

Abstract
Ubiquitination is one of the most common post-translational modifications of proteins, and mediates regulated protein degradation among other cellular processes. A fundamental question regarding the mechanism of protein ubiquitination is whether and how ubiquitin affects the biophysical nature of the modified protein. For some systems, it was shown that the position of ubiquitin within the attachment site is quite flexible and ubiquitin does not specifically interact with its substrate. Nevertheless, it was revealed that polyubiquitination can decrease the thermal stability of the modified protein in a site-specific manner because of alterations of the thermodynamic properties of the folded and unfolded states. In this study, we used detailed atomistic simulations to focus on the molecular effects of ubiquitination on the native structure of the modified protein. As a model, we used Ubc7, which is an E2 enzyme whose in vivo ubiquitination process is well characterized and known to lead to degradation. We found that, despite the lack of specific direct interactions between the ubiquitin moiety and Ubc7, ubiquitination decreases the conformational flexibility of certain regions of the substrate Ubc7 protein, which reduces its entropy and thus destabilizes it. The strongest destabilizing effect was observed for systems in which Lys48-linked tetra-ubiquitin was attached to sites used for in vivo degradation. These results reveal how changes in the configurational entropy of the folded state may modulate the stability of the protein’s native state. Overall, our results imply that ubiquitination can modify the biophysical properties of the attached protein in the folded state and that, in some proteins, different ubiquitination sites will lead to different biophysical outcomes. We propose that this destabilizing effect of polyubiquitin on the substrate is linked to the functions carried out by the modification, and in particular, regulatory control of protein half-life through proteasomal degradation.
Keywords: molecular dynamics simulations, multidomain protein, ubiquitination, native state dynamics
Introduction
Ubiquitination is a common post-translational modification (PTM) of proteins that mediates many different cellular pathways.1,2 Protein ubiquitination occurs in several steps and results in the formation of an isopeptide bond between the C-terminal Gly of ubiquitin (Ub) and a Lys residue in the substrate. Furthermore, one of the seven Lys residues (and the N-terminal Met) of Ub can be used to form an isopeptide bond with an additional Ub molecule, which results in the formation of polymeric chains of Ub.1,3 All possible linkages of the seven lysines of Ub, including mixed ones, have been observed in vivo. The topology and cellular function of these Ub chains varies depending on the position of the Lys residues involved in Ub chain formation. For example, Lys48-linked poly-Ub chains target proteins for proteasomal degradation.4 Lys63-linked chains are known to be involved mostly in DNA repair, receptor activation, and other nonproteasomal pathways.5 Monoubiquitination (mono-Ub) usually mediates nonproteasomal pathways,6,7 but it was also shown that for small proteins it can lead to proteasomal degradation.8–13
The enormous variety of different functions performed by Ub is related, first of all, to the structural diversity of its chains as determined by the different linkage positions, which are recognized by different Ub receptors (containing one or more ubiquitin binding domains, UBDs). Specificity in UBD–Ub interactions is achieved by various mechanisms, including distinct affinities for specific linkages and conformational changes during UBD–Ub interactions.14–16 Mono-Ub itself can adopt slightly different conformations depending on the type of UBD it interacts with.17
In accordance with these observations, Ub is usually considered to serve as a tagging molecule that can be recognized by other proteins involved in a certain signal pathway. Understanding this correspondence between the lysine linkages of ubiquitination and their specific function could, in principle, enable us to decipher the “ubiquitin code”.18 However, ubiquitination may result in additional effects that originate from cross-talks between the Ub and the modified protein. To understand these possible effects it is important to investigate whether and how Ub interacts with the protein to which it is attached. Ub is covalently attached to its substrate, however the interface it forms with its substrate and its actual residence on the surface of the substrate can vary.19–22 Investigating these questions first requires in vivo identification of the ubiquitination sites of the protein of interest and the solved structure of the protein, preferably with the Ub moiety conjugated. However, very few ubiquitinated protein structures have been solved,23 despite marked progress in recent years in nonenzymatic (i.e., chemical synthesis) methods for ubiquitination.24–27 Most of the effort directed at producing ubiquitinated proteins for biochemical and structural studies has focused on mono-Ub. The structures of mono-Ub proteins have been solved for proliferating cell nuclear antigen (PCNA),21,28 Ras,29 and Josephin30 proteins. Currently, no structure of a poly-ubiquitinated protein is available, but progress in synthesizing isolated poly-Ub chains24,25,31 may soon allow homogenous preparation of these molecules for in vitro characterization.
Mono-ubiquitinated PCNA is the best biophysically characterized ubiquitinated protein. Several biophysical techniques revealed alternative positions for the mono-Ub moiety on the surface of the PCNA homotrimer that are distinct from the position identified in the crystal structure.19–22 The orientation of the Ub moiety is thus likely to be dynamic as it can adopt a variety of positions relative to the substrate. Modeling and NMR analyses indicate that, in mono-UbRas, Ub is also very dynamic.32 An analysis of the noncovalent interactions of the Ub moiety with the Ras surface did not show high affinity binding to any single site of the Ras protein. In the switch region of Ras, the Ub moiety can compete for the interface with GTPase-activating proteins (GAPs). An inability to bind GAP increases the population of the GTP-bound form of mono-UbRas in vivo, thus preventing its deactivation. These results suggest that the considerable flexibility of the attached Ub may affect substrate activity. Furthermore, ubiquitination of the Ras protein at the site that is used in vivo uniquely increases the population of the GTP-bound form of mono-UbRas.32
In addition to effects stemming from changes at the interface of the modified proteins (such as the Ras–GAP example), Ub may affect the intrinsic properties of the conjugate. Studies of cross-linked33 and multidomain proteins34–42 showed that thermodynamic stability and other folding characteristics can be strongly affected by the presence of the conjugate. Computational studies also suggested that ubiquitination may alter the intrinsic biophysical properties of the modified protein. This was illustrated for the Ubc7 protein (Fig. 1), for which it was shown that the conjugation site and the type of ubiquitination determine the precise biophysical effect.43 A pronounced modification of substrate protein characteristics was observed following ubiquitination with the Lys48-linked tetra-Ub chain, which is in good agreement with the fact that Lys48-linked chains target proteins for proteasomal degradation.44–48 The dramatic destabilization observed in ubiquitination of Ubc7 at specific sites was mostly attributed to changes in the unfolded state. Furthermore, it was recently shown that in vitro attachment of Ub reduces the thermodynamic stability of the modified protein (Ub C-terminal hydrolase-L3).49
Figure 1.
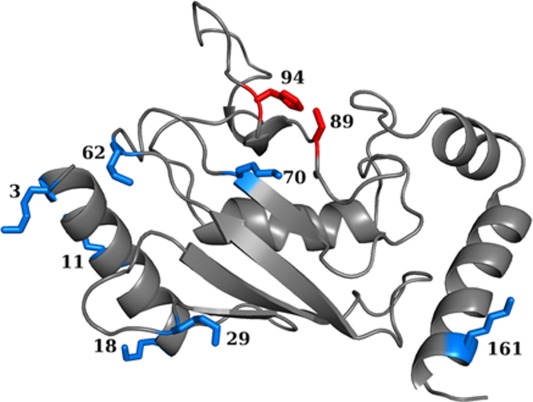
Ubiquitination sites on substrate protein Ubc7. Residues ubiquitinated in vivo (His94 and Cys89) are colored red. Other surface-exposed Lys residues capable of acting as ubiquitination sites are colored blue.
