

Abstract
Although phenotypic intratumoral heterogeneity was first described many decades ago, the advent of next-generation sequencing has provided conclusive evidence that in addition to phenotypic diversity, significant genotypic diversity exists within tumors. Tumor heterogeneity likely arises both from clonal expansions, as well as from differentiation hierarchies existent in the tumor, such as that established by cancer stem cells (CSCs) and non-CSCs. These differentiation hierarchies may arise due to genetic mutations, epigenetic alterations, or microenvironmental influences. An additional differentiation hierarchy within epithelial tumors may arise when only a few tumor cells trans-differentiate into mesenchymal-like cells, a process known as epithelial-to-mesenchymal transition (EMT). Again, this process can be influenced by both genetic and non-genetic factors. In this review we discuss the evidence for clonal interaction and cooperation for tumor maintenance and progression, particularly with respect to EMT, and further address the far-reaching effects that tumor heterogeneity may have on cancer therapy.
Keywords: cancer stem cells/CSCs, clonal evolution, epithelial-mesenchymal transition (EMT), intratumoral heterogeneity, metastasis, non-cell autonomous, tumor microenvironment
Abbreviations
- NGS
next generation sequencing
- CLL
chronic lymphoblastic leukemia
- CSC
cancer stem cell
- EMT
epithelial-to-mesenchymal transition
- hPDGF
human platelet-derived growth factor
- MMTV
mouse mammary tumor virus
- OxR
oxaliplatin resistant
- TGF-β
transforming growth factor-β
- SCLC
small cell lung cancer
- MET
mesenchymal-to-epithelial transition
- EMP
epithelial-mesenchymal plasticity
- miRNA
microRNA
- GFP
green fluorescent protein
Introduction
Increasing evidence suggests that intratumoral heterogeneity is an important aspect of the tumor landscape. Until recently, lineal evolution was widely accepted, a theory that posits that successive mutations in a single cell could result in a homogenous tumor with one dominant clone.1-3 Evidence supporting this theory arose from genetic and molecular profiling of small areas of tumors at static time-points, which was not representative of the genetic complexity of the entire tumor, and thus only a dominant clone was often detected.4,5 However, new technologies involving applications of next generation sequencing have allowed a much more refined look at the landscape of human tumors,6-8 revealing their heterogeneous nature, as multiple clones with varied genetic mutations have been found within a tumor. While intratumoral heterogeneity was first described many years ago,9 these recent data lend credence to earlier observations, and have underscored the importance of understanding both genotypic and phenotypic intratumor diversity.
Causes of intratumoral heterogeneity
Intratumoral heterogeneity can be divided into 2 main categories: genetic and non-genetic. As these types of intratumoral heterogeneity have been reviewed in detail previously,10-12 they are only briefly described below.
Genetic heterogeneity
High resolution sequencing has revealed important driver and passenger mutations within cells in the primary tumor6,8 and has also led to the finding that different cells within the same primary tumor can harbor different genetic mutations. These findings can be reconciled with the theory of clonal evolution,1,2 in which a tumor of monoclonal origin (arising from a single clone that accumulates advantageous “tumorigenic” mutations over time) may become heterogeneous due to the inherent genetic instability of the cells in the tumor (a hallmark of cancer cells13). Such instability results in divergent cell populations within the primary tumor that can compete or cooperate as the tumor progresses. It is also possible that 2 or more non-transformed cells within a tissue may accumulate independent mutations and evolve to form a malignant tumor only when they cooperate with one another. In this case, the tumor would be polyclonal in origin and the cells comprising the tumor would consist of different mutations depending both on the clone from which they arose, and on the changes in their genome over time.3 Regardless of whether tumors arise in a mono- or polyclonal manner, they can evolve to be relatively homogenous, with one dominant clone, or heterogeneous, containing multiple clones. In the latter case, sub-clones may eventually out-compete one another, depending on whether they contain more “advantageous” mutations, or can evolve in parallel, a mechanism known as “branched” evolution. Indeed, ultra-deep sequencing and multiregional genetic analysis of human tumors has confirmed that branched evolution occurs in various tumor types including chronic lymphoblastic leukemia (CLL),14,15 melanoma,16 renal cell carcinoma,8 and breast cancer.17 Branched evolution may also explain how different clones arising from the same primary tumor display wide mutation spectrums and phenotypic diversity, differing not only in cellular morphology,18 gene expression,19 and response to therapies20,21 but also in metastatic abilities.22-25 It is important to note, however, that not all genetic mutations within a tumor cell or within populations of cells have functional or advantageous phenotypic consequences. The concepts of clonal and branched evolution have been thoroughly reviewed elsewhere.15,26-28
Non-genetic heterogeneity
Not only are tumor cells inherently unstable, they are also constantly under selective pressure from the tumor microenvironment due to their interaction with the extracellular matrix and with cells in the microenvironment such as immune cells, endothelial cells, and fibroblasts. The cells in the primary tumor also face alterations in oxygen and nutrient availability and thus experience distinct selective pressures based on their location within the primary tumor (whether centrally located in the tumor, or located closer to blood vessels).29,30 This combination of internal and external pressures allows for outgrowth of subpopulations of cells that may have genetic, epigenetic, and/or phenotypic differences,21,30-32 leading to intratumoral heterogeneity.
An important non-genetic source of heterogeneity within the tumor is thought to arise from differentiation hierarchies that exist within tumor cell populations.12 For example, tumor heterogeneity may result from cells within the primary tumor existing in different states of “stem-ness," a hierarchy that can be regulated and maintained in part by the microenvironment.33-35 In this scenario, cancer stem cells (CSCs), which have the ability to self-renew as they asymmetrically divide, can propagate the tumor, whereas non-CSCs within the same tumor would not have this ability. These 2 different populations of cells (CSC and non-CSC) would then further give rise to diverse sets of cells as they accumulate mutations or epigenetic changes, and undergo selection separately, leading to increased heterogeneity within the tumor. In the CSC theory, a small population of cells within the primary tumor drives tumor progression and metastasis,36 and thus the most effective therapies should target this population as well as the bulk of the tumor. Importantly, studies have shown that CSCs within a tumor are resistant to many commonly used therapies,10,37,38 and thus when left behind, contribute to metastasis.39-41 Recently, the potential of a bidirectional relationship between CSCs and non-CSCs has been recognized in breast cancer and B cell-lymphoma among others, where cells can shift between CSC and non-CSC states throughout tumor evolution.42-47 This shift between stem cell-like and non-stem cell-like states can be spontaneous, and has been shown to be under the influence of oncogenes and the tumor microenvironment.10,42-47
The 2 concepts of clonal evolution and CSCs attempt to explain intratumoral heterogeneity and may be mutually exclusive when defined in the strictest senses. Though there are differences between these theories, specifically in terms of the existence of a differentiation hierarchy and the tumorigenic potential of only a few cells within the tumor (among others48), they do overlap. Clonal evolution and both genetic and non-genetic alterations can give rise to CSCs that have the ability to both self-renew and differentiate to further generate distinct clones within the tumor. Indeed, studies have shown that both CSCs and non-CSCs have different patterns of mutations, indicating that they have evolved from distinct clones in the primary tumor.49,50 (Fig. 1)
Figure 1.
