

Abstract
In rabbit atrial myocytes Ca signaling has unique features due to the lack of transverse (t) tubules, the spatial arrangement of mitochondria and the contribution of inositol-1,4,5-trisphosphate (IP3) receptor-induced Ca release (IICR). During excitation-contraction coupling action potential-induced elevation of cytosolic [Ca] originates in the cell periphery from Ca released from the junctional sarcoplasmic reticulum (j-SR) and then propagates by Ca-induced Ca release from non-junctional (nj-) SR toward the cell center. The subsarcolemmal region between j-SR and the first array of nj-SR Ca release sites is devoid of mitochondria which results in a rapid propagation of activation through this domain, whereas the subsequent propagation through the nj-SR network occurs at a velocity typical for a propagating Ca wave. Inhibition of mitochondrial Ca uptake with the Ca uniporter blocker Ru360 accelerates propagation and increases the amplitude of Ca transients (CaTs) originating from nj-SR. Elevation of cytosolic IP3 levels by rapid photolysis of caged IP3 has profound effects on the magnitude of subcellular CaTs with increased Ca release from nj-SR and enhanced CaTs in the nuclear compartment. IP3 uncaging restricted to the nucleus elicites ‘mini’-Ca waves that remain confined to this compartment. Elementary IICR events (Ca puffs) preferentially originate in the nucleus in close physical association with membrane structures of the nuclear envelope and the nucleoplasmic reticulum. The data suggest that in atrial myocytes the nucleus is an autonomous Ca signaling domain where Ca dynamics are primarily governed by IICR.
Keywords: atria, excitation-contraction coupling, IP3, IICR, mitochondria, nuclear calcium
Abbreviations
- 2-APB
2-aminoethoxydiphenyl borate
- AP
action potential
- CaT
Ca transient
- [Ca]i
cytosolic Ca concentration
- [Ca]mito
mitochondrial Ca concentration
- CICR
Ca-induced Ca release
- CRU
Ca release units
- CT
central
- ECC
excitation-contraction coupling
- IICR
IP3R-induced Ca release
- IP3R
Inositol-1,4,5-trisphosphate receptor
- LCC
L-type Ca channels
- MCU
mitochondrial Ca uniporter
- NE
nuclear envelope
- NFAT
nuclear factor of activated T cells
- NPR
nucleoplasmic reticulum
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- j-SR
junctional SR
- nj-SR
non-junctional SR
- SS
subsarcolemmal
- TF50
time to half-maximal amplitude
- t-tubule
transverse tubule
- TZ
transition zone
Introduction
In atrial myocytes the mechanism of excitation-contraction coupling (ECC) and action potential (AP) induced Ca release from the sarcoplasmic reticulum (SR) reveals unique features that are strikingly different from ventricular myocytes. In atrial cells the transverse (t)-tubule system, a sarcomerically organized system of surface membrane invaginations characteristic of ventricular myoctes, is either entirely lacking or poorly and irregularly developed, revealing considerable species differences.1-6 The absence of t-tubules in atrial myocytes defines 2 types of SR based on their location relative to the surface membrane. Junctional SR (j-SR) is found in the cell periphery where it is organized in peripheral couplings, i.e., the SR membrane associates closely with the surface membrane where it faces voltage-gated L-type Ca channels (LCCs), similar to the dyadic cleft in ventricular myocytes.7,8 Thus, the Ca release units9 (CRUs) of the j-SR are functionally organized like a classical couplon in the ventricle.10,11 In the latter, LCCs and RyRs form couplons throughout the entire cell that ensure spatially homogenous Ca release during the AP. In atrial myocytes, however, the quantitatively much more abundant non-junctional SR (nj-SR) is located in central regions of the cell and does not associate with the surface membrane. Both j-SR and nj-SR possess RyRs3-5,7,12-14 which are organized in a 3-dimensional array of RyR clusters or CRUs, and contribute to ECC by active Ca release.1,7,15 As a consequence of these ultrastructural arrangements AP-induced Ca release is spatially inhomogeneous1,16,17: AP-induced membrane depolarization activates Ca entry through LCCs which triggers subsarcolemmal (SS) CICR from RyRs of the j-SR. Elevation of peripheral [Ca]i propagates then by CICR in a Ca wave-like fashion in centripetal direction by a diffusion-reaction process or a ‘fire-diffuse-fire’ mechanism.15,18
The SR of atrial cells is also equipped with a second Ca release channel, the inositol-1,4,5-trisphosphate receptor (IP3R). The IP3R is far less abundant than the RyR, however, the relative IP3R expression level is higher in atrial cells compared to ventricular myocytes19,20 and is upregulated in cardiac disease.21–24 In atrial cells IP3R-induced Ca release (IICR) has positive inotropic but also proarrhythmic effects via facilitation of Ca release from RyRs.25,26 Furthermore, the nuclear envelope, a contiguous extension of the SR that engulfs the nucleus as a functional Ca store, is equipped with both RyRs and IP3Rs that face the cytosol as well as the nucleoplasm. We have shown that IP3R-mediated Ca release outweighs RyR-dependent Ca release in the nuclear compartment,27 suggesting that IICR has specific nuclear functions and targets. For example, we have demonstrated that the activation of the Ca-dependent transcription factor NFAT is directly regulated by IICR in cardiac myocytes.28
Adding to the complexity of Ca signaling during ECC in cardiac cells are mitochondria, intracellular organelles that occupy approximately one third of the volume of a cardiac myocyte24 and contribute to Ca signaling in multiple ways: as the major cellular ATP source mitochondria fuel or modulate Ca pumps, transporters and channels, they determine the redox environment of Ca handling proteins and are capable of sequestering Ca in significant amounts. While evidence for the Ca sequestering capacity of mitochondria is abundant, it has remained a matter of debate whether mitochondria are capable of shaping the cytosolic AP-dependent Ca signal on a beat-to-beat basis (for a summary of the controversy see refs.29,30).
In a recent study24 we investigated the spatio-temporal organization of SR Ca release during ECC and the role of IICR in atrial myocytes and their remodeling in heart failure. In this Addendum, we extend this study in 2 directions. 1) We investigated in details how the spatial organization of mitochondria affects the centripetal propagation of Ca release during ECC. 2) We present unique features of nuclear IICR in atrial myocytes that support the hypothesis of autonomous Ca signaling in the nuclear domain.
