

Abstract
Aim
The aim was to investigate the efficacy and safety of intravenous retosiban in women with spontaneous preterm labour.
Methods
This was a randomized, double-blind, placebo-controlled, phase 2 trial. Retosiban was administered intravenously for 48 h to women in spontaneous preterm labour between 300/7 and 356/7 weeks’ gestation with an uncomplicated singleton pregnancy in an in-patient obstetric unit. Outcome measures were uterine quiescence (primary endpoint), days to delivery, preterm delivery and safety.
Results
Uterine quiescence was achieved in 62% of women who received retosiban (n = 30) compared with 41% who received placebo (n = 34). The relative risk (RR) was 1.53 (95% credible interval [CrI] 0.98, 2.48; NS). Retosiban resulted in a significant increase in time to delivery compared with placebo (mean difference 8.2 days, 95% CrI 2.7, 13.74). This difference was consistent across all gestational ages. The proportion of preterm births in the retosiban and placebo groups was 18.7% (95% CrI 7.4%, 33.7%) and 47.2% (95% CrI 31.4%, 63.4%), respectively. The RR of preterm birth in women treated with retosiban was 0.38 (95% CrI 0.15, 0.81). There were no deliveries within 7 days in the retosiban group, but there were six (17.6%) births in the placebo group. The maternal, fetal and neonatal adverse events were comparable in the retosiban and placebo groups.
Conclusions
Intravenous administration of retosiban in women with spontaneous preterm labour was associated with a greater than 1 week increase in time to delivery compared with placebo, a significant reduction in preterm deliveries, a non-significant increase in uterine quiescence and a favourable safety profile.
Keywords: preterm birth, preterm labour, proof-of-concept study, retosiban, uterine quiescence
What is Already Known About This Subject
Preterm birth is the largest single cause of infant morbidity and mortality. Risks increase with earlier gestational age.
Current tocolytics have not been demonstrated to improve neonatal outcome.
A tocolytic that significantly prolongs pregnancy may improve neonatal and infant outcomes.
What This Study Adds
Retosiban prolonged pregnancy and reduced preterm birth.
Treatment was well tolerated and there was no indication of a safety issue for mother, fetus or newborn.
The results demonstrate proof-of-concept in the treatment of threatened spontaneous preterm labour.
Introduction
Preterm birth is the largest single cause of infant morbidity and mortality and is frequently associated with long term disability 1–4. Current tocolytics may not be effective in delaying delivery for a number of possible reasons 1,5–7, the drug target may be inappropriate, the plasma concentration may be ineffective or redundant mechanisms may allow the process of labour to continue. Clinicians remain optimistic that an effective tocolytic will be developed which can significantly prolong pregnancy and improve neonatal and infant outcomes in appropriate pregnancies.
Atosiban, a mixed vasopressin (V1a)/oxytocin receptor antagonist, is licensed in the European Union as a tocolytic for parenteral administration 8. There are no tocolytics currently approved in North America. Many therapies are used off-label throughout the world, including β-sympathomimetics, prostaglandin synthase inhibitors and calcium channel blockers 9, although none has been conclusively demonstrated to delay delivery and improve neonatal or infant outcomes. Retosiban, a specific, high affinity oxytocin receptor antagonist, is now in development for the inhibition of uterine contractions in spontaneous preterm labour. Retosiban is an oxazole diketopiperazine oxytocin antagonist with good bioavailability and nanomolar affinity for the human oxytocin receptor (Ki = 0.65 n m), with >1400-fold selectivity over the closely related vasopressin receptors 10. Nomenclature for the vasopressin and oxytocin receptors is as specified in the Guide to Receptors and Channels (GRAC), 5th edition 11.
There is evidence from in vitro and in vivo studies that retosiban inhibits spontaneous and induced uterine contractions. Phase 1 studies have demonstrated safety in non-pregnant volunteers (unpublished data on file, study OTA 105101; Michael Fossler, PharmD, PhD, FCP, Senior Director, CPMS – US, RD Projects Clinical Platforms & Sciences, GlaxoSmithKline, King of Prussia, PA, USA) and retosiban has been evaluated in pregnant women to determine the dose range and confirm proof of mechanism based on suppression of uterine contractions 12,13. The pilot dose ranging studies were done on 29 women in threatened preterm labour between 34 and 35+6 weeks’ gestation. These studies (to be published separately) demonstrated rapid absorption of retosiban with plasma concentrations consistent with non-pregnant volunteers. The safety profile was similar to placebo. Retosiban was associated with a reduction in uterine activity and a marked increase in the number of days to delivery. In the current report, proof-of-concept was further extended to confirm the efficacy and safety of intravenous retosiban in women experiencing spontaneous preterm labour between 300/7 and 356/7 weeks’ gestation with an uncomplicated singleton pregnancy.
