking site, would lead to significant stabilization of H12 and account for strong partial agonism [Bruning et al., 2007; Nettles, 2008]. It is also noteworthy, however, that both NSAIDs contact Tyr473 on the inner surface of H12 through a single carboxylate group, similar to natural PPARγ ligands such as fatty acids, oxidized lipids and PG J2 metabolites [Kliewer et al., 1995; Liberato et al., 2012]. The fact that this contact only partly recapitulates strong TZD contacts with Tyr473 may explain why NSAIDs do not always display full TZD-like agonist activity.
It is relatively straightforward to suggest reasons that NSAIDs do not bind PPARγ as well as rosiglitazone. First, as mentioned, TZDs make tight contacts with the inner surface of H12 and nearby amino acids. Although Waku and co-workers [Waku et al., 2010] suggested that indole compounds interact with site 1 with higher affinity than site 2, it is clear that NSAIDs do not replicate the spectrum of TZD-like contacts within this region of the LBP. Second, rosiglitazone binds PPARγ in a U-shaped conformation, occupying two sub-pockets of the Y-shaped LBP [Nolte et al., 1998]. NSAIDs and other smaller ligands that bind PPARγ in multiple copies can occupy similar overall LBP space to TZDs, but do not bridge multiple PPARγ LBP sub-pockets to link different regions of the LBD. Both factors may contribute to low affinity and some aspects of attenuated transcriptional response [Pochetti et al., 2007]. It is harder to assess why NSAIDs bind PPARγ with different affinities. There are few obvious differences in sulindac sulfide and indomethacin binding modes that could explain a more than ten-fold difference in PPARγ binding affinity (Figure 1A). The relatively high affinity of diclofenac is also unexpected. We speculate that hydrogen bond contacts between the diclofenac carboxylate and the Ser342 side chain at site 2 may contribute significantly to the affinity of this ligand for PPARγ. It will be interesting to test this theory in molecular dynamics simulations.
In general, NSAID effects in the PPARγ-dependent 3T3-L1 system agreed well with predictions from crystal structures. Indomethacin, ibuprofen and diclofenac promoted adipocyte differentiation, as judged by Oil Red O staining, as previously established for classic PPARγ agonists such as Rosiglitazone, which was included here as a positive control. Further, gene expression analysis after treatment of (mature) adipocytes with NSAIDs showed extensive overlaps with rosiglitazone target genes (Figure 5 and Figure 6). However, analyses of numbers of target genes regulated (Figure 5A) and individual gene expression patterns (Figure 6 and Figure 7A) indicate that NSAIDs are weaker than rosiglitazone and that indomethacin and ibuprofen are stronger partial agonists than diclofenac.
There were some instances in which we observed gene-specific NSAID activities. For example, indomethacin, ibuprofen and diclofenac activated the Fabp5 and Irs2 genes as efficiently as rosiglitazone and NSAIDs repressed transcription of several genes as effectively as rosiglitazone. Most PPARγ ligands, including compounds that bind exclusively to site 2, stabilize the β-sheet region and block an inhibitory phosphorylation at Ser273 [Choi et al., 2010; Choi et al., 2011]. Ligands that stabilize the β-sheet without stabilizing H12 can induce genes that are particularly sensitive to inhibitory effects of Ser273 phosphorylation and it is interesting to speculate that this phenomenon could explain some instances of high gene-specific NSAID efficacy.