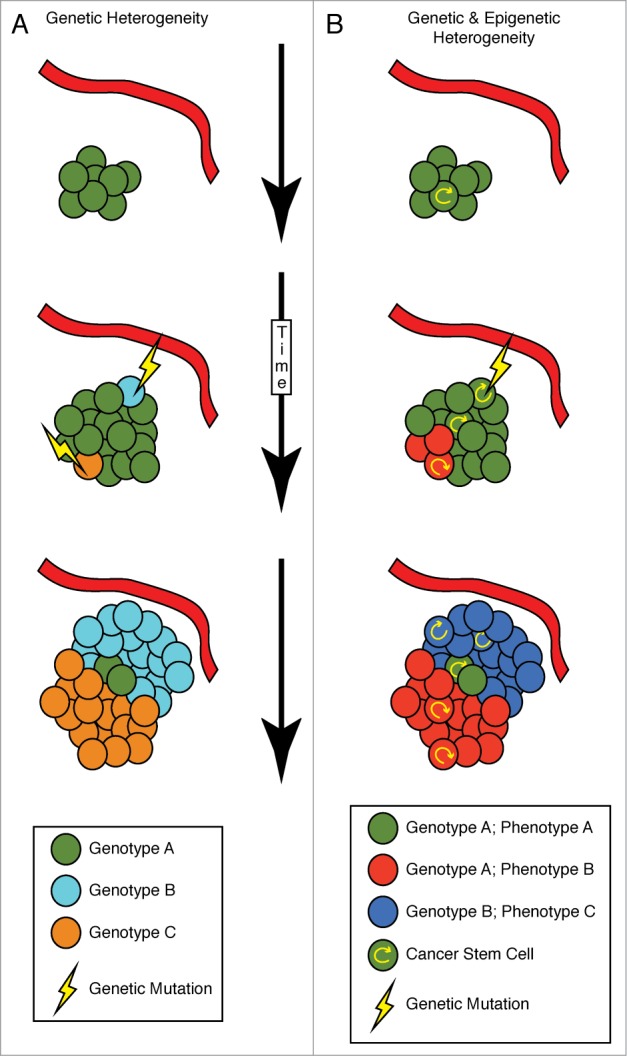
Heterogeneity can arise from both genetic and non-genetic mechanisms. Intratumoral heterogeneity can arise through a variety of mechanisms. (A) A strictly genetic mechanism can involve genetic mutations (lightening bolt), some of which may give a clone a fitness advantage over nearby tumor cells (light blue and orange genotype is more fit than green genotype). This model can involve genetic fitness being selected for by the local microenvironment. An increased oxygen and nutrient supply may select for one type of clone (light blue), while the opposite could be true for another clone (orange). This pattern of mutation can repeat over time, yielding a tumor with numerous clones. Such mutations can be stochastic in nature, can be influenced by genomic instability and can be influenced by chemo- and radiation therapy, in addition to other sources. (B) An alternative model involves cancer stem cells (CSCs), which arise due to differentiation hierarchies that may not involve genetic alterations, but may act in combination with mutations. A self-renewing tumor cell, the CSC, can undergo asymmetric division, increasing the overall number of CSC and non-CSC tumor cells. Genetic mutation of a CSC can lead to the outgrowth of a more fit genetic clone (dark blue), that may or may not be affected by the local microenvironment. Additionally, the microenvironment can trigger phenotypic changes, such as what occurs in a hypoxic environment (red cells). Importantly, if the original clone is outcompeted over time, it would not be detectable in patient biopsies.
Much discussion has been devoted to the role of CSCs in tumors as critical contributing factors to intratumoral heterogeneity.10-12,27 However, additional altered differentiation states may contribute to this heterogeneity as well. For example, in epithelial cancers, the trans-differentiation of epithelial tumor cells to more mesenchymal-like cells through an epithelial-to-mesenchymal transition (EMT) may be another critical component of the heterogeneous tumor landscape. This phenomenon can at times be linked to CSCs, as EMT has been shown to increase CSC-like characteristics.39,43,51 However, CSC and EMT do not always go hand-in-hand.52 Nonetheless, an EMT is highly associated with a gain in metastatic abilities, even when only a small proportion of the tumor has undergone this transition.53-56 Thus, EMT may be considered as a largely (although not exclusively) non-genetic contributor to intratumoral heterogeneity. The importance of EMT in the context of a heterogeneous tumor will be discussed in detail within this review.
Can clonal cooperation affect tumor initiation and progression?
A by and large unanswered question in the field with respect to intratumoral heterogeneity is whether different subpopulations of clones within the primary tumor interact to affect tumor maintenance and/or progression, or whether cells are actually recruited to the tumor and induced to be tumorigenic. Indeed, recent studies have shown that tumor maintenance may require interaction between clones in a primary tumor, as well as between tumor-initiating cells and progenitor cells in the environment.
Holland and colleagues examined this issue in a study using human platelet-derived growth factor (hPDGF)-induced murine gliomas that are thought to arise from one cell of origin (glial cells) but are heterogeneous in nature.57 Using 2 different mouse models of hPDGF-driven gliomas, along with lineage tracing studies, the authors were able to distinguish which cells in the tumor were derived from the cell of origin, versus which cells were not, and were then able to perform tumorigenic and expression analyses on the 2 different populations. The heterogeneity in these gliomas arises from the ability of the tumor cells to recruit normal neural stem cells as well as progenitor cells (that don't overexpress hPDGF), but proliferate to contribute to the tumor mass. Interestingly, when the gliomas were induced by hPDGF in mice with germline deletions of Ink4a, Arf, and/or PTEN, the recruited population (not derived from the cell of origin) was expanded such that it could dominate regions of the tumor. Importantly, transplantation studies revealed that these recruited cells could give rise to gliomas on their own, exhibiting expression profiles and genetic abnormalities that were seen in the glioma cells, but not in the normal progenitor cells. Thus, the authors demonstrated that non-cell-of-origin cells in glioma can actually be transformed non-cell autonomously by the glioma environment, providing a new paradigm for gliomagenesis.
In another such study by Gunther and colleagues, cooperation between distinct clones in a Wnt1-driven murine mammary tumor was shown to be essential for tumor maintenance.58 In this study, the mouse mammary tumor virus (MMTV)-Wnt1 murine mammary tumor model was employed, where aberrant expression of Wnt1 in the mammary epithelial cells was shown to result in bi-clonal, heterogeneous tumors consisting of basal and luminal epithelial cells. A requirement for Wnt1 in tumor maintenance was shown using an inducible Wnt1 system, since deprivation of Wnt1 led to tumor regression. Interestingly, some cells within this heterogeneous tumor cell population were found to spontaneously acquire Hras mutations. On further analysis of the Hras mutant tumors, the authors found that half of the tumors consisted of basal and luminal cells with identical Hras mutations. On the other hand, the remaining half of the tumors consisted of basal cells that harbored mutant Hras and expressed low Wnt1 levels and luminal cells that contained wild-type Hras and high Wnt1 levels. They also found that the luminal cells within the heterogeneous tumors were the main source of Wnt1 that helped in the maintenance of the tumor mass. When the tumors were deprived of the Wnt1 ligand to imitate targeted therapy, the basal cells recruited other luminal cells to provide the required Wnt1, which led to tumor recurrence. Hence, within the heterogeneous Wnt1-driven mammary tumor, the low Wnt1-expressing, mutant HRas basal cells required Wnt1 from the high-Wnt1 expressing luminal cells to maintain tumor mass, indicating that interclonal cooperation is necessary in this context for tumor maintenance.
Additional studies have provided evidence for clonal cooperativity not only in tumor maintenance, but also in tumor progression. Using a colorectal cancer model, Ellis and colleagues demonstrated that both CSC-like cells and chemoresistant cells within the primary tumor have the ability to confer chemoresistance on surrounding “chemo-naïve” cells.59 Specifically, colorectal cancer cells were made chemoresistant through chronic exposure to Oxaliplatin (OxR cells), a common chemotherapeutic agent used in the treatment of colorectal cancer. Not only did the OxR population of cells have an increased percentage of CSCs compared to the chemo-naïve parental cells, but the conditioned media from OxR cells, when placed on chemo-naïve cells, led to their increased survival both in the presence or absence of Oxaliplatin. In addition, subcutaneous injections of different ratios of OxR and parental chemo-naïve cells into mice resulted in the largest tumors when the injections contained equal numbers of both cell types (in a 1:1 ratio), as compared to injection of either pure population of cells, even though the total number of cells injected into mice in each case was the same. Since the investigators observed that the OxR cells grew at a slower rate compared to the parental cells, the larger mixed in vivo tumors suggest that the cell lines were non-cell autonomously interacting to aid tumor growth. Intriguingly, the effect of the OxR cells was shown to occur over significant distances, as injection of these cells into one flank of a mouse promoted the growth of chemo-naïve cells that were injected into the other flank of the same mouse. Thus, these studies again demonstrate that interclonal cooperation is necessary for tumor maintenance and progression.
These aforementioned studies demonstrate that once a tumor has formed, it can be composed of phenotypically and/or genotypically distinct clones that interact to the benefit of one or more clones within the tumor. Thus, while competition between clones may result in dominant clones with maximum fitness taking over the tumor,60 clonal cooperation can also occur, in which co-existence of multiple different clones can impact tumor progression positively and lead to more aggressive disease. In recent years, interclonal cooperativity has clearly been demonstrated to impinge on metastatic dissemination.