Methods
Study design
This was a double-blind, placebo-controlled study in women admitted with spontaneous preterm labour between 300/7 and 356/7 weeks’ gestation (registration number NCT00404768; http://clinicaltrials.gov/ct2/show/NCT00404768?term=be+NCT00404768&rank=1.)
Eligible women were stratified by gestational age, 300/7 to 326/7 weeks or 330/7 to 356/7 weeks and randomized 1: 1 to intravenous retosiban or placebo. Magnesium sulphate for neuroprotection and antenatal steroids were allowed.
The retosiban dosing regimen was designed to achieve a mean steady-state concentration of 75 ng ml–1 (informed by pre-clinical data, the dose-ranging study and studies in non-pregnant healthy volunteers) using a loading dose of 6 mg over 5 min and a continuous infusion of 6 mg h–1 over 48 h. At any point after 1 h of receiving the 6 mg h–1 rate, a single dose increase was permitted in women who did not respond to treatment. In this case, the infusion rate could be increased to 12 mg h–1 after an additional 6 mg loading dose. An adequate treatment response was defined as a clinically relevant reduction in the frequency of contractions without an increase in cervical dilatation. Women who did not respond to the dose increase could discontinue study medication and receive an alternative rescue tocolytic at the discretion of the investigator.
A group sequential design was used with up to three planned interim analyses (four planned cohorts of 16 women each). At each interim analysis, the study could have been stopped for success or futility based on a priori stopping rules.
Eligible women
Eligible women were 18 to 45 years of age, had a singleton pregnancy between 300/7 and 356/7 weeks’ gestation based on best available obstetric estimate, were having six or more uterine contractions per hour of at least 30 s duration by external cardiotocography (CTG) with cervical dilatation ≥1 to ≤4 cm, and had intact fetal membranes.
Excluded were women with indications for delivery, such as pre-eclampsia or fetal compromise, women with contraindications to tocolysis, such as clinically apparent intrauterine infection or placental abruption and women with comorbid conditions with the potential to complicate pregnancy and outcomes, such as hypertension, insulin-dependent diabetes or substance abuse.
Procedure
Following confirmation of eligibility, maternal examination and investigations were done (vital signs, 12-lead electrocardiogram (ECG), biochemistry, haematology and urinalysis). An ultrasound was done to determine amniotic fluid index (AFI) and a CTG for fetal heart rate monitoring. These tests were not repeated if they had been done in the 6 h before consent. Within 1 h before dosing, the contraction rate and duration were determined, a vaginal examination was done to assess cervical dilatation and fetal heart rate was recorded. Dosing began at time zero. After the start of treatment, the following assessments were conducted at specified time points, maternal blood pressure, heart rate, ECG, uterine contractions, physical examination, clinical laboratory tests, AFI and fetal heart rate. Women who discontinued study medication and their infants were followed for safety.
Study endpoints
The primary pharmacodynamic endpoint (response rate) was the proportion of women who achieved and maintained uterine quiescence, defined as four or fewer contractions per hour and <1 cm change in cervical dilatation at hour 6. The principal efficacy endpoints were days to delivery (a tertiary endpoint) and preterm births (<37 weeks). The safety endpoints were aimed at detecting adverse drug effects based on maternal monitoring (ECG, laboratory results, vital signs and adverse events), fetal monitoring (CTG, modified biophysical profile consisting of AFI and non-stress test and adverse events) and neonatal observations (Apgar scores, growth parameters at birth and follow-up, gross development and adverse events).
Follow-up
Women were discharged 6 h after the end of the infusion or at the discretion of the investigator. Hospital records were reviewed to determine gestational age at birth, Apgar scores and weight, length and head circumference at birth. Infants were assessed approximately 1 month after birth. Neonatal adverse events were determined from either neonatal records or maternal reporting.
Statistical analyses
The planned Bayesian statistical analysis declared statistical significance if the 95% credible interval (corresponding to the confidence interval) excluded 0 (for a difference) or 1 (for relative risk [RR]). Partially informative priors (probability distribution according to available data) from the dose-ranging study were used in the analyses of proportions of women achieving uterine quiescence and days to delivery. This was analogous to including data from a certain number of women from the prior study, as well as observed data from the present study, according to standard application of Bayes’ theorem 14. A non-informative prior (analogous to analysis of the observed data from the study) was used for the analysis of the proportion of preterm births and a sensitivity analysis (i.e. to evaluate the influence of the partially informative priors) for the endpoints of uterine quiescence and days to delivery. The planned sample size (n = 64) provided at least 86% power to detect a 40% absolute difference, or RR of 2.6, in the proportion of women achieving uterine quiescence.
The safety and analysis populations were defined as all women who received at least one dose of study drug. For the primary endpoint of uterine quiescence, women who stopped the study drug within 6 h of time zero were recorded as non-responders. For analyses of days to delivery and proportion of preterm births, actual birth data were used.