Importantly, our studies suggest that NSAIDs only regulate PPARγ responsive genes in this system. This raises the possibility that NSAIDs could trigger PPARγ responsive gene programs in adipocytes and, possibly, in other cell types. We certainly think that it is possible that NSAIDs could activate PPARs at pharmacologically-relevant doses. Indomethacin, a racemic (RS-mixture) of ibuprofen, sodium diclofenac and the prodrug sulindac are all used clinically [Bushra and Aslam, 2010; Davies and Anderson, 1997; Smyth et al., 2004; Thun et al., 2002]. COX inhibitors relieve pain and inflammation caused by PG synthesis [Smith et al., 1994]. However, high doses are usually required for palliation of other conditions, including rheumatoid arthritis. Here, required therapeutic plasma concentrations are up to 10 μM indomethacin, 20 μM sulindac or 300 μM ibuprofen [McEvoy, 1997] and exceed those required to inhibit COX-1 and 2 [Meade et al., 1993]. Perhaps NSAIDs activate PPARγ (or PPARα) to achieve some of these effects. Fish oil supplements, rich in unsaturated fatty acids, may also decrease the inflammatory response and reduce symptoms of rheumatoid arthritis, and docosahexanoic acid in this oil is a natural PPARγ agonist [Ye, 2011]. Another factor could favor NSAID actions through PPARγ; rosiglitazone and other NSAIDs are highly albumin bound in serum [63]. Very high doses of ibuprofen are used to treat rheumatoid arthritis and these concentrations are sufficient to escape from the inhibitory influence of serum albumin. Use of these drugs at high concentrations could result in significant levels of free drug, available for both on- and off-target interactions. In this regard, it is important to note that ligand treatments in transfection experiments described in Figure 1B were performed in 10% serum; accordingly, observed NSAID dose responses were not obtained in completely BSA-free conditions. Perhaps high concentrations of NSAID could permit escape of free drug and increased activities through PPARs.
Our findings, which indicate that NSAIDs display diverse binding modes and effects on PPAR activity, coupled with other studies which show that some NSAIDs activate PPARs while others with strong anti-inflammatory activities do not [Jaradat et al., 2001; Lehmann et al., 1997a], suggest that questions of whether particular NSAIDs modulate PPARs must be approached on a case-by-case basis. NSAIDs are attractive compounds for design of new drugs because their pharmacokinetics are extensively studied in humans, and it may be possible to engineer altered specificity for COX and PPARs into existing drugs. For example, removal of the sulindac sulfide indenyl methyl group abolishes COX inhibitory activities while retaining activity at other targets, including PPARγ [Felts et al., 2007]. It will be interesting to determine whether high doses of particular NSAIDs do result in significant PPAR activation, whether this interaction is associated with beneficial anti-inflammatory effects or harmful side effects, and whether it will be desirable to create novel analogs with PPAR-dependent activities.
Acknowledgements
This study was supported by NIH DK41482 to PW.
Abbreviations:
- NSAIDs
non-steroidal anti-inflammatory drugs
- PPARs
peroxisome proliferator activated receptors
- PPARγ
peroxisome proliferator activated receptor γ
- LBD
ligand binding domain
- LBP
ligand binding pocket
- COX
cyclooxygenase
- AA
arachidonic acid
- PGs
prostaglandins
- H
helix
- TZDs
thiazolidinediones
- DMSO
dimethyl sulfoxide
- PPRE
PPAR response element
Footnotes
The authors declare no competing interests.
References
- Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213-221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). CONTROLLING THE FALSE DISCOVERY RATE - A PRACTICAL AND POWERFUL APPROACH TO MULTIPLE TESTING. Journal of the Royal Statistical Society Series B-Methodological 57, 289-300. [Google Scholar]
- Berger J., Moller D. (2002). The mechanisms of action of PPARs. Annu Rev Med 53, 409-435. 10.1146/annurev.med.53.082901.104018 [DOI] [PubMed] [Google Scholar]
- Braissant O., Foufelle F., Scotto C., Dauça M., Wahli W. (1996). Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137, 354-366. [DOI] [PubMed] [Google Scholar]