Results and Discussion
Propagation of atrial SR Ca release: t-tubules and mitochondria
Membrane staining with the fluorescent probe WGA-Alexa 594 revealed a regular dense t-tubular network in rabbit ventricular cells (Fig. 1A, a), whereas in atrial myocytes membrane staining was limited to the cell periphery consistent with the absence of t-tubules (Fig. 1B, a). The abundance of this network in ventricular cells allows for robust synchronous and homogenous SR Ca release with no decrement in amplitude from cell periphery to the center (Fig. 1A, b, Ac). In atrial cells however, the absence of t-tubules requires that central (CT) nj-SR Ca release sites are activated by propagating CICR that results in a delayed onset and CaTs of smaller amplitude in the cell center (Fig. 1B, b, Bc). The significance of the t-tubular system for the rapid and synchronous Ca release in ventricular myocytes is underscored by the observation that CaTs in chemically (formamide) detubulated ventricular cells (Fig. 1C, a) show a remarkable resemblance to atrial CaTs with the typical ‘U-shaped’ appearance of the CaT in the confocal line scan image (Fig. 1C, b) and the reduced amplitude in the cell center (Fig. 1C, c). Interestingly, however, despite the spatially less homogenous CaT in atrial cells, the contraction strength (measured as cell shortening or change in sarcomere length; summarized in Figs. 1D, E) of ventricular and atrial myocytes were very similar (average cell shortening during a twitch: 23.2 ± 1.6 µm in ventricular (Fig. 1A, d; n = 11) vs. 20.9 ± 3.5 in atrial (Fig. 1B, d; n = 14) myocytes) whereas detubulation of ventricular cells greatly impaired contractility (Fig. 1C, d; average twitch cell shortening: 5.1 ± 3.9 µm, n = 12 cells, P < 0.05 vs. ventricular or atrial cells). The data indicate that the mere absence of t-tubules should have profound deleterious effects on contractile function which is obviously not the case in atrial cells. Thus, atrial cells appear to have developed structural and functional features that overcome the potential disadvantages of an absent t-tubule system and allow for efficient ECC.
Figure 1.

AP-induced CaTs and cell shortening in intact ventricular, atrial and detubulated ventricular cells. (A, a) membrane and t-tubule staining with WGA-Alexa 594 in a rabbit ventricular myocyte. (b) line scan image of an AP-induced CaT. The confocal line scan image was recorded along the line shown in (a). (c) subcellular CaTs recorded from 1 µm wide susbsarcolemmal (gray; SS in panel b) and central (black; CT in panel b) regions. (d) cell shortening. Arrow heads indicate time of electrical field stimulation. (B, C) same type of data recorded in an atrial (B) and detubulated ventricular (C) myocyte. Detubulation occurred by exposure to 1.5 M formamide. (D, E) average twitch-induced cell shortening (D) and change in sarcomere length (E) in intact ventricular, atrial and detubulated ventricular myocytes. (F) analysis of subcellular synchrony of AP-induced Ca release. (a, b and c) graph of time elapsed between onset of electrical stimulation and the time the local CaT has reached half-maximal amplitude (TF50) along the transverse cell axis in an intact ventricular (a), atrial (b) and detubulated ventricular (c) myocyte. (d) average standard deviation of all local TF50 values (SDTF50) at single pixel resolution across the transverse cell axis for intact ventricular, atrial and detubulated ventricular myocytes. Square brackets indicate significant differences at P < 0.05, one-way ANOVA. Numbers in columns indicate number of cells.
The major determinants of cellular contractility are synchrony and homogeneity of Ca release and reuptake.31,32 Synchrony or dyssynchrony of release refers to the temporal dispersion of Ca signals from individual CRUs within a myocyte that determines the degree of cell-wide homogeneity of the CaT induced by an AP. Synchrony data of Ca release was derived from confocal transverse line scan images by calculating the time elapsed between onset of stimulation and the time the local CaT has reached half-maximal amplitude (TF50) along the transverse cell axis. Ventricular cells show a highly synchronous and homogenous Ca release (Fig. 1F, a) which is reflected in the small local TF50 values and the narrow range in which TF50 varies across the cell. Fig. 1F, d quantifies the degree of synchrony by calculating the standard deviation of all local TF50 values (SDTF50) at single pixel resolution across the cell. SDTF50 is extremely low in ventricular cells and is due to the intact t-tubular system that assures essentially simultaneous activation of all CRUs. As expected for a cell with no t-tubules the spread of TF50 values in atrial cells is clearly larger (Fig. 1F, b), however the SDTF50 value is remarkably small and only 2–3 times larger than in ventricular cells (Fig. 1F, d). This is in stark contrast to detubulated ventricular cells that show a highly irregular TF50 profile across the cell and a massively increased SDTF50 value indicative of a high degree of dyssynchrony and spatio-temporal inhomogeneity of Ca release (Fig. 1F, c). The data shown in Fig. 1F suggest that propagating CICR in atrial cells is a highly organized process resulting in activation of nj-SR CRUs with high reliability and fidelity, whereas artificially detubulated cells contain areas with early release (possibly still associated with rudimentary remnants of the t-tubule network) adjacent to areas with very late Ca release that together lead to a patchy pattern of dyssynchronous and inhomogeneous Ca release and consequently poor development of contractile force (Fig. 1E).
To achieve homogenous and efficient centripetal propagation of CICR in atrial cells the transition of activation from the peripheral j-SR to the first array of nj-SR CRUs is critical and the underlying mechanism is controversial. It has been proposed that in rat atrial myocytes mitochondria located in this transition zone can prevent Ca propagation into deeper areas of the cell by sequestering Ca (‘mitochondrial firewall’12,16). Here, we challenge such notion based on new experimental data. The mitochondria-targeted fluorescent probe Mitycam24,33 was used to visualize mitochondria. As shown in Fig. 2A, a, atrial cells show 2 populations of mitochondria. The first appears to be a single row of mitochondria immediately adjacent to the sarcolemma, whereas the second population encompasses the bulk of cardiac mitochondria, is more centrally located, and has a spatial organization with a sarcomeric pattern. The two populations are separated by a transition zone (TZ) of approximately 1 µm width (1.02 ± 0.06 µm; n = 23 cells) that is devoid of mitochondria and not present in ventricular cells (Fig. 2A, b). We measured the centripetal propagation velocity of an AP-induced CaT in different subcellular regions and cell types. Propagation velocity was derived from TF50 analysis (Fig. 2B). The propagation velocity (Fig. 2C) was fastest in the TZ of atrial myocytes (171 ± 9 µm/s, n = 19 cells) and much faster than in the central cytosol of atrial myocytes (112 ± 6 µm/s; n = 19), in detubulated ventricular myocytes (115 ± 17 µm/s; n = 8) and than spontaneous propagating Ca waves in ventricular cells (97 ± 4 µm/s; n = 11). The comparable propagation velocities of Ca waves, AP-induced release from atrial nj-SR and in detubulated ventricular cells can be explained by the fact that in all 3 conditions activation propagates by the same reaction (CICR)-diffusion process, however the propagation mechanism across the TZ appears different.