Several studies showed that modifying a protein substrate by a covalent linkage to another molecule may affect its biophysical characteristics. For example, protein glycosylation can have a stabilizing or destabilizing effect depending mostly on the location of the glycosylation sites.50,51 The complexity of the effect of conjugation is reflected by the different effects of glycosylation and PEGylation on protein stability and folding kinetics when they are conjugated to the same sites.52 Similarly to the effect of PTMs on protein biophysical properties, tethering two proteins via their termini may affect the stability of the original proteins. It was computationally shown that protein tethering may result in intrinsic destabilization.53 Experimentally, both destabilization39,49 and stabilization34,35,54 were observed upon tethering or cross-linking.55 These effects may have different origins related to changes in the dynamics and energetics of either the folded or unfolded state.
In this study, we used detailed atomistic models to focus on the effects of ubiquitination on the folded protein structure. We compared the native-state dynamics of ubiquitinated substrates with that of their unmodified counterpart. We examined the ability of mono-Ub and Lys48-linked tetra-Ub chains to modulate the conformational stability of the substrate protein Ubc7, whose ubiquitination sites for proteasomal degradation are known. The results of the atomistic simulations were compared with those previously obtained from coarse-grained models where a full folding reaction was studied.43 Our analyses suggest that a reduction in the entropy of the folded state may serve as an additional mechanism for protein destabilization by Lys48-linked tetra-Ub. These results are intriguing, especially in light of the lack of specific formation of interactions between the Ub and the modified protein. Our findings provide additional independent evidence that ubiquitination can significantly modulate substrates in a manner that may be imperative for the substrate’s cellular degradation.
Results
Molecular characteristics of the Ub–Ubc7 interface: The Ub–Ubc7 interface is nonspecific
Ubiquitination, like other PTMs, may affect the biophysical properties of the substrate and, similarly to other PTMs, it is interesting to investigate whether these changes are linked to the cellular function associated with that PTM. Coarse-grained modeling showed that ubiquitination may destabilize the substrate protein through an entropic mechanism that causes the unfolded state to adopt a less compact ensemble of conformation.43 It was shown that this entropic effect on the unfolded state strongly depends on the location of the conjugation site. In the coarse-grained model, the interaction between the Ub moiety and the substrate was modeled as an excluded volume. Accordingly, in that simple model, the bulkiness of the Ub conjugate could affect the internal dynamics and thus the energetics of the substrate. The underlying reasoning of the coarse-grained model was that many proteins undergo ubiquitination and therefore specific interactions between Ub and its substrates are unlikely. Yet, an interface governed by nonspecific interactions might be formed between Ub and its substrate.
In the present study, we quantified the molecular properties of the interface between Ub and its substrate and their dependence on the location of the ubiquitination site and the type of Ub conjugate. Using all-atom modeling, we analyzed the effect of ubiquitination on the dynamics of the native state of the substrate protein. Atomistic modeling allowed us to understand the effect of ubiquitination on protein stability in more detail.
For the substrate protein, we used Ubc7, which is a member of the Ub-conjugating enzyme (E2) family. Degradation of this protein is mediated through a Lys48-tetra-Ub (tetra-Ub). Ubiquitination occurs in vivo at either the catalytic residue Cys89 or at position 94 when it is mutated to a Lys94 residue.56 Using the IUPred disorder predictor,57 we found that these sites are located in a relatively ordered region. This is in good agreement with the general tendency of Lys modifications, such as ubiquitination and acetylation, to occur mostly in ordered regions.58,59
To test the biophysical effects of ubiquitination at various sites in silico, we ubiquitinated the two sites that are used in vivo (89 and 94) as well as seven other surface-exposed Lys residues (3, 11, 18, 29, 62, 70, and 161) with tetra-Ub (Fig. 1). We also attached monoubiquitin (mono-Ub) at positions 18, 89, and 94 to compare the effects of poly- and monoubiquitination.
Each ubiquitination site can, in principle, accept Ub in multiple orientations and each type of Ub (here, mono-Ub or tetra-Ub) can adopt alternative orientations on the surface of the substrate at the site of attachment. To ensure a correct comparison, we compared the orientations of mono-and tetra-Ub only with respect to the Ub moiety attached to the substrate; thus, for tetra-Ub, we analyzed the angle and distance versus the substrate only with respect to the first Ub moiety. Variations in the angle between the Ub moiety and the Ubc7 substrate to which it is attached and variations in the distances between their centers of mass revealed that, at some sites, the overall position of the substituent varied considerably, whereas at others it was quite stable [Figs. 2 and 3(a)]. Within the chosen sites (89 and 94), the positions of mono-Ub were much more variable than those of tetra-Ub [Fig. 2(d)]. For example, at position 94, the variation of the rotation angles of the Ub monomer that is covalently linked to Ubc7 was about 25–28° for mono-Ub whereas in the case of tetra-Ub it was almost 10°. A lack of spatial constraints in the absence of additional moieties enabled mono-Ub to adopt a broad variety of alternative positions [Fig. 2(d)]. For position 18, high dynamics was observed for both mono- and tetra-Ub.
Figure 2.

Characteristics of the flexibility of the Ub–Ubc7 interface. (a) Two angles, α and β, are defined to evaluate the rotation of Ub relative to the substrate (Ubc7). For each angle, two vectors connecting two residues are defined (one on Ubc7 and the other one on Ub). The vectors were placed opposite and in parallel with each other (α angle) or lined up on a single line (β angle). In the case of tetra-Ub, the vector is defined based on the first Ub protein that is directly linked to Ubc7. (b) Values of the angles α for the tetra-Ub (red) and mono-Ub (blue) attached at site 94 as a function of time in the three MD runs; (c) Histograms of the values of angles α and β (solid and dashed lines, respectively) for mono-Ub (blue) and tetra-Ub (red) attached at site 94. (d) Variation in the value of angles α and β (i.e., standard deviation of the distribution of the angle values) for mono-Ub (blue) and tetra-Ub (red) at all the attachment sites examined.
Figure 3.