- Brune K., Beck W. S., Geisslinger G., Menzel-Soglowek S., Peskar B. M., Peskar B. A. (1991). Aspirin-like drugs may block pain independently of prostaglandin synthesis inhibition. Experientia 47, 257-261. 10.1007/BF01958153 [DOI] [PubMed] [Google Scholar]
- Bruning J. B., Chalmers M. J., Prasad S., Busby S. A., Kamenecka T. M., He Y., Nettles K. W., Griffin P. R. (2007). Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 15, 1258-1271. [DOI] [PubMed] [Google Scholar]
- Bushra R., Aslam N. (2010). An overview of clinical pharmacology of Ibuprofen. Oman Med J 25, 155-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Saez P., Chagoyen M., Tirado F., Carazo J. M., Pascual-Montano A. (2007). GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol 8, R3. 10.1186/gb-2007-8-1-r3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H., Banks A. S., Estall J. L., Kajimura S., Boström P., Laznik D., Ruas J. L., Chalmers M. J., Kamenecka T. M., Blüher M., et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451-456. 10.1038/nature09291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H., Banks A. S., Kamenecka T. M., Busby S. A., Chalmers M. J., Kumar N., Kuruvilla D. S., Shin Y., He Y., Bruning J. B., et al. (2011). Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477-81. 10.1038/nature10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project , N. m. (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760-763. 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- Davies N. M., Anderson K. E. (1997). Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin Pharmacokinet 33, 184-213. 10.2165/00003088-199733030-00003 [DOI] [PubMed] [Google Scholar]
- Deng T., Sieglaff D. H., Zhang A., Lyon C. J., Ayers S. D., Cvoro A., Gupte A. A., Xia X., Baxter J. D., Webb P., Hsueh W. A. (2011). A peroxisome proliferator-activated receptor gamma (PPARgamma)/PPARgamma coactivator 1beta autoregulatory loop in adipocyte mitochondrial function. J Biol Chem 286, 30723-30731. 10.1074/jbc.M111.251926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P., Kibbe W. A., Lin S. M. (2008). lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547-1548. 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126-2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- Felts A. S., Ji C., Stafford J. B., Crews B. C., Kingsley P. J., Rouzer C. A., Washington M. K., Subbaramaiah K., Siegel B. S., Young S. M., et al. (2007). Desmethyl derivatives of indomethacin and sulindac as probes for cyclooxygenase-dependent biology. ACS Chem Biol 2, 479-483. 10.1021/cb700077z [DOI] [PubMed] [Google Scholar]
- Forman B., Chen J., Evans R. (1996). The peroxisome proliferator-activated receptors: ligands and activators. Ann N Y Acad Sci 804, 266-275. 10.1111/j.1749-6632.1996.tb18621.x [DOI] [PubMed] [Google Scholar]
- Gilroy D. W., Colville-Nash P. R. (2000a). New insights into the role of COX 2 in inflammation. Journal of Molecular Medicine (Berlin, Germany) 78, 121-129. 10.1007/s001090000094 [DOI] [PubMed] [Google Scholar]
- Gilroy D. W., Colville-Nash P. R. (2000b). New insights into the role of COX 2 in inflammation. J Mol Med (Berl) 78, 121-129. 10.1007/s001090000094 [DOI] [PubMed] [Google Scholar]
- Guimarães B. G., Sanfelici L., Neuenschwander R. T., Rodrigues F., Grizolli W. C., Raulik M. A., Piton J. R., Meyer B. C., Nascimento A. S., Polikarpov I. (2009). The MX2 macromolecular crystallography beamline: a wiggler X-ray source at the LNLS. J Synchrotron Radiat 16, 69-75. 10.1107/S0909049508034870 [DOI] [PubMed] [Google Scholar]
- Jaradat M. S., Wongsud B., Phornchirasilp S., Rangwala S. M., Shams G., Sutton M., Romstedt K. J., Noonan D. J., Feller D. R. (2001). Activation of peroxisome proliferator-activated receptor isoforms and inhibition of prostaglandin H(2) synthases by ibuprofen, naproxen, and indomethacin. Biochem Pharmacol 62, 1587-1595. 10.1016/S0006-2952(01)00822-X [DOI] [PubMed] [Google Scholar]
- Jiang C., Ting A. T., Seed B. (1998). PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391, 82-86. 10.1038/35154 [DOI] [PubMed] [Google Scholar]
- Klemm D. J., Leitner J. W., Watson P., Nesterova A., Reusch J. E., Goalstone M. L., Draznin B. (2001). Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J Biol Chem 276, 28430-28435. 10.1074/jbc.M103382200 [DOI] [PubMed] [Google Scholar]