Figure 2.
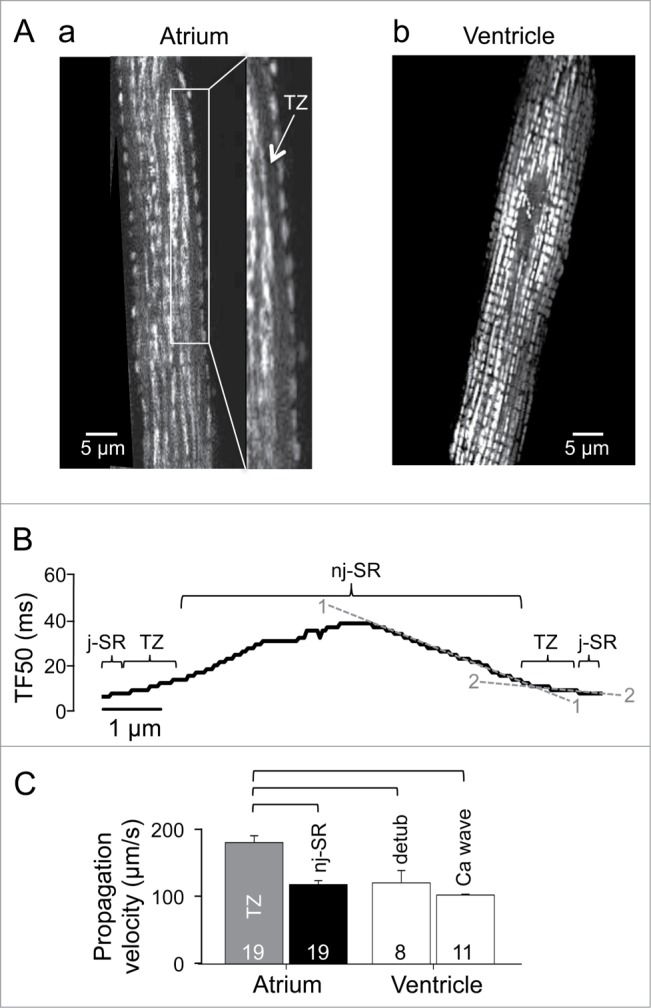
Mitochondria and Ca propagation. (A) mitochondrial distribution in atrial (a) and ventricular (b) myocytes visualized with the mitochondria targeted fluorescent probe Mitycam. TZ, transition zone. (B) TF50 profile across an atrial myocyte. The dashed lines represent the slope of the TF50 curve in the j-SR/TZ (2—2) and the nj-SR (1—1) space. The slopes represent 1/velocity of propagation of Ca release from the cell periphery to the cell center, thus a shallow slope indicates fast propagation and vice versa. (C) average propagation velocities of AP-induced Ca release in atrial (TZ vs nj-SR), detubulated ventricular myocytes and spontaneous Ca waves in ventricular myocytes. Square brackets indicate significant differences at P < 0.05, one-way ANOVA.
To explore the possibility that the lack of mitochondrial Ca uptake in the TZ is crucial for the fast and robust propagation of the subsarcolemmal Ca signal toward the nj-SR, we used Ru360, a rather specific inhibitor of the mitochondrial Ca uniporter (MCU).34 As shown in Fig. 3A, Ru360 slowed mitochondrial Ca uptake by nearly 2 orders of magnitude (time-to-peak: 0.7 ± 0.1 s in control (n = 21) vs 32 ± 2 s in Ru360 (n = 18); P < 0.05) such that mitochondrial Ca uptake on a time scale of AP-induced CaTs was essentially blocked. Mitochondrial Ca uptake was measured by exposing membrane-permeabilized myocytes expressing the mitochondrially targeted Ca-sensor Mitycam from Ca-free conditions to 5 µM Ca, a [Ca] that is close to extramitochondrial [Ca] required for half-maximal activation of MCU.35 In the presence of Ru360 overall CICR propagation in atrial myocytes was significantly slowed (Fig. 3B), however, j-SR and nj-SR Ca release were affected differently. While the propagation from j-SR across the TZ remained fast and unaltered in the presence of Ru360, propagation through the nj-SR region was significantly accelerated when mitochondrial Ca uptake was blocked (propagation velocities: 112 ± 6 µm/s in control (n = 15) vs. 142 ± 6 µm/s with Ru360 (n = 18)). Furthermore, in the presence of Ru360 subsarcolemmal CaTs were not significantly changed, but central CaTs reflecting release from nj-SR were significantly larger in amplitude (Fig. 3C). The data are consistent with the notion that the lack of mitochondria in the TZ assures a rapidly propagating Ca trigger signal that is high enough in amplitude to initiate CICR from nj-SR and that does not get compromised by mitochondrial Ca sequestration. This notion is further corroborated by the observation that FCCP, a protonophore that depolarizes mitochondria, impairs mitochondrial Ca uptake and has profound effects on Ca release and ECC36 also increased centripetal Ca propagation in cellular regions containing nj-SR (Fig. 3B; 316 ± 38 µm/s; n = 4 cells).
Figure 3.