Metastasis and intratumoral heterogeneity
Approximately 90% of cancer related deaths occur due to metastatic dissemination.56,61 There is thus an urgent need to develop better therapies to combat metastatic disease and to improve outcome, and indeed much basic research focuses on gaining a more complete understanding of the molecular mechanisms behind the metastatic cascade. During the process of metastasis, tumor cells need to gain characteristics that enable them to move out of the primary tumor, into and out of the vasculature, and into a secondary site where, perhaps after a period of dormancy, they must regain the capacity to proliferate to colonize at that site.44 Thus, a tumor cell will form clinically detectable metastases only if it is able to successfully navigate the many steps of the metastatic process. Metastasis is thus considered to be an inherently inefficient process,62 with only a small percentage of the cells within the primary tumor having the potential to colonize a secondary site. It would thus seem advantageous to bestow dissemination potential upon as many cells within the primary tumor as possible. This could be achieved through interclonal cooperation and non-cell autonomous mechanisms and would increase the likelihood of metastasis and outgrowth at the secondary site. Thus, while one individual cell may not be able to carry out each of the steps of metastasis, a neighboring cell may allow it to perform a step it normally could not on its own.
There is still some controversy over whether tumor cells innately contain the capability to metastasize, or whether metastatic ability is a phenotype that cells gain from a set of genetic and epigenetic changes that occur throughout tumor progression.25,63-68 It is likely that both these mechanisms could be at play. This question is important to address since current therapies mostly rely on the theory that metastases are similar to the primary tumor from which they arise.69-71 Indeed, studies in breast cancer have shown that primary tumor cells left behind post neo-adjuvant treatment have molecular profiles different from matched biopsy samples taken prior to the treatment. Genetic profiling of these residual tumor cells revealed molecular alterations that correlate with poor patient prognosis and might thus be similar to micro-metastases formed in these aggressive cancers.72 On the other hand, there is ample evidence that both disseminated tumor cells, as well as metastases, are made up of cells with a mutational spectrum that is different from that in the cells of the primary tumor.73
A multitude of studies have also shown that both tumor initiation and metastatic progression are influenced and often facilitated by the tumor microenvironment consisting of stromal cells, extracellular matrix and immune cells.74 However, the tumor microenvironment can also serve to negatively regulate tumor progression. For instance, p53 expression in hepatic stellate cells has been shown to non-cell autonomously curb initiation of hepatocellular carcinoma by promoting an anti-tumor microenvironment via secretion of senescence promoting factors in the murine liver.75 Fibroblasts in the tumor microenvironment have also been shown to secrete TGF-β (transforming growth factor-β) ligands, which can suppress tumor progression in the adjacent epithelia.76 But this equation is further complicated by the presence of different subpopulations of cells within the primary tumor itself, which express different genes, have varying metastatic potentials and respond differently to chemotherapy. Given that the primary tumor is populated with distinct cancer cells, it is important to understand whether the cells with metastatic potential within the primary tumor can increase the metastatic potential of the surrounding non-aggressive cells.
Clonal cooperation in metastasis
One of the first demonstrations of clonal cooperation in metastasis was reported by Fred Miller and colleagues in 1983. Using a set of syngeneic murine mammary carcinoma cell lines derived from a spontaneous tumor arising in a Balb/c mouse,23 the Miller group showed that the tumorigenic, yet non-metastatic 67nr cells metastasized to the lungs only when co-injected intravenously into mice with the highly metastatic 4t1 clones.77,78 The same group also demonstrated that an interaction occurred between the syngeneic cell lines with varying metastatic potentials when they were either cultured in vitro or orthotopically injected into the mammary fat pads of immunocompetent Balb/c mice. In some cases the more metastatic cells when injected into one flank of the mouse, inhibited the in vivo growth rate of the weakly metastatic cells injected into the other flank of the same mouse, a phenomenon that was attributed to the interaction of these cells with the immune system of the host mice.79 In another study, growing the more metastatic and weakly metastatic cells together resulted in the suppression of growth of the weakly metastatic clones.80 Around the same time, Fidler and colleagues discovered that clonal interactions between more and less metastatic clones in the B16 murine melanoma model led to stabilization of the metastatic phenotype in the less metastatic clones,81 again demonstrating the importance of interclonal interactions in metastasis. In 1993, this issue was revisited when David Tarin and colleagues demonstrated that subcutaneous co-injection of intrinsically metastatic and non-metastatic clones of the same murine fibrosarcoma line into mice, although resulting in a heterogeneous primary tumor, produced a significant number of metastases consisting only of the non-metastatic clones.82 Importantly, subcutaneous injection of only the non-metastatic clones into mice did not result in any metastases. Data from this study yet again suggested that metastatic cells must either impart metastatic characteristics on non-metastatic cells, or that they must somehow aid the non-metastatic cells in reaching the secondary site.
More than a decade later, Xiang and colleagues demonstrated that metastatic BL6-10 murine melanoma cells release exosomes, which are small vesicles derived from the endolysosomal pathway that transport various molecules between cells, and that these exosomes can increase the metastatic potential of syngeneic non-metastatic F1 melanoma cells.83 In this study, non-metastatic F1 (parental) melanoma cells metastasized efficiently to the lungs after intravenous injection only when previously cultured with exosomes derived from highly metastatic BL6–10 cells. The exosomes derived from the metastatic cells were profiled and shown to express a previously described metastatic marker Met 72 (a glycoprotein and tumor antigen expressed exclusively by the metastatic BL6–10 cells but not the F1 cells). Interestingly, Met 72 was observed to be expressed by the non-metastatic F1 cells after their treatment with exosomes released from the metastatic BL6–10 cells, which the authors concluded, enabled the F1 cells to metastasize. This study suggests that had the metastatic and non-metastatic clones co-existed in the primary tumor, the exosomes released by the metastatic clones could stably alter non-metastatic clones, making them more metastatic (Fig. 2).
Figure 2.

Non-cell autonomous interactions can increase metastatic propensity: exosomes as an example. The BL6–10 cell line is a highly metastatic subclone derived from the non-metastatic F1 cell line. Met 72, a previously identified metastasis marker, was found to be expressed on the surface of (A) BL6–10 cells, but not (B) F1 cells. Additionally, Met 72 was expressed on the surface of exosomes derived from BL6–10 cells. (C) Treating F1 cells with exosomes from BL6–10 cells caused F1 cells to become Met 72-positive and significantly increased the metastatic ability of F1 cells.83
More recently, Berns and colleagues have provided clear evidence that clonal cooperation, under some circumstances, is required for metastasis.84 These investigators found 2 populations of cells within a murine model of small cell lung cancer (SCLC): neuroendocrine small cells (NE) and mesenchymal large cells (nonNE), each with distinct gene and protein expression profiles. Importantly, this study showed that crosstalk occurred between the 2 populations of cells in vitro wherein each cell type had a proliferative advantage when cultured in the presence of the other. Surprisingly, the intrinsically non-metastatic NE cells formed metastases only when co-injected subcutaneously with nonNE cells. Single injections of neither the NE nor nonNE cells formed metastases, suggesting that cooperation between these cells was necessary to facilitate metastasis.84
Taken together, these studies provide compelling evidence that interclonal cooperativity within the primary tumor can affect metastatic outcome positively by increasing the number of cells with metastatic potential and thus the overall probability of metastatic occurrence.