- Kliewer S., Forman B., Blumberg B., Ong E., Borgmeyer U., Mangelsdorf D., Umesono K., Evans R. (1994). Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A 91, 7355-7359. 10.1073/pnas.91.15.7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S. A., Lenhard J. M., Willson T. M., Patel I., Morris D. C., Lehmann J. M. (1995). A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83, 813-819. 10.1016/0092-8674(95)90194-9 [DOI] [PubMed] [Google Scholar]
- Lehmann J., Lenhard J., Oliver B., Ringold G., Kliewer S. (1997a). Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272, 3406-3410. 10.1074/jbc.272.6.3406 [DOI] [PubMed] [Google Scholar]
- Lehmann J., Lenhard J., Oliver B., Ringold G., Kliewer S. (1997b). Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. Journal of Biological Chemistry 272, 3406-3410. 10.1074/jbc.272.6.3406 [DOI] [PubMed] [Google Scholar]
- Lehrke M., Lazar M. (2005). The many faces of PPARgamma. Cell 123, 993-999. 10.1016/j.cell.2005.11.026 [DOI] [PubMed] [Google Scholar]
- Leslie A. G. (1999). Integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 55, 1696-1702. 10.1107/S090744499900846X [DOI] [PubMed] [Google Scholar]
- Li M. D., Yang X. (2011). A Retrospective on Nuclear Receptor Regulation of Inflammation: Lessons from GR and PPARs. PPAR Res 2011, 742785. 10.1155/2011/742785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberato M. V., Nascimento A. S., Ayers S. D., Lin J. Z., Cvoro A., Silveira R. L., Martínez L., Souza P. C., Saidemberg D., Deng T., et al. (2012). Medium chain fatty acids are selective peroxisome proliferator activated receptor (PPAR) γ activators and pan-PPAR partial agonists. PLoS One 7, e36297. 10.1371/journal.pone.0036297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy, G. K. (1997). AHFS 97 Drug Information (Bethesda, MD, American Society of Health - Systems Pharmacists). [Google Scholar]
- Meade E. A., Smith W. L., DeWitt D. L. (1993). Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem 268, 6610-6614. [PubMed] [Google Scholar]
- Mukherjee A., Hale V. G., Borga O., Stein R. (1996). Predictability of the clinical potency of NSAIDs from the preclinical pharmacodynamics in rats. Inflamm Res 45, 531-540. 10.1007/BF02342223 [DOI] [PubMed] [Google Scholar]
- Nettles K. (2008). Insights into PPARgamma from structures with endogenous and covalently bound ligands. Nat Struct Mol Biol 15, 893-895. 10.1038/nsmb0908-893 [DOI] [PubMed] [Google Scholar]
- Nogales-Cadenas R., Carmona-Saez P., Vazquez M., Vicente C., Yang X., Tirado F., Carazo J. M., Pascual-Montano A. (2009). GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res 37, W317-322. 10.1093/nar/gkp416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte R., Wisely G., Westin S., Cobb J., Lambert M., Kurokawa R., Rosenfeld M., Willson T., Glass C., Milburn M. (1998). Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395, 137-143. 10.1038/25931 [DOI] [PubMed] [Google Scholar]
- Oliveros, J. C. (2007). VENNY. An interactive tool for comparing lists with venn diagrams (http://bioinfogp.cnb.csic.es/tools/venny/index.html.).
- Pingali H., Jain M., Shah S., Patil P., Makadia P., Zaware P., Sairam K. V., Jamili J., Goel A., Patel M., Patel P. (2008). Modulation of PPAR receptor subtype selectivity of the ligands: aliphatic chain vs aromatic ring as a spacer between pharmacophore and the lipophilic moiety. Bioorg Med Chem Lett 18, 6471-6475. 10.1016/j.bmcl.2008.10.062 [DOI] [PubMed] [Google Scholar]
- Pochetti G., Godio C., Mitro N., Caruso D., Galmozzi A., Scurati S., Loiodice F., Fracchiolla G., Tortorella P., Laghezza A., et al. (2007). Insights into the mechanism of partial agonism: crystal structures of the peroxisome proliferator-activated receptor gamma ligand-binding domain in the complex with two enantiomeric ligands. Journal of Biological Chemistry 282, 17314-17324. 10.1074/jbc.M702316200 [DOI] [PubMed] [Google Scholar]
- Pountos I., Giannoudis P. V., Jones E., English A., Churchman S., Field S., Ponchel F., Bird H., Emery P., McGonagle D. (2011). NSAIDS inhibit in vitro MSC chondrogenesis but not osteogenesis: implications for mechanism of bone formation inhibition in man. J Cell Mol Med 15, 525-534. 10.1111/j.1582-4934.2010.01006.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhl A. C., Bernardes A., Silveira R. L., Yuan J., Campos J. L., Saidemberg D. M., Palma M. S., Cvoro A., Ayers S. D., Webb P., et al. (2012). Mode of Peroxisome Proliferator-Activated Receptor γ Activation by Luteolin. Mol Pharmacol 81, 788-799. 10.1124/mol.111.076216 [DOI] [PubMed] [Google Scholar]