Mitochondrial effects on CaTs. (A, a) effect of rapidly increasing [Ca]i from 0 to 5 μM on mitochondrial Ca uptake in permeabilized atrial cells under control conditions and in the presence of the MCU inhibitor Ru360 (10 µM). [Ca]mito was measured with the mitochondria-targeted fluorescent probe Mitycam. (b) Average time-to-peak of the [Ca]mito signal after elevation of extramitochondrial [Ca] to 5 µM in control and in the presence of Ru360. (B, a) TF50 profile along the transverse cell axis in control and in the presence of Ru360. (b) Average Ca propagation velocities in the TZ and central cell regions occupied by nj-SR in control, and in the presence of Ru360 and FCCP (5 µM), respectively. (C) line scan images (top) and subcellular (SS, CT) CaTs (bottom) induced by electrical field stimulation in control and in the presence of Ru360. Arrow heads indicate electrical stimulation. (D) average normalized SS and CT CaT amplitudes in the presence of Ru360. Dashed line (100%) indicates normalized SS and CT CaT amplitudes under control conditions. Numbers in columns indicate number of cells. Square brackets indicate significant differences at P < 0.05, one-way ANOVA. *P < 0.05, student´s t-test.
Our results shed new light on the controversy whether mitochondria participate in shaping the CaT during ECC. We show that inhibition of the MCU increases the velocity of centripetal Ca propagation and enhances the amplitude of CaTs that arise from release from nj-SR. The observation of inhibition of mitochondrial Ca uptake to influence CaTs is not unprecedented. In atrial cells inhibition of mitochondrial function with oligomycin and antimycin enhanced cytosolic Ca release,12 and similar effects on CaTs and Ca propagation were demonstrated in ventricular myocytes.37 In summary, we show that atrial Ca signaling during ECC fundamentally differs from ventricular cells. A set of special functional and structural features (i.e., the lack of mitochondria in the TZ and highly organized centripetal propagation of CICR with little indication of local dyssynchrony of release) provide robust and fast nj-SR Ca release and allow for an efficient contraction of atrial cells despite their lack of t-tubules. In atrial cells j-SR Ca release is primarily designed to assure initiation of propagating CICR with high fidelity, whereas Ca release from nj-SR has an extended dynamic range of regulation indicative of a broad inotropic reserve to respond to contractile and hemodynamic demands. As we have shown in our previous work,24 in early stage left-ventricular heart failure, Ca release from nj-SR is indeed enhanced in atrial cells from these failing hearts. We proposed that this remodeling of atrial Ca release served the purpose of enhancing atrial contractility, improve ventricular filling (increased atrial kick) and thereby preserve cardiac output.
IICR in atrial cells: evidence for autonomous nuclear Ca signaling
Numerous effects of IICR in atrial myocytes have been reported including positive inotropic and proarrhythmic effects and regulation of Ca-dependent transcription factors. Fig. 4 shows the effect of elevating cytosolic IP3 levels on CaTs in an atrial myocyte. Photolytical release of IP3 (caged IP3, cf. Methods) leads to global whole-cell increase of the amplitude of electrically elicited CaTs but with interesting subcellular differences (Fig. 4B). While under control conditions SS CaTs exceeded CT and nuclear CaTs in amplitude, after IP3 uncaging the largest CaTs were observed in the nuclear region, whereas SS and CT CaTs became nearly equal in amplitude. Thus, the data suggest that the nuclear compartment is particularly susceptible to IICR. We therefore further explored nuclear IICR specifically.
Figure 4.
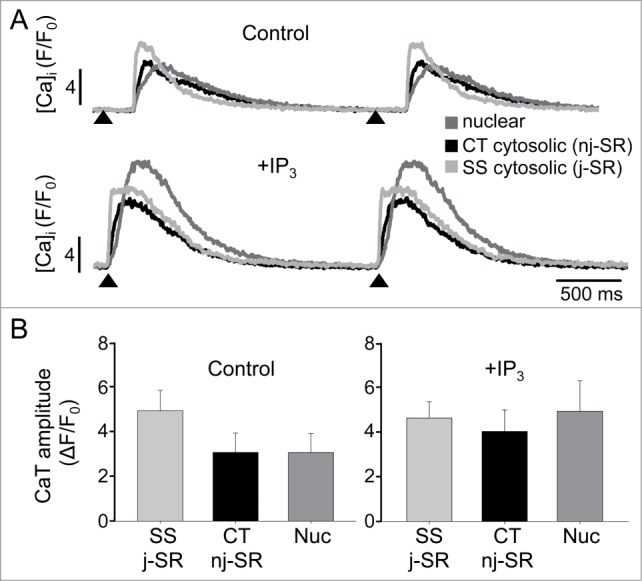
IP3-dependent effects on subcellular CaTs. (A) AP-induced subcellular CaTs under control conditions (top) and after photolysis of caged IP3 (bottom) recorded from 1 µm wide regions of interest. The traces represent subsarcolemmal (SS) cytosolic, central (CT) cytosolic and nuclear changes of [Ca]i. IP3 was uncaged by illumination of the entire cell with 405 nm laser light (2 ms). Arrow heads indicate electrical stimulation. (B) Summary data of averaged subcellular CaT amplitudes under control conditions and after IP3 photolysis (n = 4).
First, we used the uncaging technique to elevate [IP3] in the nuclear compartment (Fig. 5A). Local elevation of [IP3] in the nucleus elicited ‘mini’-Ca waves that were restricted to the nucleus and did not propagate into the neighboring cytosol, however a slow and very gradual increase in nuclear and cytosolic [Ca] was observed. The nuclear ‘mini’-Ca waves occurred at a frequency of 0.16 ± 0.01 waves/s whereas at the same time no waves were observed in the cytosolic region of interest immediately adjacent to the nucleus. When the same experiment was conducted in the presence of Ru360 to inhibit mitochondrial Ca sequestration cell-wide, nuclear IP3 uncaging, with a delay, also triggered Ca waves that originated in the cytosol, presumably by [Ca]i reaching the threshold for triggering spontaneous Ca release (data not shown).
Figure 5.