Epithelial-to-mesenchymal transition and metastasis
Epithelial-to-mesenchymal transition (EMT) is a normal developmental process that is critical for embryogenesis, but has also been observed during wound healing and carcinoma progression.55 Three types of EMT have been defined: 1) The EMT that occurs during embryogenesis and is required for processes such as gastrulation, which is referred to as Type I EMT,85,86 2) The EMT that occurs during wound healing and tissue regeneration, which is referred to as Type II EMT,86 and 3) The EMT that occurs in cancer cells, which are thought to hijack a process that shares numerous characteristics with developmental EMTs, and is referred to as Type III or “oncogenic” EMT. In EMT, epithelial cells reduce cell-cell adhesion and apical-basal polarity, enabling them to separate from each other and invade through the basement membrane. This change is accompanied by the formation of filopodia on the cells and a switch from cell-cell adhesion-promoting integrins to those that promote cell-extracellular matrix adhesions.87 In addition, remodeling of the cytoskeleton occurs, replacing the static configuration of the epithelium with a more fluid and dynamic state which allows the cells to be more motile.88 Morphologically, the more cuboidal shaped epithelial cells often become more elongated through changes to the actin-myosin cytoskeleton,88 though this morphological change may not always be observed. This change is often accompanied by downregulation of epithelial proteins such as E-cadherin and keratin 18, concordant with the upregulation of mesenchymal proteins such as N-cadherin and Vimentin. Such changes promote migration and invasion, and could thus facilitate metastasis.85-87 It should be noted that this conversion in cancer is not always thought to be complete, and in fact recent evidence suggests that carcinoma cells at the leading edge of the tumor as well as in circulation may exist in an intermediate state, expressing both epithelial and mesenchymal markers simultaneously.56 These recent findings make it easier to understand how a reversal in EMT may occur at the secondary site (a mesenchymal-to-epithelial transition or MET), to allow for outgrowth of the metastatic lesion. Indeed, the requirement of an EMT-MET axis in cancer metastasis has been shown in some models.52,89-91 Because EMT during carcinoma progression is thought to be very plastic, the term Epithelial-Mesenchymal Plasticity (EMP) is now frequently used instead of EMT.92 Importantly, EMP has been shown to occur only within a subpopulation of cells within the primary tumor, thus again demonstrating the heterogeneous nature of tumors.53-56
Cancer cells are known to utilize embryonic pathways to promote tumor progression via enhancing properties such as migration, invasion, and neovascularization.55,93,94 Thus, many of the same molecular players that control developmental EMTs also regulate oncogenic EMTs. These include signaling pathways like Wnt and TGF-β, transcription factors like Snail, Twist1, the Six family members, which promote EMT, and various microRNAs (miRNAs), including the miR-200 family members that regulate epithelial identity.55,93,95 During stages of embryogenesis such as gastrulation and cardiogenesis, ligands of signaling pathways like Wnt1 and TGF-β1, 2 and 3 signal in an autocrine and/or paracrine manner to control EMT.93,96 Different sets of cells in the primitive streak, and later the mesoderm, express the ligands or the respective receptors for each of these pathways (with some cells expressing both), and communication between these cells is essential for migration, proliferation and cell-fate determination. Thus, activation of these signaling pathways in one set of cells can non-cell autonomously influence other cells. MiRNAs are a fairly recently described class of small non-coding RNA molecules which were first discovered in C. elegans as molecules that regulate the timing of differentiation.97,98 Since then numerous miRNAs have been implicated in the process of embryogenesis,95,99 and they are known to regulate many developmental genes.100-102 Importantly, they are known to cause Type II EMTs in various systems including during kidney fibrosis and have been implicated in Type III EMTs during cancer progression.103-105 The specific role of miRNAs in non-cell autonomously causing EMT of cells during development is still unknown. The roles of these factors during development have been reviewed extensively elsewhere.55,99,106-108
In the context of cancer, many of the above mentioned signaling pathways, transcription factors, and miRNAs have been shown to cell autonomously cause oncogenic EMTs and/or to increase metastasis, and their overexpression in various cancers is often associated with lower overall survival and poor prognosis.93,109,110 Since some EMT-inducing factors like Wnt1 and TGF-β ligands are secreted molecules, it is not a surprise that secretion of these ligands from cells in the tumor microenvironment affects the tumor cells expressing the appropriate receptors i.e., that they have non-cell autonomous roles in cancer.76 However, the non-cell autonomous functions attributed to EMT-inducing factors may result not only from secretion of these molecules from cells in the microenvironment, but also from tumor cells within the heterogeneous tumor. They may also occur in response to alterations in EMT-inducing transcription factors as well as miRNAs. For example, it was recently discovered that malignant cells could release miRNAs in larger exosomes that expressed different surface markers as compared to the exosomes released from normal non-cancerous cells.111 Since studies such as the murine melanoma study outlined above, have shown that exosomes released from malignant cells can increase the metastatic properties of other cells83,112 and since exosomes are known to contain mRNA, proteins and miRNAs, it suggests that secreted miRNAs, as well as any other proteins or mRNA, contained within the exosomes can non-cell autonomously alter the properties of cancer cells. Indeed, a study done by Taylor and colleagues113 showed that tumor-derived exosomes isolated from sera of ovarian cancer patients contained a variety of miRNAs, both similar to and different from the miRNAs expressed by the tumor mass, including some that have been previously associated with increased metastasis in ovarian cancer.114
Finally, it should be noted that EMT-inducing factors exist in a complex web, which may have consequences in a heterogeneous tumor. For example, TGF-β, which activates Snail during development, can also activate and/or be activated by Snail in the context of cancer.115-117 Thus, cancer cells in which Snail has been upregulated may increase their secretion of TGF-β, leading to EMT in neighboring cells that did not originally have increased levels of Snail (but may ultimately upregulate Snail downstream of TGF-β). However, to date, no formal studies have demonstrated that the EMT-inducing transcription factors play non-cell autonomous roles in mediating tumor progression.
EMT and intratumoral heterogeneity
Although only few cells in a heterogeneous primary tumor may undergo EMT, particularly those at the leading edge of the tumor,53-56 it is possible that these few cells may non-cell autonomously increase the potential of surrounding cells to undergo EMT and/or metastasize. Alternatively, it has been proposed that the epithelial tumor cells within a heterogeneous tumor may help to maintain the mesenchymal-like state of tumor cells that have undergone EMT.3 To elaborate, it has been hypothesized that when some cells within the primary tumor undergo EMT (induced by factors secreted from within the tumor), the other surrounding tumor cells that secreted those EMT-inducing factors remain “epithelial” to continue providing the factors to maintain the EMT phenotype of the other cells.3 Thus interclonal cooperation between distinct clones in the primary tumor that have undergone EMT to become mesenchymal-like (EMT cells) and those that have stayed epithelial (non-EMT cells) may be critical for maintaining the EMT status of the tumor cells. Since studies have shown that cells that have undergone EMT also metastasize more efficiently,55,118 interclonal cooperation between EMT and non-EMT cells and its possible effect on metastasis, is important to consider. In recent years, a few studies have probed into this cooperation and have shown that it may be critical to facilitate metastasis. These studies will be discussed is brief in the following section.
Clonal cooperation in EMT and the effect on metastasis
As mentioned earlier, some EMT-inducing factors have the ability to act non-cell autonomously to cause EMT during development. Many studies have shown that a tumor's microenvironment can affect the EMT status of the tumor cells (reviewed more thoroughly in),74,119,120 and factors like TGF-β1, secreted by the microenvironment, have been shown to non-cell autonomously regulate EMT of cells in the primary tumor.121 Few, if any, studies have asked if a mixed population of cells within the tumor itself, some of which have undergone EMT, can cause the surrounding cells to undergo EMT to affect metastatic progression. However, a number of studies have demonstrated cooperativity between tumor cells that have undergone EMT and those that have not.
Studies first done by Lyons and colleagues looked at interclonal cooperativity between cells that underwent EMT (EMT cells) and cells that remained epithelial (non-EMT cells) within a carcinoma cell population.122-124 Using BC1 rat mammary carcinoma cells consisting of a mixed population of epithelial (non-EMT) and metaplastic (EMT) cells, this group demonstrated that the non-EMT cells induced production of proteinases by the EMT cells which enabled efficient degradation of extracellular matrix. The non-EMT cells also secreted factors that increased growth and attachment of the EMT cells.122,124 The EMT cells reciprocated by producing secreted proteins that conferred multidrug resistance benefiting both cell types.123 Hence, this interclonal interaction in the heterogeneous BC1 cell line benefitted both populations of cells.
An elegant study by Tsuji and colleagues recently demonstrated that, at least in some models, cooperation between EMT and non-EMT cells is necessary to mediate metastasis.125 In this study, a downstream effector of TGF-β signaling, p12, was used to induce EMT of hamster cheek pouch carcinoma cells. p12 expression increased the invasive phenotype and dissemination of these cells, but did not enable metastatic colonization, regardless of whether the cells were injected into mice subcutaneously, to first form primary tumors, or intravenously, to bypass early metastatic steps including invasion and intravasation into the vasculature. On the other hand, the non-EMT parental cells were able to metastasize, but only when injected into mice intravenously, bypassing the early steps of metastasis. Importantly, when the non-EMT cells were subcutaneously injected into mice along with the EMT cells, both EMT and non-EMT cells could be found in the blood stream (differentiated by differentially labeling the cells with GFP or DsRed), whereas only the non-EMT cells could be found in the lungs as metastases. These data strongly suggest cooperativity between EMT and non-EMT cells, whereby EMT cells facilitate invasion through the basement membrane and intravasation into the vasculature, which allows the non-EMT cells to access circulation. Once the non-EMT cells are in circulation, they are themselves able to extravasate and grow at the secondary site. Thus, different clonal populations, as represented by EMT and non-EMT cells, were able to interact to produce a metastatic outcome.
