

Abstract
We recently reported the development of a computational method for the design of coassembling multicomponent protein nanomaterials. While four such materials were validated at high-resolution by X-ray crystallography, low yield of soluble protein prevented X-ray structure determination of a fifth designed material, T33-09. Here we report the design and crystal structure of T33-31, a variant of T33-09 with improved soluble yield resulting from redesign efforts focused on mutating solvent-exposed side chains to charged amino acids. The structure is found to match the computational design model with atomic-level accuracy, providing further validation of the design approach and demonstrating a simple and potentially general means of improving the yield of designed protein nanomaterials.
Keywords: computational protein design, crystal structure, solubility, coassembly, symmetry, tetrahedral, nanomaterial
Introduction
Symmetric homomeric and heteromeric protein complexes perform a broad range of functions in biological systems1,2. Inspired by these natural protein-based molecular machines and materials, many efforts have been undertaken to design novel supramolecular protein structures.3–19 We recently described a design strategy that combines symmetric modeling with protein–protein interface design to generate novel protein assemblies with atomic-level accuracy.7,16 Using this approach we were able to successfully design five novel tetrahedral protein nanomaterials formed through the coassembly of multiple copies of two distinct protein subunits.16
All five designs were confirmed to yield co-assembled nanoparticles of the expected size and shape by analytical size exclusion chromatography (SEC) and negative stain electron microscopy (EM). Crystal structures of four of the nanomaterials were found to match the design models with high accuracy, but we were unable to attempt crystallization of the fifth design, termed T33-09, because of the low yield of soluble protein. In addition to the limited soluble yield of T33-09, the majority of unsuccessful designs exhibited low or undetectable amounts of soluble expression. This observation, combined with a lack of discernible differences in the calculated metrics of interface quality for successful and unsuccessful design models, indicated that developing methods to increase soluble expression of the designs is likely to be important for improving our design approach.
With this motivation, we designed and experimentally characterized variants of T33-09 in which a subset of the solvent-exposed side chains on each subunit were mutated to either positively or negatively charged amino acids. This approach, referred to as “supercharging” when taken to an extreme, has previously been shown to be effective at increasing protein solubility20,21 and is an enticing option for improving our designed nanomaterials as it avoids the need to mutate core or interface residues, which are generally less tolerant of mutations than surface residues. Using a quick and simple cell lysate-based screen, this approach led to the successful production of a design variant with significantly increased soluble yield and to the determination of a high-resolution structure of the redesigned material. As intended, the designed interface and the overall structure of the nanomaterial were not changed during the redesign process, and were found to match closely with the experimentally determined structure.
Results and Discussion
T33-09 is comprised of multiple copies of two distinct protein subunits, referred to as A and B, each about 110 amino acids in length. Both subunit types are naturally trimeric, and the introduction of a de novo designed protein–protein interface between the two types of subunits gives rise to a symmetric, tetrahedral assembly comprised of four trimers of each type.16 In an attempt to rescue the low solubility of this designed material, one positively charged and one negatively charged version of each protein subunit were designed using the Rosetta macromolecular modeling software package as follows.22,23 Using the original T33-09 design model as the starting point, with the same treatment of the backbone and rigid body DOFs as published previously,16 side chains with greater than 28 Å2 of solvent accessible surface area, and not already possessing the desired charge state, were selected as designable positions. Two new design models were generated, one in which all designable residues in subunit A were allowed to mutate to aspartate or glutamate, while those in subunit B were allowed to mutate to arginine or lysine, and another in which all designable residues in subunit A were allowed to mutate to arginine or lysine, while those in subunit B were allowed to mutate to aspartate or glutamate. The resulting designs were refined and selected for experimental characterization based on Rosetta score metrics and visual inspection in PyMOL,24 yielding four new variants with four to eight mutations per subunit compared with the original design [Supporting Information Table S1].
Synthetic genes encoding the four designed variants were cloned into the pET29b vector (Novagen) for inducible expression in Eschericia coli and the level of soluble expression and assembly state of all nine possible pairwise combinations of original, negatively, or positively charged A and B subunits was then assessed by mixing cell lysates containing the individually expressed subunits and analyzing the resulting soluble and insoluble fractions by polyacrylamide gel electrophoresis (PAGE). One combination of subunits, with a negatively charged A subunit and the original B subunit, was found to significantly increase the yield of the assembled state in the soluble fraction [Supporting Information Figure 1S]. We named this new design variant, which contains five mutations in the A component relative to the original design, T33-31 [Fig. 1(A)].