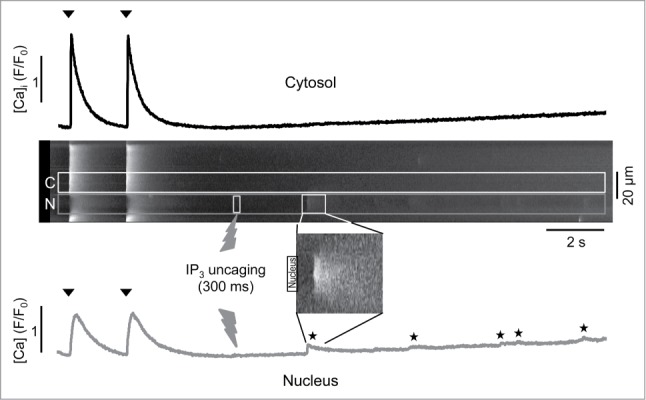
Differential effects of IP3 on cytosolic and nuclear [Ca]. From top to bottom: cytosolic CaT recorded from a cytosolic (C) region immediately adjacent to the nucleus (N), longitudinal line scan image with C and N regions of interest marked and time, duration and location where IP3 uncaging occurred (flash symbol), image of a ‘mini’-Ca wave that was restricted to the nuclear compartment at higher magnification and intensity setting, and nuclear CaT. Arrow heads indicate electrical stimulation. Nuclear ‘mini’-Ca waves are marked by stars.
The observation of spatially restricted nuclear ‘mini’-Ca waves raised the intriguing possibility that nuclear IICR can occur independently from cytosolic IP3-Ca signaling. Therefore, we set out to detect elementary IICR events termed Ca puffs24,38,39 in the nucleus. For this purpose we exposed membrane permeabilized atrial myocytes first to tetracaine (4 mM) to eliminate any RyR-mediated Ca release events (Ca sparks). Subsequently, cells were exposed to IP3 (5 µM) to elicit Ca puffs. The origin of Ca puffs from IP3Rs was confirmed by the application of the IP3R blocker 2-aminoethoxydiphenyl borate (2-APB; 10 µM) which abolished puff activity. Fig. 6A shows confocal line scan images of elementary Ca release events (Ca sparks and Ca puffs) in control conditions, after addition of tetracaine, and after exposure to IP3, which significantly enhanced the frequency of elementary release events, presumably from IP3Rs (Ca puffs) since RyR-mediated release was blocked. In the presence of tetracaine and IP3 Ca puff activity was about twice as high in the nucleus compared to the central cytosol (Fig. 6B: 0.7 ± 0.4 Ca puffs s−1 * (100 µm)−1 in the nucleus vs. 0.4 ± 0.2 Ca puffs s−1 * (100 µm)−1s in the central cytosolic region). Fig. 6C summarizes average frequencies of elementary Ca release events observed in the nuclear compartment under control conditions, after addition of tetracaine to eliminate RyR activity, followed by stimulation of IICR with IP3 and finally inhibition of IICR with 2-APB.
Figure 6.

Ca puff activity. (A) protocol to isolate IICR-mediated Ca puffs in atrial myocytes. Longitudinal line scan images from a permeabilized atrial myocyte under control (CTRL) conditions (a), after application of 4 mM tetracaine (b), tetracaine + IP3 (5 μM) (c), and tetracaine +IP3 + 2-APB (10 μM) (d). (B) Ca puff frequency in the presence of teracaine + IP3 in the cytosol vs. nuclear compartment. (C) average frequencies of nuclear elementary Ca release events under control conditions, and in the presence of tetracaine, tetracaine + IP3, and tetracaine + IP3 + 2-APB. (n = 15 cells). (D) 2-D (x-y, left) and line scan (x-t, right) confocal image of the nuclear and perinuclear regions of an atrial myocytes loaded with X-rhod-1/AM. The line in the x-y image marks the position of the scan line. The horizontal black lines in the x-t image indicate the nuclear envelope (NE) and membrane structures of the nucleoplasmic reticulum (NPR). (E) simultaneously recorded line scan images of X-rhod-1 (left) and fluo-4 (middle) fluorescence signals from a permeabilized atrial myocyte in the presence of tetracaine + IP3. Right: F/F0 image of a Ca puff originating from the nuclear envelope. F0 represents resting cellular Fluo-4 fluorescence measured at the beginning of the recording. (F) Average frequencies of nuclear Ca puffs measured close to (<1 µm) and distant (>1 µm) from nuclear membrane structures (n = 15 cells).
It has been proposed that nuclear IP3Rs associate not only with the nuclear envelope but also with the nucleoplasmic reticulum,40,41 a branched network of invaginations and protrusions that extends the nuclear envelope into the nucleoplasm.42 We therefore investigated whether nuclear Ca puffs were associated with nuclear membrane structures. For this purpose permeabilized cells were also loaded with a membrane-permeable high-affinity Ca dye (X-rhod-1/AM) with distinctly different spectral properties than fluo-4 that compartmentalizes into the SR and nuclear envelope and creates a bright fluorescence signal from these compartments since the dye was fully saturated with Ca given the sub-millimolar [Ca] within the SR (Fig. 6D). With this approach Ca puffs could be co-localized with the reticular network structures that extend the nuclear envelope Ca store into the nucleoplasm (Fig. 6E). Ca puffs occurred with a nearly four-fold higher frequency (Fig. 6F) in close vicinity to nuclear envelope extensions and invaginations or the nuclear envelope itself (1.8 ± 0.8 Ca puffs s−1 * (100 µm)−1 in <1 µm distance from nuclear membranes vs. 0.5 ± 0.4 Ca puffs s−1 * (100 µm)−1 in >1 µm distance). Considering the image formation by a confocal microscope the calculated frequency of non-membrane Ca puffs might be underestimated. Ca puffs can originate from membrane locations outside the focal plane but by no more than ∼1 µm in the axial dimension. In such case membrane structure from where the Ca signal originates would be poorly resolved or even escape detection, whereas a Ca puff originating from such a site is likely to be detected because of the diffusional spread of Ca into the focal plane.