Additional studies performed in prostate and bladder cancer models have recently provided further evidence that EMT and non-EMT cells cooperate to affect metastatic outcome. In this study by Thomson and colleagues,126 the authors found that in contrast to previous studies where the more mesenchymal cells have increased tumor initiating capacities,39 the more epithelial (non-EMT) cells exhibited a CSC-like gene signature and were less invasive, but more metastatic when injected into mice, as compared to the more mesenchymal-like (EMT) cells. Co-culturing the non-EMT and EMT cells resulted in increasing the in vitro invasive properties of the non-EMT cells. It was also observed that orthotopic co-injection of the non-EMT and EMT clones into mice diminished the growth rate of the primary tumor, compared to injection of the non-EMT clone only, but accelerated the metastatic rate of the metastatic non-EMT cells. Acceleration of metastasis was seen when the EMT and non-EMT cells were co-injected into mice intramuscularly, as well as intravenously. It should be noted that while the non-EMT metastatic cells appeared more epithelial in vitro, these cells were able to undergo partial EMT in vivo as demonstrated by upregulation of the mesenchymal marker fibronectin and downregulation of the epithelial marker E-cadherin in the primary tumor. These data support the idea that cellular plasticity may be the critical factor that enables metastatic dissemination and that this plasticity may be transferred non-cell autonomously to other cells, to ultimately affect metastatic progression.
Together, these studies show that cells that have undergone EMT, while not always metastatic in nature, are able to influence those cells in the tumor that have not undergone EMT (or vice versa).
Consequences of clonal cooperativity
The interaction between different clones in the tumor has been hypothesized to occur through adjoining matrix connections or through secretion of diffusible factors by one or both kinds of clones.127 If interactions occur via adjoining matrices, the effects could be expected to be more proximal than if a diffusible factor was involved. In addition, any factors’ effects on neighboring cells would be expected to be inversely proportional to the distance of the recipient cells from the cell(s) secreting the factors.3,127 Given that diffusible factors that can alter the properties of neighboring cells within the primary tumor have been identified, only a few cells would need to undergo EMT and/or gain metastatic potential in order to increase overall metastasis.
It is clear from the studies summarized above that crosstalk occurs between the different cell populations comprising the primary tumor. Not only are the intrinsically metastatic clones within the tumor capable of making the surrounding cells more metastatic, the non-aggressive cells also often aid in altering the metastatic outcome.
It is unlikely, given the sequencing studies carried out in various cancers, that one single clone will take over the entire tumor, since multiple clones with varied genetic mutations have been identified within the same tumor. It seems beneficial for both the aggressive and non-aggressive clones to have the other present. Whether the cooperation between the different clones results in positive tumor progression and increased metastasis, or whether it results in the stunting of tumor progression, it is likely under the regulation of many factors and probably heavily influenced by the tumor microenvironment. Microenvironmental changes and pressures have been shown to be partly responsible for the outgrowth of these different subpopulations of cells30 and could possibly play a role in making one direction of interaction between these cells more dominant than the other.
It is also unknown whether the result of this cooperation is permanent. Aggressive cells could only temporarily change the non-aggressive cells; just enough to allow them to escape into the blood stream and colonize at a secondary site. Thus, these cells with “pseudo” metastatic potential might revert to their non-aggressive versions in the metastatic site. These cells could then potentially play a role in aiding the aggressive cells to colonize at the secondary site. It is known that cancer cells can be dormant at the secondary site for long periods of time, and microenvironmental changes are known to be responsible for bringing cancer cells out of dormancy.128,129 Alternatively, it is possible that these intrinsically non-aggressive, yet cancerous, cells that have metastasized to the secondary sites secrete factors that form a more habitable niche for the metastatic cells. This niche could enable the cells to transition from dormant to active metastases.
The possibility that cells with more metastatic potential can increase the metastatic ability of the surrounding cells suggests that in order to curb the overall incidence of metastasis, it will be important to identify factors that facilitate this conversion and inhibit their activity. Indeed, intratumoral heterogeneity can have dire consequences on patient survival, as studies have shown that different populations of cells within the tumor react differently to chemotherapeutic drugs. Many studies have also shown that CSCs are resistant to chemotherapy. However, if CSCs can cause the conversion of non-CSCs to CSCs, or if these cells are also capable of non-cell autonomously rendering neighboring cells resistant to chemotherapy, then a larger percentage of the tumor than anticipated may not respond to chemotherapy. Eradication of such heterogeneous tumors would require strategies to both purge the CSCs and curb their ability to convert other surrounding cells. Since non-genetic factors can contribute to heterogeneity by generation of CSCs12 or alterations in EMT-status, and since CSC/EMT cells are often chemoresistant, one could envision a scenario in which we try to shift the population of cells toward a homogenous chemosensitive12 and/or differentiated state by targeting factors released by the microenvironment or released by the CSC and/or EMT cells. A homogenous tumor made of similar cells would then be easier to target (Fig. 3).
Figure 3.
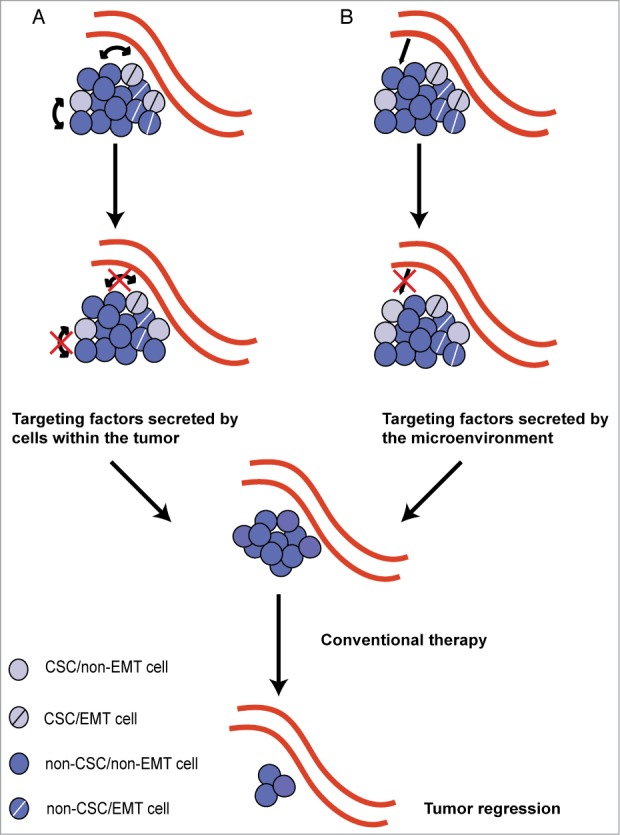
Targeting heterogeneous tumors. The differentiation hierarchy existing within an epithelial tumor, making it heterogeneous in nature, can be due to both the presence of cancer stem cells (CSCs) and non-CSCs as well as cells that have undergone an epithelial-to-mesenchymal transition (EMT; EMT cells) and those that remain epithelial (non-EMT cells). Current evidence suggests that bidirectional conversion between non-CSCs and CSCs is possible, and that CSCs do not always go hand-in-hand with EMT cells. Additionally, intratumoral heterogeneity can arise from both (A) non-cell autonomous interactions between cells in the tumors and (B) signals from the microenvironment. Future therapeutics should focus on identifying and targeting molecules that function non-cell autonomously within the tumor and are secreted from the microenvironment, which will aid in driving the tumor toward homogeneity. Conventional therapy may then be used on the resulting homogenous tumor leading to tumor regression.
Current therapies rarely take intratumoral heterogeneity into account. Since this heterogeneity is now known to be influenced by both genetic and non-genetic factors, and since clones in the tumor that cooperate with one another could harbor different advantageous mutations, more attempts at combination therapies may increase our chances of successfully inhibiting tumor progression.