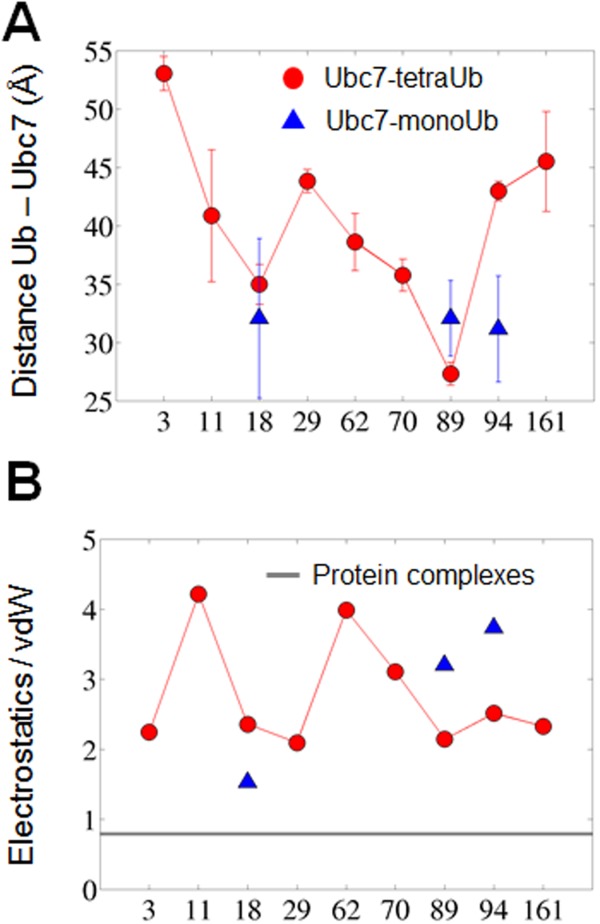
Structural and energetic characterization of the Ub–Ubc7 interface. (a) Distance (Å) between the centers of mass of Ubc7 and the attached Ub moiety at different positions. Mono-Ub and tetra-Ub are designated by blue triangles and red circles, respectively; (b) Ratio of electrostatics (columbic) to van der Waals (vdW) interactions within the Ub–Ubc7 interface at each ubiquitination site. The average ratio is also given for six representative protein–protein complexes from (PDB codes: 1acb, 1dvf, 1mct, 1tgs, 2sni, and 3sgb; solid gray line).
The energetics of the Ub–Ubc7 interface may also shed light on its structural stability. Figure 3(b) shows the ratio between the Coulombic and van der Waals interactions within the interface between Ub and Ubc7 for each possible ubiquitination site. This ratio was also estimated using MD simulations for six representative protein–protein complexes from Ref.60 (PDB codes: 1acb, 1dvf, 1mct, 1tgs, 2sni, and 3sgb). The interface formed between the Ub moiety and Ubc7 is governed by charged–charged interactions whereas, for other complexes, the interface is much more hydrophobic. The highly electrostatic characteristics of the interface between Ub and Ubc7 indicate weak specificity which is expected to be manifested in higher mobility. To examine the implications of the different ratio between the electrostatic and the vdW energies on the interface, we measured all the pairwise distances between the interfacial residues of each complex (defined based on the crystal structures or the modeled ubiquitinated Ubc7) along the simulations. Figure 4 shows the standard deviation of all the interfacial residues, SDij, of selected systems, indicating much higher dynamics for the interfaces of the ubiquitinated systems than for the interfaces of x-ray resolved structures that are more hydrophobic.
Figure 4.
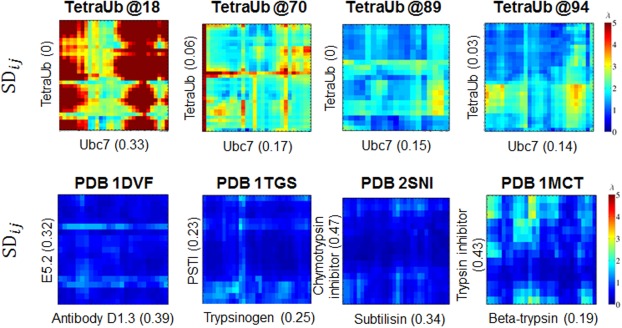
Dynamics of the interfaces in the transient complexes of Ub–Ubc7 (top raw) and in stable complexes (bottom raw). The x- and y- axes of each panel correspond to the interfacial residues in the two proteins of each complex (ordered based on their residue index). These interfacial residues were defined using the modeled structured for the ubiquitinated Ubc7 complexes or using the crystal structures for the protein complexes (PDB IDs 1dvf, 1tgs, 2sni, and 1mct). The numbers in brackets estimated the hydrophobicity of the corresponding patch (estimated by the fraction of hydrophobic residues A, V, L, I, M, F, W, and Y). The matrices show the value of the standard deviation of the pairwise distance between residue i and j from the two proteins that comprise the complexes. The more reddish the color is the more fluctuating is the corresponding pairwise distance.
To further investigate the degree of specificity between Ub and Ubc7, we analyzed the size of Ub–Ubc7 interface. The size of the interface was calculated as the difference between the accessible surface areas (ASAs) of the two separate proteins and the ASA of its complexes. We compared the values of the ASAs obtained for all the Ub–Ubc7 complexes with the values obtained in a bioinformatic analysis for 46 interfaces in protein–protein complexes and for 173 crystal packing interfaces60 [Fig. 5(a)]. For all the Ub–Ubc7 complexes we studied (using mono-Ub and tetra-Ub), the interface area was similar to or lower than the crystal packing area. Some interfaces in the ubiquitinated systems were similar to those of protein complexes, yet they were lower than the average interface area of protein complexes (1970 Å2). The size of the Ub–Ubc7 interfaces thus reflects nonspecific interactions.61 Uniquely, when ubiquitination occurred at the in vivo Lys89 site, the Ub–Ubc7 interface was larger and resembled the interface in protein–protein complexes, which may be related to its biological function. The large interface observed for this system correlates with the relatively short distance between the tetra-Ub–Ubc7 centers of mass [Fig. 3(a)]. As discussed above, Lys48-linked chains may alter the intrinsic biophysical properties of the modified protein. A large interface between Ub and its substrate may provide a possibility for such alterations.
Figure 5.
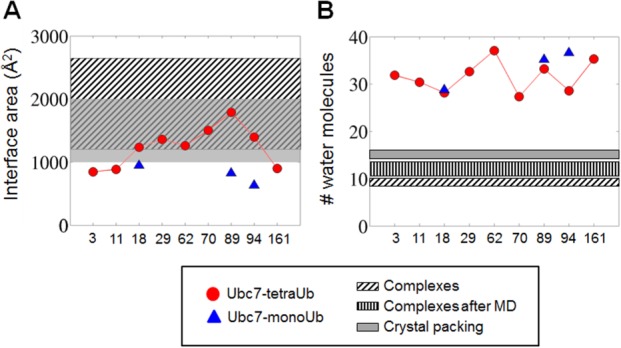
Geometry and hydration of the Ub–Ubc7 interface: (a) Area (Å2) of the interface between Ubc7 and the tetra-Ub (red circles) or mono-Ub (blue triangles) attached to it at different sites. (b) Number of water molecules per 1000 Å2 interfacial area for tetra-Ub –Ubc7 (red circles) and for mono-Ub–Ubc7 (blue triangles). For comparison with the interfaces of other protein–protein interfaces, panels (a) and (b) indicate the interfacial area and hydration found in a bioinformatics survey of protein complexes.60 In (a) and (b), slanted stripes represent data for interfaces in protein–protein complexes and gray represents interfaces found in crystal packing (see main text). The vertical stripes in (b) represent data for the interfacial area in protein–protein complexes following a molecular dynamic simulation.