Figure 1.

Experimental characterization of designed protein assembly T33-31 by SDS-PAGE, analytical SEC, and electron microscopy. A: Close-up of the original subunit A and negatively charged subunit A (Aneg) from the T33-09 (white) and T33-31 (green and blue) design models. The five surface residues mutated in T33-31 compared to T33-09 are labeled and shown as sticks. B: SDS-PAGE analysis of whole cell and clarified lysates from cells expressing the original subunit A or the redesigned, negatively charged subunit A (ANeg). A strong band is observed near the expected molecular weight of 12.5 kDa in the clarified lysate of ANeg, but is only faintly visible in the subunit A sample. C: SEC chromatograms of purified designs and wild-type oligomeric proteins from which they are derived. The A and B subunits are derived from Protein Data Bank entries 1nza and 1ufy, respectively. The designed proteins elute near the expected volume for the target tetrahedral assembly (‘24mers’, arrow), while the wild-type proteins elute as trimers (‘3mers’, arrow). The T33-09 sample was produced from coexpressed subunits, whereas the T33-31 sample was produced through in vitro mixing as described in the Materials and Methods. D: SDS-PAGE analysis of SEC-purified T33-31. Two bands, with approximately equal intensity, are observed near the expected molecular weights of 12.5 and 14.5 kDa. E: Negative stain electron micrograph of in vitro-mixed, SEC-purified T33-31.
SDS-PAGE analysis of individually expressed subunits showed a clear increase in the soluble expression of the redesigned, negatively charged subunit A compared with the original design [Fig. 1(B)]. In addition, gel filtration of individually expressed subunits purified by nickel affinity chromatography showed a substantial reduction of apparent soluble aggregate in the negatively charged subunit A sample compared with the original design [Supporting Information Figure 2S(B)], suggesting that the negatively charged variant has less of a tendency to self-associate. Purified T33-31, obtained by nickel affinity chromatography and size exclusion chromatography of coexpressed (data not shown) or in vitro-mixed hexahistidine-tagged subunits, yielded a dominant peak by analytical SEC near the same elution volume as T33-09, matching the expected size of approximately 24 subunits [Fig. 1(C)]. SDS-PAGE analysis of the SEC peak fractions yielded two bands of approximately equal intensity near the expected molecular weights for subunits A and B [Fig. 1(D)]. Negative-stain electron microscopy of the purified assembly fractions revealed fields of monodisperse particles that closely resemble the design model at low resolution and are indistinguishable from previously obtained electron micrographs of T33-09 [Fig. 1(E)].16 Taken together, these data provide strong evidence that T33-31 coassembles to form a structure of similar size and shape to our design model and with the expected one to one stoichiometry of subunits A and B.
Facilitated by the increased yield, purified T33-31 was subsequently characterized by X-ray crystallography to confirm the accuracy of the design at high-resolution. T33-31 crystallized readily, leading to the determination of a 3.4 Å structure (Fig. 2). The asymmetric unit of the crystal comprises one complete tetrahedron. The backbone atoms of the three subunits composing the interface in the design model (two subunits from component A and one subunit from component B) have an average root mean square deviation (r.m.s.d.) of 0.6 Å compared with the twelve noncrystallographically-related instances of the equivalent atoms in the crystal structure. The r.m.s.d. over all backbone atoms in the 24 subunits compared with the design model is only slightly higher at 0.7 Å (Fig. 2). At positions where the electron density permitted side chain placement, the T33-31 design model also matches the crystal structure with high accuracy. While the backbone and side chain conformations do not match as well at the redesigned positions (W43E, Q44E, H62D, A73E, and T78E), this is not surprising because: (1) the backbone degrees of freedom (DOFs) were held fixed during the computational design protocol despite many of the mutated residues residing in loop regions and (2) the side chains are highly exposed to solvent and expected to be able to adopt many conformations. Other than the five mutated side chains in subunit A and several additional non-mutated surface residues, the T33-09 and T33-31 design models are nearly identical and thus the original T33-09 design model matches the crystal structure equally well over both the backbone and the core and interface side chain conformations (Fig. 2).
Figure 2.