In closing, the evidence of intratumoral heterogeneity has added an additional layer to an already complex problem of how to target cancers, as we become cognizant that not only is every person's tumor different (intertumoral heterogeneity), but that there are also vast differences present within a person's tumor (intratumoral heterogeneity), as well as in some cases, between the primary tumor and metastases. These findings will have far-reaching implications for prognosis and therapeutics.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
HLF is funded by grants from the National Cancer Institute, specifically R01-CA095277 which is related to EMT and metastasis, the topic of this review.
References
- 1. Nowell PC. The clonal evolution of tumor cell populations. Science 1976; 194:23-8; PMID:959840; http://dx.doi.org/ 10.1126/science.959840 [DOI] [PubMed] [Google Scholar]
- 2. Fialkow PJ. Clonal origin of human tumors. Biochim Biophys Acta 1976; 458:283-321; PMID:1067873 [DOI] [PubMed] [Google Scholar]
- 3. Lyons JG, Lobo E, Martorana AM, Myerscough MR. Clonal diversity in carcinomas: its implications for tumour progression and the contribution made to it by epithelial-mesenchymal transitions. Clin Exp Metastasis 2008; 25:665-77; PMID:18071912; http://dx.doi.org/ 10.1007/s10585-007-9134-2 [DOI] [PubMed] [Google Scholar]
- 4. Parsons BL. Monoclonal tumor origin is an underlying misconception of the RESIC approach. Proc Natl Acad Sci U S A 2011; 108:E15; author reply E6; PMID:21239682; http://dx.doi.org/ 10.1073/pnas.1017998108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parsons BL. Many different tumor types have polyclonal tumor origin: evidence and implications. Mutat Res 2008; 659:232-47; PMID:18614394; http://dx.doi.org/ 10.1016/j.mrrev.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 6. Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011; 469:356-61; PMID:21160474; http://dx.doi.org/ 10.1038/nature09650 [DOI] [PubMed] [Google Scholar]
- 7. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. Tumour evolution inferred by single-cell sequencing. Nature 2011; 472:90-4; PMID:21399628; http://dx.doi.org/ 10.1038/nature09807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366:883-92; PMID:22397650; http://dx.doi.org/ 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res 1978; 38:2651-60; PMID:354778 [PubMed] [Google Scholar]
- 10. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem 2013; 59:168-79; PMID:23220226; http://dx.doi.org/ 10.1373/clinchem.2012.184655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138:822-9; PMID:19737509; http://dx.doi.org/ 10.1016/j.cell.2009.08.017 [DOI] [PubMed] [Google Scholar]
- 12. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 2012; 12:323-34; PMID:22513401; http://dx.doi.org/ 10.1038/nrc3261 [DOI] [PubMed] [Google Scholar]
- 13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 14. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013; 152:714-26; PMID:23415222; http://dx.doi.org/ 10.1016/j.cell.2013.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013; 501:338-45; PMID:24048066; http://dx.doi.org/ 10.1038/nature12625 [DOI] [PubMed] [Google Scholar]
- 16. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell 2012; 150:251-63; PMID:22817889; http://dx.doi.org/ 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012; 486:395-9; PMID:22495314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosai J, Ackerman LV. The pathology of tumors, part III: grading, staging & classification. CA: Cancer J Clin 1979; 29:66-77; PMID:219944; http://dx.doi.org/ 10.1017/S0009838800035175 [DOI] [PubMed] [Google Scholar]
- 19. Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 2012; 109:3041-6; PMID:22323597; http://dx.doi.org/ 10.1073/pnas.1114033109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heppner GH, Dexter DL, DeNucci T, Miller FR, Calabresi P. Heterogeneity in drug sensitivity among tumor cell subpopulations of a single mammary tumor. Cancer Res 1978; 38:3758-63; PMID:698935 [PubMed] [Google Scholar]
- 21. Kreso A, O'Brien CA, van Galen P, Gan OI, Notta F, Brown AM, Ng K, Ma J, Wienholds E, Dunant C, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013; 339:543-8; PMID:23239622; http://dx.doi.org/ 10.1126/science.1227670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, Croul S, Bouffet E, Fults DW, Eberhart CG, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012; 482:529-33; PMID:22343890; http://dx.doi.org/ 10.1038/nature10825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992; 52:1399-405; PMID:1540948 [PubMed] [Google Scholar]
- 24. Harbst K, Lauss M, Cirenajwis H, Winter C, Howlin J, Torngren T, Kvist A, Nodin B, Olsson E, Hakkinen J, et al. Molecular and genetic diversity in the metastatic process of melanoma. J Pathol 2014; 233:39-50; PMID:24399611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC, Kaiser MF, Mirabella F, Lopez-Corral L, Humphray S, Murray L, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014; 28:384-90; PMID:23817176; http://dx.doi.org/ 10.1038/leu.2013.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013; 501:328-37; PMID:24048065; http://dx.doi.org/ 10.1038/nature12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012; 481:306-13; PMID:22258609; http://dx.doi.org/ 10.1038/nature10762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crockford A, Jamal-Hanjani M, Hicks J, Swanton C. Implications of intratumour heterogeneity for treatment stratification. J Pathol 2014; 232:264-73; PMID:24115146; http://dx.doi.org/ 10.1002/path.4270 [DOI] [PubMed] [Google Scholar]
- 29. Spremulli EN, Dexter DL. Human tumor cell heterogeneity and metastasis. J Clin Oncol 1983; 1:496-509; PMID:6366142 [DOI] [PubMed] [Google Scholar]
- 30. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013; 501:346-54; PMID:24048067; http://dx.doi.org/ 10.1038/nature12626 [DOI] [PubMed] [Google Scholar]
- 31. Marusyk A, Polyak K. Cancer. Cancer cell phenotypes, in fifty shades of grey. Science 2013; 339:528-9; PMID:23372002; http://dx.doi.org/ 10.1126/science.1234415 [DOI] [PubMed] [Google Scholar]
- 32. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 2010; 1805:105-17; PMID:9931353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, Holland EC. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010; 6:141-52; PMID:20144787; http://dx.doi.org/ 10.1016/j.stem.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12:468-76; PMID:20418870; http://dx.doi.org/ 10.1038/ncb2048 [DOI] [PubMed] [Google Scholar]
- 35. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 2011; 121:3804-9; PMID:21965337; http://dx.doi.org/ 10.1172/JCI57099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fabian A, Vereb G, Szollosi J. The hitchhikers guide to cancer stem cell theory: markers, pathways and therapy. Cytometry A 2013; 83:62-71; PMID:22997049; http://dx.doi.org/ 10.1002/cyto.a.22206 [DOI] [PubMed] [Google Scholar]
- 37. Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med 2013; 2:3; PMID:23369605; http://dx.doi.org/ 10.1186/2001-1326-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tomao F, Papa A, Strudel M, Rossi L, Lo Russo G, Benedetti Panici P, Ciabatta FR, Tomao S. Investigating molecular profiles of ovarian cancer: an update on cancer stem cells. J Cancer 2014; 5:301-10; PMID:24723972; http://dx.doi.org/ 10.7150/jca.8610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704-15; PMID:18485877; http://dx.doi.org/ 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol 2012; 198:281-93; PMID:22869594; http://dx.doi.org/ 10.1083/jcb.201202014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kang Y. Analysis of cancer stem cell metastasis in xenograft animal models. Methods Mol Biol 2009; 568:7-19; PMID:19582418; http://dx.doi.org/ 10.1007/978-1-59745-280-9_2 [DOI] [PubMed] [Google Scholar]
- 42. Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 2007; 449:473-7; PMID:17851532; http://dx.doi.org/ 10.1038/nature06159 [DOI] [PubMed] [Google Scholar]