The nucleoplasm represents a distinct subcellular domain with specific tasks including DNA and RNA signaling, nuclear receptor-mediated signal transduction and gene transcription. It has become increasingly clear that nuclear Ca signaling controls important processes such as cell differentiation, growth, survival and death (for recent reviews on nuclear Ca signaling see references20,43,44). Nuclear Ca is determined by passive Ca fluxes (cf. Fig. 4A) through the nuclear pore complex, however there is growing evidence that the nucleus is also equipped for active Ca release from the nuclear envelope and the nucleoplasmic reticulum Ca store.40,41 This raises the intriguing question whether nuclear Ca signaling is, at least in part, autonomously regulated and the nucleus constitutes a distinct and independent Ca signaling domain.45,46 We have shown previously that isolated cardiac nuclei are capable of Ca release via RyRs as well as IP3Rs.27 Both receptors are localized to both the inner and outer leaflet of the nuclear envelope such that release can be directed into the nucleoplasm as well as the cytosol. At variance with the SR in cardiac myocytes where RyRs outnumber IP3Rs by 1–2 orders of magnitude,21,47 nuclear Ca release is dominated by IICR. Here we present additional evidence for an autonomous nuclear Ca signaling domain. Well-defined IICR release signals in form of ‘mini’-Ca waves can be elicited in and stay confined to the nuclear compartment (see also27,45). Furthermore, we demonstrate for the first time in cardiac myocytes nuclear elementary IICR events in form of Ca puffs that are typically associated with membrane structures of the nucleoplasmic reticulum and the nuclear envelope. This is direct evidence that in cardiac myocytes a fully functional nuclear IICR machinery is present.
Methods
Myocyte isolation and cell culture
Left atrial and ventricular myocytes were isolated from New Zealand White rabbits (Harlan Laboratories, Indianapolis, IN, USA). Rabbits were anesthetized with pentobarbital sodium (100 mg/kg). Excised hearts were retrogradely perfused with nominally Ca-free Tyrode solution for 5 min followed by minimal essential medium Eagle (MEM) solution containing 20 µM Ca and 22.5 µg/ml Liberase Blendzyme TH (Roche Applied Science, Indianapolis, IN) for 20 min at 37°C. The left atria and left ventricle were removed from the heart and digested for an additional 5 min in the enzyme solution at 37°C. The tissue was minced, filtered, and washed in MEM solution containing 50 µM Ca and 10 mg/ml BSA, and kept in MEM solution with 50 µM Ca at room temperature (20–24°C) until experimentation. All animal protocols and procedures were approved by the Institutional Animal Care and Use Committee of Rush University Chicago, and comply with US regulations on animal experimentation.
For mitochondrial [Ca] ([Ca]mito) measurements with the mitochondrially targeted inverse pericam Ca biosensor Mitycam (excitation 488 nm, emission 530 ± 15 nm)24,33 atrial myocytes were kept in short-term (up to 48 hrs) culture in Cornig Cellgro Medium 199 (VWR, USA) containing penicillin and streptomycin (50 µg/ml). Mitycam was expressed by adenoviral gene transfer in isolated atrial cells (multiplicity of infection of 500).
Solutions and experimental conditions
All chemicals and reagents were obtained from Sigma-Aldrich (St Louis, MO, USA), unless noted otherwise. Tyrode solution contained (in mM): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 D-glucose, 10 Hepes; pH 7.4 with NaOH. For experiments on permeabilized myocytes, saponin (0.01%) was added to an ‘internal solution’ containing (in mM): 135 KCl, 5 NaCl, 20 HEPES, 5 pyruvate, 2 glutamic acid, 2 malic acid, 0.5 KH2PO4, 1 MgCl2, 5 EGTA. Free [Ca]i of this solution was zero (sub-nanomolar) or 5 µM (as determined with Maxchelator). Cells were plated on laminin-coated glass coverslips and transferred to 2 mM Ca Tyrode for experimentation. To prevent cell movement during confocal imaging, Blebbistatin (10 µM, Tocris Bioscience, Bristol, UK) or BDM (15 mM) were added to all Tyrode based solutions. Mitochondrial Ca uptake was inhibited with Ru360 (10 µM for 3 minutes; EMD Chemicals, USA) or FCCP (5 µM for 12 minutes). Experiments were conducted at room temperature (20–24°C).
Detubulation of ventricular mycoytes and assessment of t-tubular structure
Rabbit ventricular myocytes were detubulated by adding Formamide (1.5 M) to the cell suspension for 15 minutes. Detubulation48 was achieved by osmotic shock resulting from a subsequent 15 minute washing step in Tyrode solution.
For visualization of t-tubules and surface membranes cells were loaded with WGA-Alexa 594 (1 mg/ml; Molecular Probes/Life Technologies, Grand Island, NY, USA) for 35 minutes at 37°C. WGA-Alexa 594 fluorescence was excited with the 543 nm line of a HeNe laser and emission was measured at >600 nm.
[Ca]i measurements and IICR studies
Fluo-4 was used for confocal [Ca]i measurements with a Nikon A1R laser scanning microscope. Intact cells were loaded with the Ca indicator Fluo-4/AM (5 µM; Molecular Probes/Life Technologies; excitation at 488 nm, emission at 515 nm) for 20 min. Confocal [Ca]i measurements were acquired from intact myocytes stimulated at 0.5 Hz in line scan mode (512 lines s−1). The scan line was placed along the transverse axis of the cell with a pixel size of 0.02 - 0.12 μm. Action potentials and global CaTs were elicited by electrical field stimulation of intact atrial myocytes using a pair of platinum electrodes (voltage set at ∼50% above the threshold for contraction). Changes in [Ca]i in intact paced myocytes are expressed a F/F0 where F represents cellular Fluo-4 fluorescence and F0 is diastolic Fluo-4 fluorescence.
For nuclear Ca puff experiments in permeabilized cells Fluo-4 pentapotassium salt (50 µM; Molecular Probes/Life Technologies) was added to the internal solution and cells were previously loaded with X-rhod-1/AM (Molecular Probes/Life Technologies) to visualize the nuclear envelope and nucleoplasmic reticulum. X-rhod-1 fluorescence was excited at 543 nm, and emission was measured at >600 nm. To stimulate IICR and Ca puffs D-myo-inositol 1,4,5-trisphosphate hexapotassium salt (5 µM Tocris Bioscience, Bristol, UK) was added to the internal solution.
Elevation of [IP3]i in intact myocytes was achieved by photolysis of caged IP3 (cag-IP3 PM; SiChem, Bremen, Germany). Cells were loaded with 100 nM cag-IP3 for 20 min, and liberation of biologically active IP3 was achieved by 405 nm laser illumination in user-defined cellular regions using the Mosaic illumination tool integrated in the Nikon A1R confocal imaging system.49
Cell shortening
Cell contractility was measured as changes in cell length by video-edge detection50 or as sarcomere shortening using an IonOptix MyoCam-S video camera and the IonWizard data acquisition and analysis software (Ionoptix, Milton, MA, USA.)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health grants HL62231, HL80101 and HL101235 and the Leducq Foundation (to LAB).