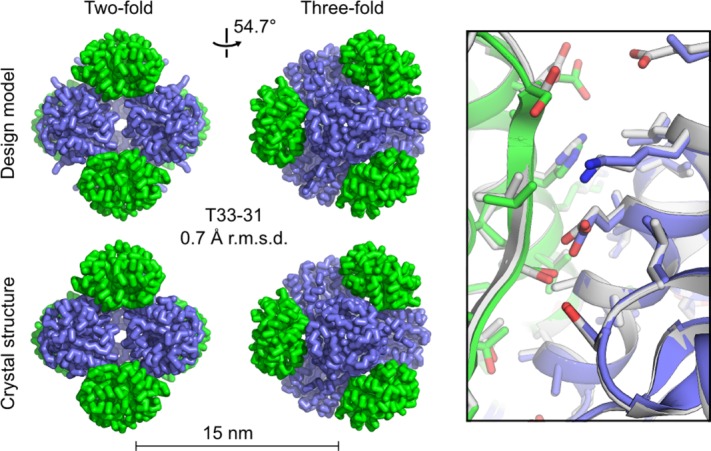
T33-31 crystal structure and design model. At left, views along the two-fold and three-fold symmetry axes are shown for the T33-31 computational design model (top) and crystal structure (bottom, PDB ID 4ZK7, scale bar: 15 nm). The r.m.s.d. was calculated using the backbone atoms in all 24 chains of the design model compared to the asymmetric unit of the crystal structure. At right, an overlay is shown of the designed interface in the design model (white) and crystal structure (green and blue). Poor electron density prevented modeling beyond the beta or delta carbon for some amino acid side chains in the crystal structure. The subunits involved in the interface shown are represented by protein chains S, A, and U in the deposited PDB structure. In the amino acid side chains shown, oxygen atoms are red, nitrogen atoms are blue, and sulfur atoms are orange. An interactive view of the crystal structure, T33-31 design model, and T33-09 design model is available in the electronic version of the article.
There are many possible reasons for the success of the T33-31 variant compared with the other combinations of subunits experimentally tested. SDS-PAGE analysis and gel filtration of the original A and B subunits showed that, when individually expressed, subunit A appeared to self-associate to form soluble aggregates whereas subunit B appeared to exist primarily as free trimer [Supporting Information Figure 2S]. Given this lower yield of soluble, nonaggregated subunit A compared to subunit B in the original design, it is perhaps not surprising that the best redesigned variant involved changes to subunit A, which decreased the tendency of the subunit to self-associate. It is at present less clear why the negatively charged version of subunit A worked better than the positively charged version or why the original version of subunit B worked better in combination with the negatively charged A subunit than the other versions of subunit B. It is possible that the greater total number of mutations in the positively charged variants (8 and 7 mutations for APos and BPos, respectively, compared to 5 and 4 for ANeg and BNeg, respectively), including a greater number of positions with switched charged states (e.g., a mutation from a glutamate to lysine), disrupted native interactions that stabilize the structures of the subunits. It is also possible that the behavior of the positively charged variants is complicated by interactions with cellular polyanions, such as nucleic acids.
The results presented here provide further validation of our approach to designing novel supramolecular protein complexes and highlight the potential utility of including residues distant from the protein–protein interface in the design process. In this sense, the present work demonstrates how experimental characterization of computationally designed proteins generates valuable feedback that can be used to improve the computational design methods. The results also demonstrate the modularity and tunability of the designed materials; it is possible to change particular features of the designs, such as solubility, by modifying the different protein subunits (A or B) and/or different regions of the protein subunits (e.g. surface, core, or interface positions) independently of one another. In this case, five surface mutations to subunit A were sufficient to significantly increase the soluble yield of T33-09 without changing the overall structure of the design. This surface redesign approach bypasses the difficulties of adjusting sensitive interfaces and core interactions, providing a relatively simple means of improving the solubility of these materials. Given the many possible applications of designed protein nanomaterials, additional experiments and methods development aimed at improving solubility and other desirable properties of the designs are merited. The genetic basis and modular nature of this class of nanomaterials, combined with the wealth of previously developed methods for protein modification,25,26 should facilitate these efforts. In conjunction with computational redesign approaches, such as the one used in the present study, the development and utilization of methods for directed evolution27–29 of protein nanostructures30–32 should provide particularly powerful tools to help tailor these new nanomaterials for a wide variety of features and target applications.
Materials and Methods
Computational design
Design calculations were performed as described above using a customized build of Rosetta (version 619f5568a4b8aeb131ee63d41be80c1f2220ae6b) available upon request.