The Ub–Ubc7 interface is highly hydrated
We next compared the number of interfacial water molecules in Ub–Ubc7 systems, protein–protein complexes, and crystal packing interfaces [Fig. 5(b)]. We defined interfacial water molecules as those within 4.5 Å of the interfacial atoms of both Ubc7 and Ub, where interfacial atoms were those that had lost at least 50% of their ASA value. This comparison showed that the Ub–Ubc7 interface is well hydrated and that the average number of interfacial water molecules associated with each type of ubiquitinated Ubc7 is 2–3 times higher than the number commonly observed in protein–protein complexes and in crystal packing interfaces.60 Figure 6 illustrates the hydration of the interface of Ub–Ubc7 linked at Lys94. We point out that our simulations used a definition for interfacial water in Ub–Ubc7 complexes that was identical to that used in Ref.60 to analyze the interfaces in protein–protein complexes and crystal packing. For a more appropriate comparison between the interfaces of Ub–Ubc7 and protein–protein complexes, we also studied the number of interfacial water molecules in six representative protein–protein complexes from Ref.60 (PDB codes: 1acb, 1dvf, 1mct, 1tgs, 2sni, and 3sgb) using MD simulations. This may allow a fair comparison to the Ub–Ubc7 interface, which was also characterized after MD simulations. The results showed that the number of interfacial water molecules in these protein–protein complexes was higher than we found earlier, because of the dynamics, but still involved fewer interfacial water molecules than the Ub–Ubc7 interface. [Fig. 5(b), vertically striped bar]. The highly hydrated interface [Fig. 5(b)] together with the small interfacial interaction area [Fig. 5(a)] in the Ub–Ubc7 systems suggest that Ub and Ubc7 do not interact specifically with each other.
Figure 6.
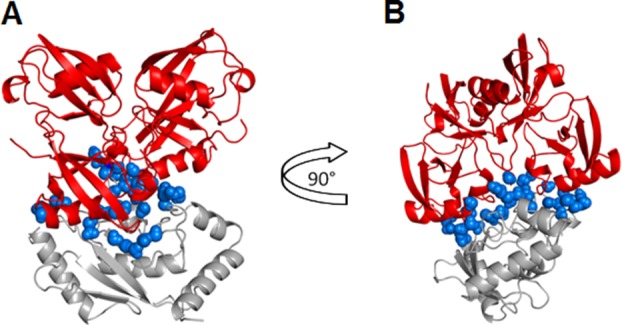
Water molecules in the interface between Ubc7 and Lys48-linked tetra-Ub at Lys94 are shown as blue spheres. The image in (b) is rotated 90° relative to the image in (a). Tetra-Ub is depicted in red and Ubc7 is in gray. We defined interfacial water molecules as those within 4.5 Å of the interface atoms of both Ubc7 and tetra-Ub, where interface atoms were those that had lost at least 50% of the value of their accessible surface area.
Ubc7 is destabilized by a reduction in its conformational entropy
Although the above analysis suggested the absence of strong specific interactions between Ub and Ubc7, we hypothesized that the attachment of Ub may affect the stability of Ubc7. To characterize the structural effect of ubiquitination at different positions on the conformational ensemble of Ubc7, we analyzed the matrices of inter-residue distances in the substrate protein (Ubc7). The matrices were calculated from the average distance between each pair of substrate residues in the ubiquitinated system compared with unmodified Ubc7 (see Methods). A difference in the distances between any pair of residues in ubiquitinated compared with unmodified Ubc7 reflects the compression or expansion of some parts of ubiquitinated Ubc7 relative to the unmodified system. Analyses conducted on Lys48-tetra-Ub–Ubc7 showed that, in most positions, the attachment of tetra-Ub results in local disruptions to the relative positions of some residue groups [Fig. 7(a–c)]. For example, for most of the ubiquitinated variants, the orientation of the loop that includes residues 95–105 is different in the modified compared with the unmodified variant. For mono-ubiquitinated Ubc7, this effect is much less pronounced (Fig. 8).
Figure 7.
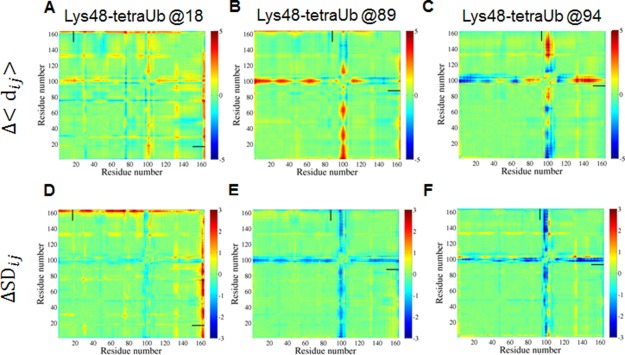
Top panel: Effect of ubiquitination with tetra-Ub on the internal dynamics of Ubc7. Difference distance matrices (Ubc7ubiquitinated - Ubc7unmodified) for Lys48-linked tetra-Ub attached at: (a) Lys18; (b) Lys89; (c) Lys94. Bottom panel: Differences between ubiquitinated and unmodified Ubc7 with respect to the standard deviation of their mean inter-residue distances. Lys48-linked tetra-Ub attached at: (d) Lys18; (e) Lys89; (f) Lys94. In both panels, color indicates the difference distances (variations in distance) in Å, with blue representing the compression (reduced variation) and red representing the expansion (higher variation) of some parts of ubiquitinated Ubc7 relative to the unmodified system. Short horizontal and vertical bars indicate the location (residue number) of the ubiquitination site.
Figure 8.
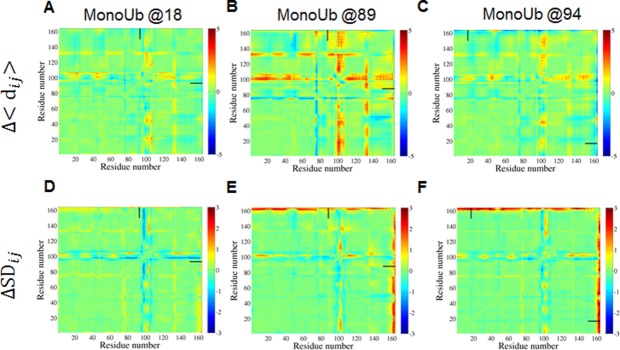
Similar to Figure 7 but for ubiquitination with mono-Ub.
These disruptions in the native conformation of Ubc7 may affect the stability of the folded state and may shift the equilibrium to the unfolded state.43 A more detailed analysis of the flexibility of the studied systems revealed an additional effect of ubiquitination. Differences between ubiquitinated and unmodified Ubc7 with respect to the standard deviation of their mean inter-residue distances illustrated changes in conformational flexibility [Fig. 7(d–f)]. The most pronounced reduction in the flexibility of Ubc7 was observed for the loop containing residues 95–100. Again, mono-Ub does not affect the flexibility of Ubc7 as much as tetra-Ub. These results suggest that Lys48-linked tetra-Ub can reduce the conformational flexibility of the substrate protein and thus lower thermodynamics by restricting the entropy of the native state.