References
- 1.Huser J, Lipsius SL, Blatter LA.. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol 1996; 494 (Pt 3):641-51; PMID:8865063; http://dx.doi.org/ 10.1113/jphysiol.1996.sp021521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cordeiro JM, Spitzer KW, Giles WR, Ershler PE, Cannell MB, Bridge JH.. Location of the initiation site of calcium transients and sparks in rabbit heart Purkinje cells. J Physiol 2001; 531:301-14; PMID:11310434; http://dx.doi.org/ 10.1111/j.1469-7793.2001.0301i.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackenzie L, Bootman MD, Berridge MJ, Lipp P.. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol 2001; 530:417-29; PMID:11158273; http://dx.doi.org/ 10.1111/j.1469-7793.2001.0417k.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyrnias I, Mair W, Harzheim D, Walker SA, Roderick HL, Bootman MD.. Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium 2010; 47:210-23; PMID:20106523; http://dx.doi.org/ 10.1016/j.ceca.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 5.Bootman MD, Higazi DR, Coombes S, Roderick HL.. Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J Cell Sci 2006; 119:3915-25; PMID:16988026; http://dx.doi.org/ 10.1242/jcs.03223 [DOI] [PubMed] [Google Scholar]
- 6.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM.. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J pPysiol Heart Circ Physiol 2011; 301:H1996-2005; PMID:21841013; http://dx.doi.org/ 10.1152/ajpheart.00284.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, Blatter LA.. Activation and propagation of Ca(2+) release during excitation-contraction coupling in atrial myocytes. Biophys J 2001; 81:2590-605; PMID:11606273; http://dx.doi.org/ 10.1016/S0006-3495(01)75903-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNutt NS, Fawcett DW.. The ultrastructure of the cat myocardium. II. Atrial muscle. J Cell Biol 1969; 42:46-67; PMID:5786989; http://dx.doi.org/ 10.1083/jcb.42.1.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franzini-Armstrong C, Jorgensen AO.. Structure and development of E-C coupling units in skeletal muscle. Annu Rev Physiol 1994; 56:509-34; PMID:8010750; http://dx.doi.org/ 10.1146/annurev.ph.56.030194.002453 [DOI] [PubMed] [Google Scholar]
- 10.Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, Ríos E.. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol 1999; 113:469-89; PMID:10051521; http://dx.doi.org/ 10.1085/jgp.113.3.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scriven DR, Asghari P, Moore ED.. Microarchitecture of the dyad. Cardiovasc Res 2013; 98:169-76; PMID:23400762; http://dx.doi.org/ 10.1093/cvr/cvt025 [DOI] [PubMed] [Google Scholar]
- 12.Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD.. The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci 2004; 117:6327-37; PMID:15561771; http://dx.doi.org/ 10.1242/jcs.01559 [DOI] [PubMed] [Google Scholar]
- 13.Woo SH, Cleemann L, Morad M.. Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes. Circ Res 2003; 92:e1-11; PMID:12522129; http://dx.doi.org/ 10.1161/01.RES.0000051887.97625.07 [DOI] [PubMed] [Google Scholar]
- 14.Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ Jr, Vaghy PL, Meissner G, Ferguson DG.. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J Cell Biol 1995; 129:672-82; PMID:7730403; http://dx.doi.org/ 10.1083/jcb.129.3.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shkryl VM, Blatter LA.. Ca(2+) release events in cardiac myocytes up close: insights from fast confocal imaging. PloS One 2013; 8:e61525; PMID:23637847; http://dx.doi.org/ 10.1371/journal.pone.0061525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bootman MD, Smyrnias I, Thul R, Coombes S, Roderick HL.. Atrial cardiomyocyte calcium signalling. Biochimica et Biophysica Acta 2011; 1813:922-34; PMID:21295621; http://dx.doi.org/ 10.1016/j.bbamcr.2011.01.030 [DOI] [PubMed] [Google Scholar]
- 17.Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J, Lipsius SL.. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol 2003; 546:19-31; PMID:12509476; http://dx.doi.org/ 10.1113/jphysiol.2002.025239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keizer J, Smith GD, Ponce-Dawson S, Pearson JE.. Saltatory propagation of Ca2+ waves by Ca2+ sparks. Biophys J 1998; 75:595-600; PMID:9675162; http://dx.doi.org/ 10.1016/S0006-3495(98)77550-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA.. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol 2008; 294:H596-604; PMID:18055509; http://dx.doi.org/ 10.1152/ajpheart.01155.2007 [DOI] [PubMed] [Google Scholar]
- 20.Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD.. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol 2008; 45:128-47; PMID:18603259; http://dx.doi.org/ 10.1016/j.yjmcc.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM.. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005; 97:1314-22; PMID:16269653; http://dx.doi.org/ 10.1161/01.RES.0000194329.41863.89 [DOI] [PubMed] [Google Scholar]
- 22.Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR.. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest 1995; 95:888-94; PMID:7860772; http://dx.doi.org/ 10.1172/JCI117739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi H, Sano H, Iizuka K, Okada H, Kudo T, Kageyama K, Muramoto S, Murakami T, Okamoto H, Mochizuki N, et al.. Phosphatidylinositol metabolism in hypertrophic rat heart. Circ Res 1993; 72:966-72; PMID:8477530; http://dx.doi.org/ 10.1161/01.RES.72.5.966 [DOI] [PubMed] [Google Scholar]
- 24.Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, Lonchyna VA, Blatter LA.. Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. J Physiol 2015; 593:1459-77; PMID:25416623; http://dx.doi.org/ 10.1113/jphysiol.2014.283226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zima AV, Blatter LA.. Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias. J Physiol 2004; 555:607-15; PMID:14754996; http://dx.doi.org/ 10.1113/jphysiol.2003.058529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Zima AV, Sheikh F, Blatter LA, Chen J.. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res 2005; 96:1274-81; PMID:15933266; http://dx.doi.org/ 10.1161/01.RES.0000172556.05576.4c [DOI] [PubMed] [Google Scholar]