- 43. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008; 3:e2888; PMID:18682804; http://dx.doi.org/ 10.1371/journal.pone.0002888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011; 108:7950-5; PMID:21498687; http://dx.doi.org/ 10.1073/pnas.1102454108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A 2011; 108:1397-402; PMID:21220315; http://dx.doi.org/ 10.1073/pnas.1018898108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu CC, Lo WL, Chen YW, Huang PI, Hsu HS, Tseng LM, Hung SC, Kao SY, Chang CJ, Chiou SH. Bmi-1 regulates snail expression and promotes metastasis ability in head and neck squamous cancer-derived ALDH1 positive cells. J oOcol 2011; 2011:pii: 609259; PMID:20936121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, Lee YJ. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 2013; 25:961-9; PMID:23333246; http://dx.doi.org/ 10.1016/j.cellsig.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martelotto LG, Ng CK, Piscuoglio S, Weigelt B, Reis-Filho JS. Breast cancer intra-tumor heterogeneity. Breast Cancer Res 2014; 16:210; http://dx.doi.org/ 10.1186/bcr3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007; 11:259-73; PMID:17349583; http://dx.doi.org/ 10.1016/j.ccr.2007.01.013 [DOI] [PubMed] [Google Scholar]
- 50. Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011; 469:362-7; PMID:21248843; http://dx.doi.org/ 10.1038/nature09733 [DOI] [PubMed] [Google Scholar]
- 51. May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 2011; 13:202; PMID:21392411; http://dx.doi.org/ 10.1186/bcr2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012; 22:709-24; PMID:23201163; http://dx.doi.org/ 10.1016/j.ccr.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 53. Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011; 19:372-86; PMID:21397860; http://dx.doi.org/ 10.1016/j.ccr.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanzawa M, Semba S, Hara S, Itoh T, Yokozaki H. WNT5A is a key regulator of the epithelial-mesenchymal transition and cancer stem cell properties in human gastric carcinoma cells. Pathobiology 2013; 80:235-44; PMID:23615002; http://dx.doi.org/ 10.1159/000346843 [DOI] [PubMed] [Google Scholar]
- 55. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 56. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013; 339:580-4; PMID:23372014; http://dx.doi.org/ 10.1126/science.1228522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fomchenko EI, Dougherty JD, Helmy KY, Katz AM, Pietras A, Brennan C, Huse JT, Milosevic A, Holland EC. Recruited cells can become transformed and overtake PDGF-induced murine gliomas in vivo during tumor progression. PloS one 2011; 6:e20605; PMID:21754979; http://dx.doi.org/ 10.1371/journal.pone.0020605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014; 508:113-7; PMID:24695311; http://dx.doi.org/ 10.1038/nature13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bose D, Zimmerman LJ, Pierobon M, Petricoin E, Tozzi F, Parikh A, Fan F, Dallas N, Xia L, Gaur P, et al. Chemoresistant colorectal cancer cells and cancer stem cells mediate growth and survival of bystander cells. British journal of cancer 2011; 105:1759-67; PMID:22045189; http://dx.doi.org/ 10.1038/bjc.2011.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 2010; 1805:105-17; PMID:19931353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer 2006; 6:449-58; PMID:16723991; http://dx.doi.org/ 10.1038/nrc1886 [DOI] [PubMed] [Google Scholar]
- 62. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2:563-72; PMID:12154349; http://dx.doi.org/ 10.1038/nrc865 [DOI] [PubMed] [Google Scholar]
- 63. Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5:591-602; PMID:16056258; http://dx.doi.org/ 10.1038/nrc1670 [DOI] [PubMed] [Google Scholar]
- 64. Havens AM, Pedersen EA, Shiozawa Y, Ying C, Jung Y, Sun Y, Neeley C, Wang J, Mehra R, Keller ET, et al. An in vivo mouse model for human prostate cancer metastasis. Neoplasia 2008; 10:371-80; PMID:18392141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Omholt K, Karsberg S, Platz A, Kanter L, Ringborg U, Hansson J. Screening of N-ras codon 61 mutations in paired primary and metastatic cutaneous melanomas: mutations occur early and persist throughout tumor progression. Clin Cancer Res 2002; 8:3468-74; PMID:12429636 [PubMed] [Google Scholar]
- 66. Bernards R, Weinberg RA. A progression puzzle. Nature 2002; 418:823; PMID:12192390; http://dx.doi.org/ 10.1038/418823a [DOI] [PubMed] [Google Scholar]
- 67. Weiss L, Holmes JC, Ward PM. Do metastases arise from pre-existing subpopulations of cancer cells? Br J Cancer 1983; 47:81-9; PMID:6571784; http://dx.doi.org/ 10.1038/bjc.1983.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977; 197:893-5; PMID:887927; http://dx.doi.org/ 10.1126/science.887927 [DOI] [PubMed] [Google Scholar]
- 69. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415:530-6; PMID:11823860; http://dx.doi.org/ 10.1038/415530a [DOI] [PubMed] [Google Scholar]
- 70. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet 2003; 33:49-54; PMID:12469122; http://dx.doi.org/ 10.1038/ng1060 [DOI] [PubMed] [Google Scholar]
- 71. McGuire WL, Clark GM. Prognostic factors and treatment decisions in axillary-node-negative breast cancer. N Engl J Med 1992; 326:1756-61; PMID:1594018; http://dx.doi.org/ 10.1056/NEJM199206253262607 [DOI] [PubMed] [Google Scholar]
- 72. Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sanchez V, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 2014; 4:232-45; PMID:24356096; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stoecklein NH, Klein CA. Genetic disparity between primary tumours, disseminated tumour cells, and manifest metastasis. Int J Cancer 2010; 126:589-98; PMID:19795462; http://dx.doi.org/ 10.1002/ijc.24916 [DOI] [PubMed] [Google Scholar]
- 74. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; http://dx.doi.org/ 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. Non-cell-autonomous tumor suppression by p53. Cell 2013; 153:449-60; PMID:23562644; http://dx.doi.org/ 10.1016/j.cell.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004; 303:848-51; PMID:14764882; http://dx.doi.org/ 10.1126/science.1090922 [DOI] [PubMed] [Google Scholar]
- 77. Miller FR. Tumor subpopulation interactions in metastasis. Invasion Metastasis 1983; 3:234-42; PMID:6677628 [PubMed] [Google Scholar]
- 78. Miller FR, Heppner GH. Cellular interactions in metastasis. Cancer Metastasis Rev 1990; 9:21-34; PMID:2208566; http://dx.doi.org/ 10.1007/BF00047586 [DOI] [PubMed] [Google Scholar]
- 79. Miller BE, Miller FR, Leith J, Heppner GH. Growth interaction in vivo between tumor subpopulations derived from a single mouse mammary tumor. Cancer Res 1980; 40:3977-81; PMID:7471048 [PubMed] [Google Scholar]
- 80. Miller BE, Miller FR, Wilburn D, Heppner GH. Dominance of a tumor subpopulation line in mixed heterogeneous mouse mammary tumors. Cancer Res 1988; 48:5747-53; PMID:3167832 [PubMed] [Google Scholar]
- 81. Poste G, Doll J, Fidler IJ. Interactions among clonal subpopulations affect stability of the metastatic phenotype in polyclonal populations of B16 melanoma cells. Proc Natl Acad Sci U S A 1981; 78:6226-30; PMID:6947225; http://dx.doi.org/ 10.1073/pnas.78.10.6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Baban D, Matsumura Y, Kocialkowski S, Tarin D. Studies on relationships between metastatic and non-metastatic tumor cell populations using lineages labeled with dominant selectable genetic markers. Int J Dev Biol 1993; 37:237-43; PMID:8507566 [PubMed] [Google Scholar]
- 83. Hao S, Ye Z, Li F, Meng Q, Qureshi M, Yang J, Xiang J. Epigenetic transfer of metastatic activity by uptake of highly metastatic B16 melanoma cell-released exosomes. Exp Oncol 2006; 28:126-31; PMID:16837903 [PubMed] [Google Scholar]
- 84. Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R, Berns A. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011; 19:244-56; PMID:21316603; http://dx.doi.org/ 10.1016/j.ccr.2010.12.021 [DOI] [PubMed] [Google Scholar]
- 85. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420-8; PMID:19487818; http://dx.doi.org/ 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Drasin DJ, Robin TP, Ford HL. Breast cancer epithelial-to-mesenchymal transition: examining the functional consequences of plasticity. Breast Cancer Res 2011; 13:226; PMID:22078097; http://dx.doi.org/ 10.1186/bcr3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hay ED, Zuk A. Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis 1995; 26:678-90; PMID:7573028; http://dx.doi.org/ 10.1016/0272-6386(95)90610-X [DOI] [PubMed] [Google Scholar]
- 88. Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mechanisms Dev 2003; 120:1351-83; PMID:14623443; http://dx.doi.org/ 10.1016/j.mod.2003.06.005 [DOI] [PubMed] [Google Scholar]