This effect is illustrated by a comparison of the flexibilities of loops 95–105 in unmodified Ubc7, mono-Ub–Ubc7, and tetra-Ub–Ubc7 and by estimating the change in entropy because of ubiquitination (Fig. 9). Tetra-Ub attached at Lys94 reduces the possible orientations of this loop by introducing spatial constraints. Mono-Ub is much more flexible and can adopt various positions. As a result, it cannot introduce any constant spatial constraint that restricts the conformational flexibility in Ubc7.
Figure 9.
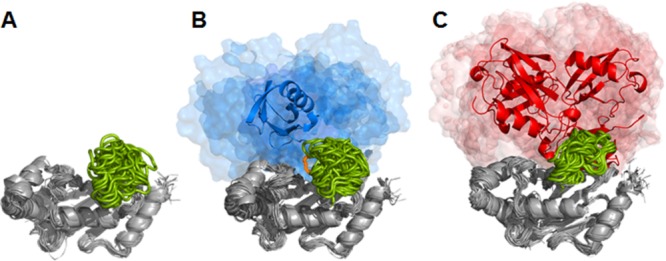
Structure and conformational dynamics of ubiquitinated Ubc7. (a) Unmodified Ubc7. (b) Mono-Ub at Lys94. (c) Lys48-linked tetra-Ub at Lys94. Each system is represented by 31 aligned snapshots from three 100 ns simulations sampled every 10 ns from each simulation. Ubc7 is depicted in gray, loop residues 95–105 are depicted in green. The Lys94 residue of Ubc7 and the C-terminal Gly76 of the attached Ub moiety are depicted in orange. The initial position of Ub is shown in the cartoon representation (mono-Ub, blue; Lys48-tetra-Ub, red). Other positions of Ub during the simulation are shown in the surface representation (as blue and red “clouds”).
To quantify the change in the internal dynamics because of ubiquitination, we estimated the conformational entropy by the Schlitter entropy using the covariance matrix.62 Comparing the contribution of entropy to the free energy (ΔTSconf=TSubiquitinated − TSunmodified) of ubiquitinated Ubc7 relative to unmodified Ubc7 revealed a significant destabilizing effect because of Ub attachment at several sites (Fig. 10). A negative ΔTSconf contributes to the overall destabilization of the system. The values of ΔTSconf clearly indicate that the most pronounced destabilization occurs at the in vivo ubiquitination sites for degradation (89, 94). Ubiquitination with tetra-Ub at some of the other positions (3, 11, 29) can also destabilize Ubc7 quite considerably by reducing the conformation flexibility of some regions. The entropy contribution to the free energy of the folded state is quite substantial (up to ∼−0.25 kcal/mol per residue). We believe that the effect of ubiquitination on the entropy of the folded state of the protein might be reduced by other effects. For example, it can be compensated by the enthalpy gain from Ub–Ubc7 interactions.
Figure 10.
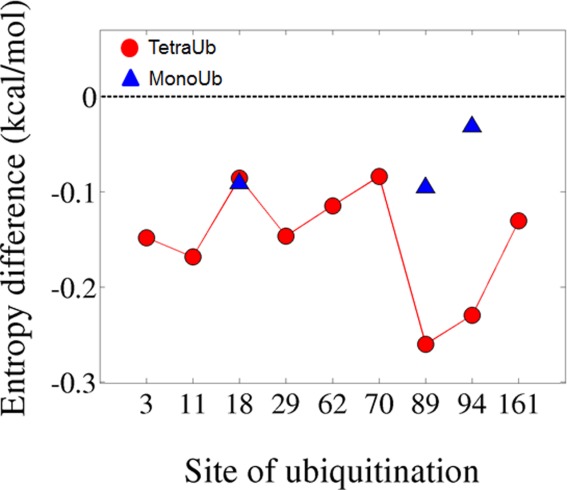
Comparison of the configurational entropic contributions to the free energy (normalized by the number of residues in the substrate, kcal/mol) in ubiquitinated and unmodified Ubc7. (ΔTSconf=TSconfubiquitinated − TSconfunmodified). The configurational entropy was estimated using the Schlitter approximation.
Discussion
Fine-tuned and efficient degradation of proteins is essential for proper cell functioning. While most proteins are marginally stable, the ability to achieve rapid and efficient unfolding and degradation varies among various proteasomes63,64 and can be influenced by the characteristics of the proteins that are to be unfolded.65 Disordered stretches of a certain length are needed to initiate degradation,66 and the structural properties of the site from which the unfolding is initiated affects degradation efficiency.65 Thus, the level of intrinsic disorder within proteins can significantly affect their proteasomal degradation,67,68 much like many other cellular processes,69–73 however, since not all proteins contain disordered regions, other factors might be needed to assist in inducing unfolding, including the Ub attachment itself.43,58
Here, we focus on the effects exerted by a Ub attachment on the stability and dynamics of the folded state of the modified protein. Our results show that the ubiquitination-induced effect of an entropic destabilization of the native-state of the protein substrate depends on the ubiquitination site, the type of Ub attachment (mono-Ub versus tetra-Ub), and the orientation of the Ub conjugate at the site of attachment. Ubiquitination at sites that are known to be modified in vivo leads to considerable destabilization of the folded state in Ubc7. Ubiquitination at other sites (Lys residues that were only ubiquitinated in silico, but that are not known to be used in vivo for ubiquitination) leads to a lesser degree of destabilization. The entropy reduction is caused by a decrease in the conformational flexibility of certain regions (particularly loop 95–105), which is sterically possible only if the Ub chain is sufficiently bulky (as is tetra-Ub) and if it is in the right position relative to the substrate. This last observation suggests that the efficiency of ubiquitination at certain sites depends on the type and size of the Ub chain, which might have implications for the ability to conjugate the Ub chain sequentially (one Ub at a time) versus in a single step (through the attachment of an entire chain at once).
The flexibility of Ub at its attachment site and the lack of strong interactions with the substrate observed in our study are in good agreement with previously discussed experimental results for ubiquitinated PCNA20,21,32) and Ras20,21,32. We show that the interface between Ub and its substrate is often much more hydrated than other interfaces of weak protein–protein interaction. It would be interesting to generalize the interaction mode of Ub chains with Ubc7 to other ubiquitinated systems and to further explore the effect of ubiquitination on protein stability and the function associated with the ubiquitination, however this is beyond the scope of the current work. The fact that Ub sites are sometimes highly conserved across many species, while in other systems ubiquitination sites seem not well-conserved74 gives further evidence for the notion that in some cases a specific interface might be formed and play an important role in the ubiquitination of specific systems, while in other only nonspecific interfaces are formed.