- 27.Zima AV, Bare DJ, Mignery GA, Blatter LA.. IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J Physiol 2007; 584:601-11; PMID:17761776; http://dx.doi.org/ 10.1113/jphysiol.2007.140731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rinne A, Blatter LA.. Activation of NFATc1 is directly mediated by IP3 in adult cardiac myocytes. Am J Physiol 2010; 299:H1701-H7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Rourke B, Blatter LA.. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol 2009; 46:767-74; PMID:19162034; http://dx.doi.org/ 10.1016/j.yjmcc.2008.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dedkova EN, Blatter LA.. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol 2013; 58:125-33; PMID:23306007; http://dx.doi.org/ 10.1016/j.yjmcc.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinzel FR, MacQuaide N, Biesmans L, Sipido K.. Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J Mol Cell Cardiol 2011; 50:390-400; PMID:21075114; http://dx.doi.org/ 10.1016/j.yjmcc.2010.11.008 [DOI] [PubMed] [Google Scholar]
- 32.Hohendanner F, Ljubojevic S, MacQuaide N, Sacherer M, Sedej S, Biesmans L, Wakula P, Platzer D, Sokolow S, Herchuelz A, et al.. Intracellular dyssynchrony of diastolic cytosolic [Ca(2)(+)] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ Res 2013; 113:527-38; PMID:23825358; http://dx.doi.org/ 10.1161/CIRCRESAHA.113.300895 [DOI] [PubMed] [Google Scholar]
- 33.Kettlewell S, Cabrero P, Nicklin SA, Dow JA, Davies S, Smith GL.. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J Mol Cell Cardiol 2009; 46:891-901; PMID:19249308; http://dx.doi.org/ 10.1016/j.yjmcc.2009.02.016 [DOI] [PubMed] [Google Scholar]
- 34.Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, et al.. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem 1998; 273:10223-31; PMID:9553073; http://dx.doi.org/ 10.1074/jbc.273.17.10223 [DOI] [PubMed] [Google Scholar]
- 35.Sedova M, Dedkova EN, Blatter LA.. Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am J Physiol Cell Physiol 2006; 291:C840-50; PMID:16723510; http://dx.doi.org/ 10.1152/ajpcell.00619.2005 [DOI] [PubMed] [Google Scholar]
- 36.Zima AV, Pabbidi MR, Lipsius SL, Blatter LA.. Effects of mitochondrial uncoupling on Ca(2+) signaling during excitation-contraction coupling in atrial myocytes. Am J Physiol Heart Circ Physiol 2013; 304:H983-93; PMID:23376829; http://dx.doi.org/ 10.1152/ajpheart.00932.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seguchi H, Ritter M, Shizukuishi M, Ishida H, Chokoh G, Nakazawa H, Spitzer KW, Barry WH.. Propagation of Ca2+ release in cardiac myocytes: role of mitochondria. Cell Calcium 2005; 38:1-9; PMID:15993240; http://dx.doi.org/ 10.1016/j.ceca.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 38.Yao Y, Choi J, Parker I.. Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J Physiol 1995; 482 (Pt 3):533-53; PMID:7738847; http://dx.doi.org/ 10.1113/jphysiol.1995.sp020538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huser J, Blatter LA.. Elementary events of agonist-induced Ca2+ release in vascular endothelial cells. Am J Physiol 1997; 273:C1775-82; PMID:9374666 [DOI] [PubMed] [Google Scholar]
- 40.Echevarria W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH.. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat Cell Biol 2003; 5:440-6; PMID:12717445; http://dx.doi.org/ 10.1038/ncb980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guatimosim S, Amaya MJ, Guerra MT, Aguiar CJ, Goes AM, Gomez-Viquez NL, Rodrigues MA, Gomes DA, Martins-Cruz J, Lederer WJ, et al.. Nuclear Ca2+ regulates cardiomyocyte function. Cell Calcium 2008; 44:230-42; PMID:18201761; http://dx.doi.org/ 10.1016/j.ceca.2007.11.016 [DOI] [PubMed] [Google Scholar]
- 42.Malhas A, Goulbourne C, Vaux DJ.. The nucleoplasmic reticulum: form and function. Trends Cell Biol 2011; 21:362-73; PMID:21514163; http://dx.doi.org/ 10.1016/j.tcb.2011.03.008 [DOI] [PubMed] [Google Scholar]
- 43.Bootman MD, Fearnley C, Smyrnias I, MacDonald F, Roderick HL.. An update on nuclear calcium signalling. J Cell Sci 2009; 122:2337-50; PMID:19571113; http://dx.doi.org/ 10.1242/jcs.028100 [DOI] [PubMed] [Google Scholar]
- 44.Hohendanner F, McCulloch AD, Blatter LA, Michailova AP.. Calcium and IP3 dynamics in cardiac myocytes - Experimental and computational perspectives and approaches. Front Pharmacol 2014; 5:35; PMID:24639654; http://dx.doi.org/ 10.3389/fphar.2014.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo D, Yang D, Lan X, Li K, Li X, Chen J, Zhang Y, Xiao RP, Han Q, Cheng H.. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium 2008; 43:165-74; PMID:17583790; http://dx.doi.org/ 10.1016/j.ceca.2007.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abrenica B, Gilchrist JS.. Nucleoplasmic Ca(2+)loading is regulated by mobilization of perinuclear Ca(2+). Cell Calcium 2000; 28:127-36; PMID:10970769; http://dx.doi.org/ 10.1054/ceca.2000.0137 [DOI] [PubMed] [Google Scholar]
- 47.Moschella MC, Marks AR.. Inositol 1,4,5-trisphosphate receptor expression in cardiac myocytes. J Cell Biol 1993; 120:1137-46; PMID:8382205; http://dx.doi.org/ 10.1083/jcb.120.5.1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brette F, Despa S, Bers DM, Orchard CH.. Spatiotemporal characteristics of SR Ca(2+) uptake and release in detubulated rat ventricular myocytes. J Mol Cell Cardiol 2005; 39:804-12; PMID:16198369; http://dx.doi.org/ 10.1016/j.yjmcc.2005.08.005 [DOI] [PubMed] [Google Scholar]
- 49.Shkryl VM, Maxwell JT, Blatter LA.. A novel method for spatially complex diffraction-limited photoactivation and photobleaching in living cells. J Physiol 2012; 590:1093-100; PMID:22183727; http://dx.doi.org/ 10.1113/jphysiol.2011.223446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dedkova EN, Seidlmayer LK, Blatter LA.. Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J Mol Cell Cardiol 2013; 59:41-54; PMID:23388837; http://dx.doi.org/ 10.1016/j.yjmcc.2013.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]