Protein expression, lysate screening, and purification
Codon-optimized genes encoding the designed variants of subunit A and B were purchased (Integrated DNA Technologies) and cloned into the pET29b expression vector between the NdeI and XhoI restriction endonuclease sites for individual expression. Two co-expression constructs were also generated in the pET29b expression vector, one expressing the negatively charged subunit A together with the positively charged subunit B and one expressing the positively charged subunit A together with the negatively charged subunit B. The pairs of genes for these coexpression constructs were cloned between the NdeI and XhoI restriction sites and connected by an intergenic region derived from the pETDUET-1 vector as described previously.16 The pET29b encoded hexahistidine tag was appended to the C-terminus of each individual expression construct and to subunit B in the co-expression constructs. Expression constructs for the wild-type proteins and the original T33-09 design were generated as described previously.16
Expression plasmids were transformed into BL21 Star (DE3) E. coli. Cells were grown in LB medium supplemented with 50 mg L−1 of kanamycin at 37°C until an OD600 of 0.8 was reached. Protein expression was induced by addition of 1.0 mM isopropyl-thio-β-d-galactopyranoside and allowed to proceed for 3 h at 37°C before cells were harvested by centrifugation. Cells were lysed by sonication in 50 mM TRIS pH 8.0, 250 mM NaCl, 1 mM DTT, 20 mM imidazole supplemented with 1 mM phenylmethanesulfonyl fluoride.
For lysate-based screening experiments, a portion of the crude lysates of the original, negatively and positively charged versions of subunits A and B were mixed in all nine possible pairwise combinations in one-to-one volumetric ratios. Mixed and unmixed lysates were incubated at 4°C for 1 hour followed by 22°C for an additional hour. Insoluble material was then cleared by centrifugation and the samples analyzed by denaturing and non-denaturing PAGE. For comparison, the samples were analyzed together with clarified lysates of the unmixed subunits, the wild-type subunits, and coexpressed subunits of the original T33-09 design, negatively charged subunit A and positively charged subunit B, and positively charged subunit A and negatively charged subunit B.
For purification of T33-31, in vitro-mixed samples were obtained by mixing cells prior to lysis and subsequently incubating the crude lysates at 4°C for 1 hour with gentle rocking followed by incubation at 22°C for 1 hour with gentle rocking. Crude lysates of these in vitro-mixed samples, coexpressed T33-09 subunits, and individually expressed wild-type subunits were cleared by centrifugation and filtered through 0.22 μm filters. The filtered supernatants were purified by nickel affinity chromatography and eluted using a linear gradient of imidazole. Fractions containing pure protein(s) of interest were pooled, concentrated, and further purified on a Superdex 200 10/300 gel filtration column using 25 mM TRIS pH 8.0, 150 mM NaCl, 1 mM DTT as running buffer. Gel filtration fractions containing pure protein in the desired assembly state were pooled, concentrated, and stored at room temperature or 4°C for subsequent use in analytical size exclusion chromatography, electron microscopy, and X-ray crystallography.
Analytical size exclusion chromatography
Analytical SEC was performed on a Superdex 200 30/100 gel filtration column using the running buffer described above. Wild-type proteins and designed materials were each loaded onto the column at a concentration of 50 μM. The apparent molecular weights of the designed proteins were estimated by comparison with the corresponding wild-type proteins and previously determined nanocage standards.
Negative stain electron microscopy
Three microliters of SEC purified T33-31 at 0.1 mg mL−1 was applied to glow discharged, carbon coated 200-mesh copper grids (Ted Pella, Inc.), washed with Milli-Q water and stained with 0.75% uranyl formate as described previously.33 Grids were visualized on a 120 kV Tecnai Spirit T12 transmission electron microscope (FEI, Hillsboro, OR). All images were recorded using a bottom-mount Teitz CMOS 4k camera at 60,000x magnification at the specimen level. The contrast of all micrographs was enhanced in Fiji.34
Crystallization
T33-31 was crystallized using the hanging drop vapor diffusion method at room temperature. Crystals grew in hanging drops containing 0.11 µL of protein at 13 mg mL−1 and 0.1 µL of a 100 mL well solution containing 100 mM HEPES buffer at pH 7.5, 9% (w/v) polyethylene glycol 8000, and 11.7% (v/v) ethylene glycol. Crystals with tetrahedral or octahedral morphology grew over the course of about two to three days and reached dimensions of about 50–100 μm. For X-ray data collection a crystal was cryoprotected using the well solution augmented with 33% glycerol.