The phenomenon whereby a change in the configurational entropy of a protein’s native folded state affects the stability of that state is not unique to this system. In a previous study, using a combination of experimental and computational techniques, we showed that native protein structure can be stabilized by increasing the entropy of the folded state.75 The increased stability was observed upon modifying a loop region of the enzyme acylphosphatase. In the present study, we observe the opposite situation: a decrease in the entropy of the folded state (mainly by restricting the flexibility of the loop) that results in destabilization of the native structure. The configurational entropy of the folded state can also be affected by the flexibility of the side chains of the protein residues. It was earlier argued using a bioinformatic and simulation study that the excess of positively charged lysine residues at the expense of arginine residues, which is common to hyperthermophilic proteins, may explain higher stability. This linkage between arginine to lysine replacement and higher thermodynamic stability can be explained by the greater number of accessible rotamers of lysines that may have larger contribution to the native-state entropy.76 Taken together, these studies illustrate that the thermodynamic properties of proteins can be significantly modulated by controlling the entropy of the folded state.
The atomistic simulations presented in this study suggest that Ub may affect the native state dynamics and so they may complement an earlier study using coarse-grained simulations, which suggested that, under some conditions, Ub may affect the properties of the substrate mostly by affecting the biophysical nature of the unfolded state.43 Both studies support the idea that Ub may serve not only as a molecular tag but also as a substituent that affects the biophysics of the protein in a manner that is important to its cellular half-life. This idea is supported by the various reports on the effects of PTMs on protein stability, even when no specific interface is formed (i.e., in glycosylation and PEGylation). Since recent proteomics and bioinformatics studies have greatly expanded the known repertoire of ubiquitinated proteins and ubiquitination sites,77–79 further studies can shed light on how ubiquitination affects other systems.
Materials and Methods
All-atom modeling
In this in silico study, we ubiquitinated Ubc7 at nine sites (Fig. 1) to explore the molecular details of the interface formed between the Ub moiety and its substrate. Ubc7 (pdb: 2ucz) was ubiquitinated at various locations using a single Ub (pdb: 1ubq) or a Ub -chain tetramer internally linked at Lys48 (pdb: 2o6v). Tetramers were used as the polyubiquitin, as it was shown that they are the minimal unit needed for efficient recognition and degradation in the proteasome.80 We minimized these structures using 300 steps of steepest descent followed by 500 steps of conjugated gradient algorithms in CHARMM.81
To study the functional activity of Ub substrate complexes conjugated at different sites, we used all-atom modeling. The molecular dynamics simulations were performed using GROMACS Version 4.5.4.82 After energy minimization, we equilibrated the system in two phases: (1) under an NVT ensemble; and (2) under an NPT ensemble (100 ps/phase). Next, we performed long molecular dynamics simulations. For all Ub–Ubc7 complexes, as well as for unmodified Ubc7, we preformed three simulations of 100 ns each. We used an AMBER99SB-ILDN force field83 that was modified to simulate isopeptide bonding between Ub moieties and between Ub and Ubc7. The LINCS algorithm84 was used to control bonds during the simulation. The leapfrog algorithm was employed with steps of 2 fs. All the simulations were performed at constant pressure and temperature (NPT ensemble). Temperature was controlled at 300 K, with a modified Berendsen thermostat.85 The molecular system was solvated in a box with periodic boundary conditions containing pre-equilibrated TIP3P water molecules.86 Na+ and Cl− ions were added to maintain overall system neutrality.
Differences between the dynamics
The dynamics of unmodified and ubiquitinated Ubc7 was analyzed by plotting difference distance matrices (Δij) and difference standard deviation matrices (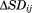 ). The former were calculated using the average distance (dij) between each pair of residues in Ubc7ubiquitinated
). The former were calculated using the average distance (dij) between each pair of residues in Ubc7ubiquitinated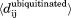 compared with Ubc7unmodified
compared with Ubc7unmodified
where <dij> is the mean pairwise distance between the Cα atoms of residues i and j derived from an analysis of 3000 snapshots observed during three 100 ns runs.
Difference standard deviation matrices were calculated using the standard deviation of the distance between each pair of residues in Ubc7ubiquitinated (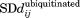 ) compared with Ubc7unmodified (
) compared with Ubc7unmodified (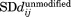 ).
).
where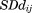 is the standard deviation for the distance between residues i and j.
is the standard deviation for the distance between residues i and j.
Configurational entropy calculations of the Ubc7 were performed based on covariance matrices of the atomic fluctuations observed in the MD trajectories, because of the Quasi Harmonic approximation.62 The presented configurational entropy is the average of the independent entropies that were independently estimated for each run.
Analysis of the Ub–Ubc7 interface
The interface formed between Ub and Ubc7 was analyzed by both geometric and energetic measures. Energetically, the ratio between the total electrostatic and vdW energies of the interfacial residues was estimated. Geometrically, several measured were calculated: the distance between the center of mass of Ubc7 and the tethered Ub, the pairwise distances between the interfacial residues of Ub and Ubc7, the rotation of the Ub moiety relative to Ubc7, and the size of the interface and the degree of its hydration. Because the Ub–Ubc7 interface is expected to be nonspecific and dynamic, we followed the fluctuations of some of the geometrical characteristics by focusing on the variance rather than on the average value. To calculate the accessible surface area (ASA) and to define the interface area for Ub–Ubc7 complexes, we used the NACCESS program by S. Hubbard (University College, London), which implements the Lee and Richards algorithm.87 The angles between Ub and Ubc7 proteins were calculated using GROMACS tools.82 Angles were calculated between vectors connecting the Cα atoms of two residues in the attached Ub moiety and Ubc7. Initially, vectors were placed parallel to each other or on one line (see the scheme in Fig. 2).
The size of the formed interfaces and their degree of hydration were compared to other protein-protein interfaces reported in the literature. Two types of interfaces were used for this comparison: in stable protein complexes and in crystal packing (46 and 173 interfaces, respectively). To compare the fluctuation of the interfaces of stable protein complexes with that of the Ub–Ubc7 complexes, selected six complexes (from the 46 reported structures) and simulated each for 100 ns. The selected complexes correspond to PDB codes: 1acb, 1dvf, 1mct, 1tgs, 2SNI, and 3sgb.
Acknowledgments
This work was supported by the Kimmelman Center for Macromolecular Assemblies. We would like to thank Yevgeniya Korotinsky for assistance with preparing Figure 9. Y.L. is The Morton and Gladys Pickman professional chair in Structural Biology.