- 89. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012; 22:725-36; PMID:23201165; http://dx.doi.org/ 10.1016/j.ccr.2012.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res 2006; 66:11271-8; PMID:17145872; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2044 [DOI] [PubMed] [Google Scholar]
- 91. Chaffer CL, Thompson EW, Williams ED. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 2007; 185:7-19; PMID:17587803; http://dx.doi.org/ 10.1159/000101298 [DOI] [PubMed] [Google Scholar]
- 92. Ford HL, Thompson EW. Mammary gland studies as important contributors to the cause of epithelial mesenchymal plasticity in malignancy. J Mammary Gland Biol Neoplasia 2010; 15:113-5; PMID:20544376; http://dx.doi.org/ 10.1007/s10911-010-9182-0 [DOI] [PubMed] [Google Scholar]
- 93. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 2010; 15:117-34; PMID:20490631; http://dx.doi.org/ 10.1007/s10911-010-9178-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med 2009; 60:167-79; PMID:19630570; http://dx.doi.org/ 10.1146/annurev.med.59.053006.104707 [DOI] [PubMed] [Google Scholar]
- 95. Bhaskaran M, Mohan M. MicroRNAs: History, Biogenesis, and Their Evolving Role in Animal Development and Disease. Vet Path 2013; 51:759-774; PMID:24045890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Brade T, Manner J, Kuhl M. The role of Wnt signalling in cardiac development and tissue remodelling in the mature heart. Cardiovasc Res 2006; 72:198-209; PMID:16860783; http://dx.doi.org/ 10.1016/j.cardiores.2006.06.025 [DOI] [PubMed] [Google Scholar]
- 97. Erson AE, Petty EM. MicroRNAs in development and disease. Clin Genet 2008; 74:296-306; PMID:18713256; http://dx.doi.org/ 10.1111/j.1399-0004.2008.01076.x [DOI] [PubMed] [Google Scholar]
- 98. Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev 2011; 91:827-87; PMID:21742789; http://dx.doi.org/ 10.1152/physrev.00006.2010 [DOI] [PubMed] [Google Scholar]
- 99. Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet 2006; 38 Suppl:S20-4; PMID:16736020; http://dx.doi.org/ 10.1038/ng1803 [DOI] [PubMed] [Google Scholar]
- 100. Hornstein E, Mansfield JH, Yekta S, Hu JK, Harfe BD, McManus MT, Baskerville S, Bartel DP, Tabin CJ. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature 2005; 438:671-4; PMID:16319892; http://dx.doi.org/ 10.1038/nature04138 [DOI] [PubMed] [Google Scholar]
- 101. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell 2006; 11:441-50; PMID:17011485; http://dx.doi.org/ 10.1016/j.devcel.2006.09.009 [DOI] [PubMed] [Google Scholar]
- 102. Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004; 304:594-6; PMID:15105502; http://dx.doi.org/ 10.1126/science.1097434 [DOI] [PubMed] [Google Scholar]
- 103. Srivastava SP, Koya D, Kanasaki K. MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. BioMed Res Int 2013; 2013:125469; PMID:24089659; http://dx.doi.org/ 10.1155/2013/125469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhao X, Lu Y, Nie Y, Fan D. MicroRNAs as critical regulators involved in regulating epithelial- mesenchymal transition. Curr Cancer Drug Targets 2013; 13:935-44; PMID:24168189; http://dx.doi.org/ 10.2174/15680096113136660099 [DOI] [PubMed] [Google Scholar]
- 105. Hao J, Zhang Y, Deng M, Ye R, Zhao S, Wang Y, Li J, Zhao Z. MicroRNA control of epithelial-mesenchymal transition in cancer stem cells. Int J Cancer 2014; 135:1019-27; PMID:24500893 [DOI] [PubMed] [Google Scholar]
- 106. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 2009; 16:329-43; PMID:19289080; http://dx.doi.org/ 10.1016/j.devcel.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 107. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004; 20:781-810; PMID:15473860; http://dx.doi.org/ 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- 108. Christensen KL, Patrick AN, McCoy EL, Ford HL. The six family of homeobox genes in development and cancer. Adv Cancer Res 2008; 101:93-126; PMID:19055944; http://dx.doi.org/ 10.1016/S0065-230X(08)00405-3 [DOI] [PubMed] [Google Scholar]
- 109. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 2010; 107:15449-54; PMID:20713713; http://dx.doi.org/ 10.1073/pnas.1004900107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Garg M. Epithelial-mesenchymal transition - activating transcription factors - multifunctional regulators in cancer. World J Stem Cells 2013; 5:188-95; PMID:24179606; http://dx.doi.org/ 10.4252/wjsc.v5.i4.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Palma J, Yaddanapudi SC, Pigati L, Havens MA, Jeong S, Weiner GA, Weimer KM, Stern B, Hastings ML, Duelli DM. MicroRNAs are exported from malignant cells in customized particles. Nucleic Acids Res 2012; 40:9125-38; PMID:22772984; http://dx.doi.org/ 10.1093/nar/gks656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Demory Beckler M, Higginbotham JN, Franklin JL, Ham AJ, Halvey PJ, Imasuen IE, Whitwell C, Li M, Liebler DC, Coffey RJ. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics 2013; 12:343-55; PMID:23161513; http://dx.doi.org/ 10.1074/mcp.M112.022806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol 2008; 110:13-21; PMID:18589210; http://dx.doi.org/ 10.1016/j.ygyno.2008.04.033 [DOI] [PubMed] [Google Scholar]
- 114. Xu CX, Xu M, Tan L, Yang H, Permuth-Wey J, Kruk PA, Wenham RM, Nicosia SV, Lancaster JM, Sellers TA, et al. MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog. J Biol Chem 2012; 287:34970-8; PMID:22927443; http://dx.doi.org/ 10.1074/jbc.M112.374611 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 2010; 107:15449-54; PMID:20713713; http://dx.doi.org/ 10.1073/pnas.1004900107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PloS One 2011; 6:e26514; PMID:22028892; http://dx.doi.org/ 10.1371/journal.pone.0026514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Smith AP, Verrecchia A, Faga G, Doni M, Perna D, Martinato F, Guccione E, Amati B. A positive role for Myc in TGFbeta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene 2009; 28:422-30; PMID:18978814; http://dx.doi.org/ 10.1038/onc.2008.395 [DOI] [PubMed] [Google Scholar]
- 118. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013; 27:2192-206; PMID:24142872; http://dx.doi.org/ 10.1101/gad.225334.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci 2011; 1:29; PMID:21880137; http://dx.doi.org/ 10.1186/2045-3701-1-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res 2012; 72:4883-9; PMID:23002209; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Junk DJ, Cipriano R, Bryson BL, Gilmore HL, Jackson MW. Tumor microenvironmental signaling elicits epithelial-mesenchymal plasticity through cooperation with transforming genetic events. Neoplasia 2013; 15:1100-9; PMID:24027434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Martorana AM, Zheng G, Crowe TC, O'Grady RL, Lyons JG. Epithelial cells up-regulate matrix metalloproteinases in cells within the same mammary carcinoma that have undergone an epithelial-mesenchymal transition. Cancer Res 1998; 58:4970-9; PMID:9810007 [PubMed] [Google Scholar]
- 123. Zheng G, Lyons JG. Cyclosporin A improves the selection of cells transfected with the puromycin acetyltransferase gene. BioTechniques 2002; 33:32, 34, 36; PMID:12139252 [DOI] [PubMed] [Google Scholar]
- 124. Lyons JG, Siew K, O'Grady RL. Cellular interactions determining the production of collagenase by a rat mammary carcinoma cell line. Int J Cancer 1989; 43:119-25; PMID:2536004; http://dx.doi.org/ 10.1002/ijc.2910430123 [DOI] [PubMed] [Google Scholar]
- 125. Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, Hu GF. Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res 2008; 68:10377-86; PMID:19074907; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Celia-Terrassa T, Meca-Cortes O, Mateo F, de Paz AM, Rubio N, Arnal-Estape A, Ell BJ, Bermudo R, Diaz A, Guerra-Rebollo M, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest; 2012; 122:1849-68; PMID:22505459; http://dx.doi.org/ 10.1172/JCI59218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A 2006; 103:13474-9; PMID:16938860; http://dx.doi.org/ 10.1073/pnas.0606053103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007; 7:834-46; PMID:17957189; http://dx.doi.org/ 10.1038/nrc2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bragado P, Sosa MS, Keely P, Condeelis J, Aguirre-Ghiso JA. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res 2012; 195:25-39; PMID:22527492 [DOI] [PMC free article] [PubMed] [Google Scholar]