Crystallographic data collection, structure determination, and refinement
Diffraction data sets were collected at the Advanced Photon Source (APS) beamline 24-ID-C equipped with a Pilatus-6M detector. All data were collected at 100 K. Data were collected at a detector distance of 602 mm, with 0.5° oscillations, and at 0.979100 Å wavelength. The crystals showed diffraction to 3.25 Å. The XDS/XSCALE package35 was used to integrate, reduce, and scale the data. The data were reduced in P212121 space group symmetry. Based on the crystal symmetry, it was expected that the asymmetric unit of the crystal would contain a complete tetrahedral assembly composed of 24 peptide chains, corresponding with a Matthews coefficient of 2.44 Å3/Da and a 49.5% solvent content in the crystal. We used the PHASER program36 to determine the structure by molecular replacement, with the full model of the designed tetrahedron as the search model. Molecular replacement yielded a single solution with log-likelihood (LLG) 334. The symmetry axes of the tetrahedron do not overlap with the symmetry axes of the space group. After the solution was obtained, the structure was refined in iterative runs using the BUSTER.37–40 program. In each run, a single translation libration screw-motion (TLS) group was assigned per peptide chain and TLS was switched on for the first and third big-cycles (TLSbasic). We also used the automatic setup for noncrystallographic symmetry (autoncs), and limited the refinement resolution range to 100–3.4 Å. At each step, the quality of the refined model was assessed by COOT,41 and adjustments were made when there was support based on Fo-Fc difference maps. The limited resolution did not support the addition of any bound water molecules during refinement. The final R and Rfree values were 18.9% and 23.9%. The molecular replacement solution was further confirmed using omit maps (following simulated annealing in torsion angle space) generated around several regions of the protein using PHENIX.42 Omit maps were calculated around the following regions: residues 18–25 in chains A–L, residues 32–51 in chains A–L, residues 11–25 in chains M–X, residues 31–61 in chains M–X, residues 15–25 in chains A–L, and 11–25 in chains M–X. These fragments were chosen to be either in the core of one of the protein subunits, or at the designed interface between two proteins. In all cases, the density came back for each of the deleted fragments, validating the molecular replacement solution. Coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4ZK7.
Acknowledgments
TOY and YL acknowledge support from the BER program of the DOE Office of Science. The authors thank Michael Collazo for assistance with protein crystallization and the staff at NECAT beamline 24 ID of the Argonne National Laboratory APS for assistance with X-ray data collection.
Glossary
- APS
advanced photon source
- DOFs
degrees of freedom
- EM
electron microscopy
- PAGE
polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
- TLS
translation libration screw-motion
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- Janin J, Bahadur RP, Chakrabarti P. Protein-protein interaction and quaternary structure. Q Rev Biophys. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- Lai YT, King NP, Yeates TO. Principles for designing ordered protein assemblies. Trends Cell Biol. 2012;22:653–661. doi: 10.1016/j.tcb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- King NP, Lai YT. Practical approaches to designing novel protein assemblies. Curr Opin Struct Biol. 2013;23:632–638. doi: 10.1016/j.sbi.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Sinclair JC. Constructing arrays of proteins. Curr Opin Chem Biol. 2013;17:946–951. doi: 10.1016/j.cbpa.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Brodin JD, Ambroggio XI, Tang C, Parent KN, Baker TS, Tezcan FA. Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat Chem. 2012;4:375–382. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, Gonen T, Yeates TO, Baker D. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanci CJ, MacDermaid CM, Kang SG, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG. Computational design of a protein crystal. Proc Natl Acad Sci U S A. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranges PB, Machius M, Miley MJ, Tripathy A, Kuhlman B. Computational design of a symmetric homodimer using beta-strand assembly. Proc Natl Acad Sci U S A. 2011;108:20562–20567. doi: 10.1073/pnas.1115124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair JC, Davies KM, Venien-Bryan C, Noble ME. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- Lai YT, Cascio D, Yeates TO. Structure of a 16-nm cage designed by using protein oligomers. Science. 2012;336:1129. doi: 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J Am Chem Soc. 2012;134:375–385. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Harniman RL, Barnes FR, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, MJ Miles, RB Sessions, P Verkade, DN Woolfson. Self-assembling cages from coiled-coil peptide modules. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AL, Bromley EH, Bartlett GJ, Sessions RB, Sharp TH, Williams CL, Curmi PM, Forde NR, Linke H, Woolfson DN. Squaring the circle in peptide assembly: from fibers to discrete nanostructures by de novo design. J Am Chem Soc. 2012;134:15457–15467. doi: 10.1021/ja3053943. [DOI] [PubMed] [Google Scholar]
- Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO, Baker D. Accurate design of co-assembling multi-component protein nanomaterials. Nature. 2014;510:103–108. doi: 10.1038/nature13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradisar H, Bozic S, Doles T, Vengust D, Hafner-Bratkovic I, Mertelj A, Webb B, Sali A, Klavzar S, Jerala R. Design of a single-chain polypeptide tetrahedron assembled from coiled-coil segments. Nat Chem Biol. 2013;9:362. doi: 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YT, Reading E, Hura GL, Tsai KL, Laganowsky A, Asturias FJ, Tainer JA, Robinson CV, Yeates TO. Structure of a designed protein cage that self-assembles into a highly porous cube. Nat Chem. 2014;6:1065–1071. doi: 10.1038/nchem.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet AR, Noguchi H, Addy C, Simoncini D, Terada D, Unzai S, Park SY, Zhang KY, Tame JR. Computational design of a self-assembling symmetrical beta-propeller protein. Proc Natl Acad Sci U S A. 2014;111:15102–15107. doi: 10.1073/pnas.1412768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der BS, Kluwe C, Miklos AE, Jacak R, Lyskov S, Gray JJ, Georgiou G, Ellington AD, Kuhlman B. Alternative computational protocols for supercharging protein surfaces for reversible unfolding and retention of stability. PLoS One. 2013;8:e64363. doi: 10.1371/journal.pone.0064363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, IW Davis, S Cooper, A Treuille, DJ Mandell, F Richter, YE Ban, SJ Fleishman, JE Corn, DE Kim, S Lyskov, M Berrondo, S Mentzer, Z Popovic, JJ Havranek, J Karanicolas, R Das, J Meiler, T Kortemme, JJ Gray, B Kuhlman, D Baker, P Bradley. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F, Leaver-Fay A, Bradley P, Baker D, Andre I. Modeling symmetric macromolecular structures in Rosetta3. PLoS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System. 2012. 1.5: Schrödinger, LLC.
- Stephanopoulos N, Francis MB. Choosing an effective protein bioconjugation strategy. Nat Chem Biol. 2011;7:876–884. doi: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- Spicer CD, Davis BG. Selective chemical protein modification. Nat Commun. 2014;5:4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- Badran AH, Liu DR. In vivo continuous directed evolution. Curr Opin Chem Biol. 2015;24:1–10. doi: 10.1016/j.cbpa.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currin A, Swainston N, Day PJ, Kell DB. Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem Soc Rev. 2015;44:1172–1239. doi: 10.1039/c4cs00351a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsdorfer B, Woycechowsky KJ, Hilvert D. Directed evolution of a protein container. Science. 2011;331:589–592. doi: 10.1126/science.1199081. [DOI] [PubMed] [Google Scholar]
- Song WJ, Tezcan FA. A designed supramolecular protein assembly with in vivo enzymatic activity. Science. 2014;346:1525–1528. doi: 10.1126/science.1259680. [DOI] [PubMed] [Google Scholar]
- Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- Nannenga BL, Iadanza MG, Vollmar BS, Gonen T. 2013. Overview of electron crystallography of membrane proteins: crystallization and screening strategies using negative stain electron microscopy. Curr Protoc Protein Sci Chapter 17:Unit17 15.
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, JY Tinevez, DJ White, V Hartenstein, K Eliceiri, P Tomancak, A Cardona. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G. Bayesian statistical viewpoint on structure determination: basic concepts and examples. Methods Enzymol. 1997;276:361–423. doi: 10.1016/S0076-6879(97)76069-5. [DOI] [PubMed] [Google Scholar]
- Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr. 2004;60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- Roversi P, Blanc E, Vonrhein C, Evans G, Bricogne G. Modelling prior distributions of atoms for macromolecular refinement and completion. Acta Crystallogr D Biol Crystallogr. 2000;56:1316–1323. doi: 10.1107/s0907444900008490. [DOI] [PubMed] [Google Scholar]
- Bricogne G. Direct phase determination by entropy maximization and likelihood ranking: status report and perspectives. Acta Crystallogr D Biol Crystallogr. 1993;49:37–60. doi: 10.1107/S0907444992010400. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, AJ McCoy, NW Moriarty, R Oeffner, RJ Read, DC Richardson, JS Richardson, TC Terwilliger, PH Zwart. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information