References
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. The ubiquitin system, an immense realm. Annu Rev Biochem. 2012;81:167–176. doi: 10.1146/annurev-biochem-051910-094049. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Molecular Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan G-L, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Sigismund S, Polo S, Di Fiore PP. Signaling through monoubiquitination. Signalling from Internalized Growth Factor Receptors. 2004;286:149–185. doi: 10.1007/978-3-540-69494-6_6. [DOI] [PubMed] [Google Scholar]
- Shaeffer JR. Monoubiquitinated α globin is an intermediate in the ATP-dependent proteolysis of α globin. J Biol Chem. 1994;269:22205–22210. [PubMed] [Google Scholar]
- Shaeffer JR, Kania MA. Degradation of Monoubiquitinated .alpha.-Globin by 26S Proteasomes. Biochemistry-Us. 1995;34:4015–4021. doi: 10.1021/bi00012a020. [DOI] [PubMed] [Google Scholar]
- Boutet SC, Disatnik MH, Chan LS, Iori K, Rando TA. Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell. 2007;130:349–362. doi: 10.1016/j.cell.2007.05.044. [DOI] [PubMed] [Google Scholar]
- Shabek N, Herman-Bachinsky Y, Ciechanover A. Ubiquitin degradation with its substrate, or as a monomer in a ubiquitination-independent mode, provides clues to proteasome regulation. Proc Natl Acad Sci U S A. 2009;106:11907–11912. doi: 10.1073/pnas.0905746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabek N, Herman-Bachinsky Y, Buchsbaum S, Lewinson O, Haj-Yahya M, Hejjaoui M, Lashuel HA, Sommer T, Brik A, Ciechanover A. The Size of the Proteasomal Substrate Determines Whether Its Degradation Will Be Mediated by Mono- or Polyubiquitylation. Mol Cell. 2012;48:87–97. doi: 10.1016/j.molcel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Stanhill A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim Biophys Acta. 2014;1843:86–96. doi: 10.1016/j.bbamcr.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains—From structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, Dikic I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- Roscoe BP, Bolon DNA. Systematic Exploration of Ubiquitin Sequence, E1 Activation Efficiency, and Experimental Fitness in Yeast. J Mol Biol. 2014;426:2854–2870. doi: 10.1016/j.jmb.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Freudenthal BD, Gakhar L, Ramaswamy S, Washington MT. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat Struct Mol Biol. 2010;17:479–484. doi: 10.1038/nsmb.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Van Wynsberghe AW, Freudenthal BD, Weinacht CP, Gakhar L, Washington MT, Zhuang Z, Tainer JA, Ivanov I. Solution X-ray scattering combined with computational modeling reveals multiple conformations of covalently bound ubiquitin on PCNA. Proc Natl Acad Sci U S A. 2011;108:17672–17677. doi: 10.1073/pnas.1110480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbert RG, Sixma TK. Intrinsic flexibility of ubiquitin on proliferating cell nuclear antigen (PCNA) in translesion synthesis. J Biol Chem. 2012;287:39216–39223. doi: 10.1074/jbc.M112.389890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang S, Lin SH, Wang X, Wu L, Lee EY, Lee MY. Structure of monoubiquitinated PCNA: implications for DNA polymerase switching and Okazaki fragment maturation. Cell Cycle. 2012;11:2128–2136. doi: 10.4161/cc.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggiano S, Pastore A. The challenge of producing ubiquitinated proteins for structural studies. Cells. 2014;3:639–656. doi: 10.3390/cells3020639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda C, Liu J, Chaturvedi A, Nowicka U, Cropp TA, Fushman D. Nonenzymatic assembly of natural polyubiquitin chains of any linkage composition and isotopic labeling scheme. J Am Chem Soc. 2011;133:17855–17868. doi: 10.1021/ja207220g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spasser L, Brik A. Chemistry and biology of the ubiquitin signal. Angew Chem Int Ed Engl. 2012;51:6840–6862. doi: 10.1002/anie.201200020. [DOI] [PubMed] [Google Scholar]
- Haj-Yahya M, Fauvet B, Herman-Bachinsky Y, Hejjaoui M, Bavikar SN, Karthikeyan SV, Ciechanover A, Lashuel HA, Brik A. Synthetic polyubiquitinated alpha-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc Natl Acad Sci U S A. 2013;110:17726–17731. doi: 10.1073/pnas.1315654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemantha HP, Bavikar SN, Herman-Bachinsky Y, Haj-Yahya N, Bondalapati S, Ciechanover A, Brik A. Nonenzymatic polyubiquitination of expressed proteins. J Am Chem Soc. 2014;136:2665–2673. doi: 10.1021/ja412594d. [DOI] [PubMed] [Google Scholar]
- Freudenthal B, Gakhar L, Ramaswamy S, Washington M. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nature Struct Biol Mol Biol. 2010;17:479–484. doi: 10.1038/nsmb.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker R, Lewis SM, Sasaki AT, Wilkerson EM, Locasale JW, Cantley LC, Kuhlman B, Dohlman HG, Campbell SL. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat Struct Mol Biol. 2013;20:46–52. doi: 10.1038/nsmb.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggiano S, Menon RP, Kelly GP, McCormick J, Todi SV, Scaglione KM, Paulson HL, Pastore A. Enzymatic production of mono-ubiquitinated proteins for structural studies: The example of the Josephin domain of ataxin-3. FEBS Open Bio. 2013;3:453–458. doi: 10.1016/j.fob.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller CE, Pilkerton ME, Chatterjee C. Chemical strategies to understand the language of ubiquitin signaling. Biopolymers. 2014;101:144–155. doi: 10.1002/bip.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker R, Lewis SM, Sasaki AT, Wilkerson EM, Locasale JW, Cantley LC, Kuhlman B, Dohlman HG, Campbell SL. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat Struct Mol Biol. 2012;20:46–52. doi: 10.1038/nsmb.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Stites WE. Effects of Excluded Volume upon Protein Stability in Covalently Cross-Linked Proteins with Variable Linker Lengths†. Biochemistry-Us. 2008;47:8804–8814. doi: 10.1021/bi800297j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Batey S, Nickson AA, Teichmann SA, Clarke J. The folding and evolution of multidomain proteins. Nat Rev Mol Cell Biol. 2007;8:319–330. doi: 10.1038/nrm2144. [DOI] [PubMed] [Google Scholar]
- Batey S, Nickson AA, Clarke J. Studying the folding of multidomain proteins. Hfsp J. 2008;2:365–377. doi: 10.2976/1.2991513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Sasai M. Cooperativity, connectivity, and folding pathways of multidomain proteins. P Natl Acad Sci USA. 2008;105:13865–13870. doi: 10.1073/pnas.0804512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora M, Cieplak M. Mechanical stability of multidomain proteins and novel mechanical clamps. Proteins. 2011;79:1786–1799. doi: 10.1002/prot.23001. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chu XK, Suo ZC, Wang EK, Wang J. Multidomain Protein Solves the Folding Problem by Multifunnel Combined Landscape: Theoretical Investigation of a Y-Family DNA Polymerase. J Am Chem Soc. 2012;134:13755–13764. doi: 10.1021/ja3045663. [DOI] [PubMed] [Google Scholar]
- Bandi S, Singh SM, Mallela KM. The C-terminal domain of the utrophin tandem calponin-homology domain appears to be thermodynamically and kinetically more stable than the full-length protein. Biochemistry-Us. 2014;53:2209–2211. doi: 10.1021/bi500120e. [DOI] [PubMed] [Google Scholar]