- Rao P., Knaus E. E. (2008). Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci 11, 81s-110s. [DOI] [PubMed] [Google Scholar]
- Ricote M., Huang J., Fajas L., Li A., Welch J., Najib J., Witztum J. L., Auwerx J., Palinski W., Glass C. K. (1998a). Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A 95, 7614-7619. 10.1073/pnas.95.13.7614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M., Li A. C., Willson T. M., Kelly C. J., Glass C. K. (1998b). The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79-82. 10.1038/34178 [DOI] [PubMed] [Google Scholar]
- Ricote M., Welch J. S., Glass C. K. (2000). Regulation of macrophage gene expression by the peroxisome proliferator-activated receptor-gamma. Horm Res 54, 275-280. 10.1159/000053271 [DOI] [PubMed] [Google Scholar]
- Sastre M., Dewachter I., Rossner S., Bogdanovic N., Rosen E., Borghgraef P., Evert B. O., Dumitrescu-Ozimek L., Thal D. R., Landreth G., et al. (2006). Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A 103, 443-448. 10.1073/pnas.0503839103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonjans K., Martin G., Staels B., Auwerx J. (1997). Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr Opin Lipidol 8, 159-166. 10.1097/00041433-199706000-00006 [DOI] [PubMed] [Google Scholar]
- Smith W. L., Meade E. A., DeWitt D. L. (1994). Interactions of PGH synthase isozymes-1 and -2 with NSAIDs. Annals of the New York Academy of Sciences 744, 50-57. 10.1111/j.1749-6632.1994.tb52723.x [DOI] [PubMed] [Google Scholar]
- Smyth, G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3. [DOI] [PubMed]
- Smyth J. M., Collier P. S., Darwish M., Millership J. S., Halliday H. L., Petersen S., McElnay J. C. (2004). Intravenous indometacin in preterm infants with symptomatic patent ductus arteriosus. A population pharmacokinetic study. Br J Clin Pharmacol 58, 249-258. 10.1111/j.1365-2125.2004.02139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B., Koenig W., Habib A., Merval R., Lebret M., Torra I. P., Delerive P., Fadel A., Chinetti G., Fruchart J. C., et al. (1998). Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 393, 790-793. 10.1038/31701 [DOI] [PubMed] [Google Scholar]
- Tabas-Madrid D., Nogales-Cadenas R., Pascual-Montano A. (2012). GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res 40, W478-483. 10.1093/nar/gks402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R. D. C. (2011). R: A language and environment for statistical computing (Vienna, Austria, R Foundation for Statistical Computing). [Google Scholar]
- Thun M. J., Henley S. J., Patrono C. (2002). Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst 94, 252-266. 10.1093/jnci/94.4.252 [DOI] [PubMed] [Google Scholar]
- Tontonoz P., Hu E., Graves R. A., Budavari A. I., Spiegelman B. M. (1994a). mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8, 1224-1234. 10.1101/gad.8.10.1224 [DOI] [PubMed] [Google Scholar]
- Tontonoz P., Hu E., Spiegelman B. (1994b). Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79, 1147-1156. 10.1016/0092-8674(94)90006-X [DOI] [PubMed] [Google Scholar]
- Vane J. R., Bakhle Y. S., Botting R. M. (1998). Cyclooxygenases 1 and 2. Annual Review of Pharmacology and Toxicology 38, 97-120. 10.1146/annurev.pharmtox.38.1.97 [DOI] [PubMed] [Google Scholar]
- Waku T., Shiraki T., Oyama T., Maebara K., Nakamori R., Morikawa K. (2010). The nuclear receptor PPARγ individually responds to serotonin- and fatty acid-metabolites. EMBO J 29, 3395-3407. 10.1038/emboj.2010.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick M., Hurteau G., Dessev C., Chan D., Geraci M. W., Winn R. A., Heasley L. E., Nemenoff R. A. (2002). Peroxisome proliferator-activated receptor-gamma is a target of nonsteroidal anti-inflammatory drugs mediating cyclooxygenase-independent inhibition of lung cancer cell growth. Mol Pharmacol 62, 1207-1214. 10.1124/mol.62.5.1207 [DOI] [PubMed] [Google Scholar]
- Ye J. (2011). Challenges in Drug Discovery for Thiazolidinedione Substitute. Acta Pharmaceutica Sinica B 1, 137-142. 10.1016/j.apsb.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]