- Giri Rao VV, Gosavi S. In the multi-domain protein adenylate kinase, domain insertion facilitates cooperative folding while accommodating function at domain interfaces. Plos Comput Biol. 2014;10:e1003938. doi: 10.1371/journal.pcbi.1003938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanami T, Terada TP, Sasai M. Folding pathway of a multidomain protein depends on its topology of domain connectivity. P Natl Acad Sci USA. 2014;111:15969–15974. doi: 10.1073/pnas.1406244111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang C, Wang EK, Wang J. PolyUbiquitin Chain Linkage Topology Selects the Functions from the Underlying Binding Landscape. Plos Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagai T, Levy Y. Ubiquitin not only serves as a tag but also assists degradation by inducing protein unfolding. Proc Natl Acad Sci USA. 2010;107:2001–2006. doi: 10.1073/pnas.0912335107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD. The discovery of ubiquitin-dependent proteolysis. Proc Natl Acad Sci U S A. 2005;102:15280–15282. doi: 10.1073/pnas.0504842102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule at atomic resolution. Nat Struct Mol Biol. 2008;15:1238–1240. doi: 10.1038/nsmb1208-1238. [DOI] [PubMed] [Google Scholar]
- Wojciechowski M, Szymczak P, Carrion-Vazquez M, Cieplak M. Protein Unfolding by Biological Unfoldases: Insights from Modeling. Biophys J. 2014;107:1661–1668. doi: 10.1016/j.bpj.2014.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro MF, Carmody L, Romo-Fewell O, Lokensgard ME, Love JJ. Characterizing Substrate Selectivity of Ubiquitin C-Terminal Hydrolase-L3 Using Engineered alpha-Linked Ubiquitin Substrates. Biochemistry-Us. 2014 doi: 10.1021/bi5006317. [DOI] [PubMed] [Google Scholar]
- Shental-Bechor D, Levy Y. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci USA. 2008;105:8256–8261. doi: 10.1073/pnas.0801340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D Shental-Bechor, Y Levy. Folding of glycoproteins: toward understanding the biophysics of the glycosylation code. Curr Opin Struc Biol. 2009;19:524–533. doi: 10.1016/j.sbi.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Lawrence PB, Gavrilov Y, Matthews SS, Langlois MI, Shental-Bechor D, Greenblatt HM, Pandey BK, Smith MS, Paxman R, Torgerson CD, Merrell JP, Ritz CC, Prigozhin MB, Levy Y, Price JL. Criteria for Selecting PEGylation Sites on Proteins for Higher Thermodynamic and Proteolytic Stability. J Am Chem Soc. 2014;136:17547–17560. doi: 10.1021/ja5095183. [DOI] [PubMed] [Google Scholar]
- Arviv O, Levy Y. Folding of multidomain proteins: Biophysical consequences of tethering even in apparently independent folding. Proteins. 2012;80:2780–2798. doi: 10.1002/prot.24161. [DOI] [PubMed] [Google Scholar]
- Batey S, Randles LG, Steward A, Clarke J. Cooperative folding in a multi-domain protein. J Mol Biol. 2005;349:1045–1059. doi: 10.1016/j.jmb.2005.04.028. [DOI] [PubMed] [Google Scholar]
- Kim YH, Stites WE. Effects of excluded volume upon protein stability in covalently cross-linked proteins with variable linker lengths. Biochemistry-Us. 2008;47:8804–8814. doi: 10.1021/bi800297j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, Hochstrasser M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat Cell Biol. 2007;9:422–427. doi: 10.1038/ncb1558. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Hagai T, Azia A, Toth-Petroczy A, Levy Y. Intrinsic disorder in ubiquitination substrates. J Mol Biol. 2011;412:319–324. doi: 10.1016/j.jmb.2011.07.024. [DOI] [PubMed] [Google Scholar]
- Duttler S, Pechmann S, Frydman J. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol Cell. 2013;50:379–393. doi: 10.1016/j.molcel.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Bahadur RP, Chakrabarti P, Janin J. Hydration of protein–protein interfaces. Proteins. 2005;60:36–45. doi: 10.1002/prot.20478. [DOI] [PubMed] [Google Scholar]
- Janin J. Specific versus non-specific contacts in protein crystals. Nature Struct Biol. 1997;4:973–974. doi: 10.1038/nsb1297-973. [DOI] [PubMed] [Google Scholar]
- Andricioaei I, Karplus M. On the calculation of entropy from covariance matrices of the atomic fluctuations. J Chem Phys. 2001;115:6289–6292. [Google Scholar]
- Koodathingal P, Jaffe NE, Kraut DA, Prakash S, Fishbain S, Herman C, Matouschek A. ATP-dependent proteases differ substantially in their ability to unfold globular proteins. J Biol Chem. 2009;284:18674–18684. doi: 10.1074/jbc.M900783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inobe T, Matouschek A. Paradigms of protein degradation by the proteasome. Curr Opin Struct Biol. 2014;24:156–164. doi: 10.1016/j.sbi.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- Inobe T, Fishbain S, Prakash S, Matouschek A. Defining the geometry of the two-component proteasome degron. Nat Chem Biol. 2011 doi: 10.1038/nchembio.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70:1357–1366. doi: 10.1002/prot.21614. [DOI] [PubMed] [Google Scholar]
- van der Lee R, Lang B, Kruse K, Gsponer J, Sanchez de Groot N, Huynen MA, Matouschek A, Fuxreiter M, Babu MM. Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep. 2014;8:1832–1844. doi: 10.1016/j.celrep.2014.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea CA, High AA, Obenauer JC, Mishra A, Park CG, Punta M, Schlessinger A, Ma J, Rost B, Slaughter CA, Kriwacki RW. Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. J Proteome Res. 2009;8:211–226. doi: 10.1021/pr800308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu MM, Kriwacki RW, Pappu RV. Structural biology. Versatility from protein disorder. Science. 2012;337:1460–1461. doi: 10.1126/science.1228775. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Toth-Petroczy A, Kraut DA, Matouschek AT, Lim RY, Xue B, Kurgan L, Uversky VN. Disordered proteinaceous machines. Chem Rev. 2014;114:6806–6843. doi: 10.1021/cr4007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Multisteric regulation by structural disorder in modular signaling proteins: an extension of the concept of allostery. Chem Rev. 2014;114:6715–6732. doi: 10.1021/cr4005082. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signaling and regulation. Nature Reviews Molecular Cell Biology. 2015;16:18–25. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagai T, Toth-Petroczy A, Azia A, Levy Y. The origins and evolution of ubiquitination sites. Molec BioSyst. 2012;8:1865–1877. doi: 10.1039/c2mb25052g. [DOI] [PubMed] [Google Scholar]
- Dagan S, Hagai T, Gavrilov Y, Kapon R, Levy Y, Reich Z. Stabilization of a protein conferred by an increase in folded state entropy. Proc Natl Acad Sci U S A. 2013;110:10628–10633. doi: 10.1073/pnas.1302284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovsky IN, Chen WW, Choi PJ, Shakhnovich EI. Entropic stabilization of proteins and its proteomic consequences. Plos Comput Biol. 2005;1:e47. doi: 10.1371/journal.pcbi.0010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Bork P, Krogan NJ, van Noort V. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol. 2013;9:714. doi: 10.1002/msb.201304521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Song J, Zhang Z. Structural propensities of human ubiquitination sites: accessibility, centrality and local conformation. Plos One. 2013;8:e83167. doi: 10.1371/journal.pone.0083167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhou Y, Zhang Z, Song J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief Bioinform. 2014 doi: 10.1093/bib/bbu031. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL. Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO. Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry. 1997;18:1463–1472. [Google Scholar]
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]