Uterine quiescence response rates for each treatment group and the RRs (defined as the ratio of retosiban to placebo response rates) along with 95% credible intervals (CrI) were estimated by using a Bayesian formulation of Fisher’s exact test. The proportion of women delivering preterm was similarly analyzed. The mean difference in days to delivery between treatment groups, along with the 95% credible interval, was estimated from a Bayesian formulation of an analysis of covariance model with gestational age at entry fit as a continuous covariate. No other hypothesis tests were performed, according to the protocol. All statistical analyses were performed with SAS/IML® (SAS Software, Cary, NC, USA) or the R open-source software environment for statistical computing.
Details of ethics approval
Written informed consent was obtained from all women. The study was conducted in compliance with Good Clinical Practice and the Declaration of Helsinki (2008) and was approved by the following institutional review boards: CPP Ile-de-France XI, Saint-Germain-en-Laye 78105, France, ref. 174142/063858, St Joseph’s Hospital and Medical Center Institutional Review Board for Human Research, Phoenix, AZ 85013, USA, ref. 105422/034702, Sandhills Multi-Institutional Review Board, Pinehurst, NC 28374, USA, ref. 135920/048485, University of Tennessee Graduate School of Medicine Institutional Review Board, Knoxville TN 37920, USA, ref. 129343/045389, Asan Medical Center IRB, Seoul 138-736, Korea, ref. 170633/061966; Singhealth Office of Research169611, Singapore, ref. 136524/048769, Western Institutional Review Board, Olympia, WA 98502, USA, ref. 184592/069282, TMC Institutional Review, Tucson, AZ 85712, USA, ref. 166865/061308, Comite de Etica en Investigacion del Hospital Universitario Clinica San Rafael, Bogota/Cundinamarca, Colombia, ref. 205896/078327, Ethics Committee for Multicenter Trials, Sofia 1504, Bulgaria, ref. 171264/061999, Severance Hospital IRB, Seodaemun-gu, Seoul 120-752, Korea, ref. 170632/062795, Comite Etico de Investigacion Clinica, Hospital Universitario Vall d’Hebron 08035, Barcelona, Spain, ref. 171272/062160, CPP Ile-de-France XI, Saint-Germain-en-Laye 78105, France, ref 174154/063864, Forsyth Medical Center Institutional Review Board, Winston-Salem, NC 27103, USA, ref. 037174/048486, Comite de Etica Medica en Investigacion, Bogota, Colombia, ref. 205915/078324, ArrowHead Regional Medical Center IRB, Colton, CA 92324, USA, ref. 120625/040718, Ethics Committee for Multicenter Trials, Sofia 1303, Bulgaria, ref. 171265/062002 and University of Texas Medical Branch IRB, Galveston, TX 77555, USA, ref. 126172/034700.
Results
Seventeen centres enrolled, randomized and treated 64 women (Figure1). One additional subject was randomized but not dosed because of labour progression. Her data are not included in this report. Six women did not complete the retosiban infusion due to lack of response (n = 2) or a decision on the part of the subject or investigator (n = 4). The principal reasons for discontinuation of the infusion in the placebo group (n = 12) were lack of response (n = 7), adverse event (n = 3) or subject or investigator decision (n = 2).
Figure 1.
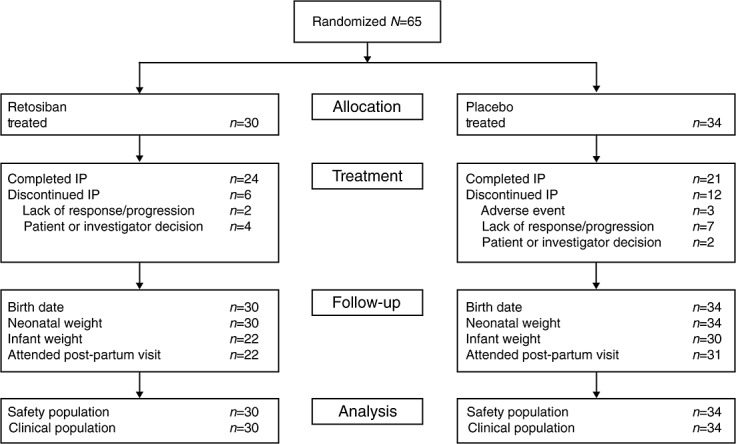
Disposition of women in proof-of-concept study (IP, investigational product)
Demographic and baseline characteristics of women participating in the study are summarized in Table1. The groups were well matched. Patients were primarily White women in their mid-to-late twenties, and ranging in age from 18 to 41 years. There was a slight imbalance in randomization, with fewer retosiban patients (9/30 vs. 15/34) randomized to the earlier gestational age group, although the increased rate of discontinuation of drug in the placebo group resulted in a similar number of patients completing the infusion in each study arm (22 and 20 for retosiban and placebo, respectively). A protocol amendment was introduced after the first 12 women were enrolled for stratification by gestational age to ensure future balanced randomization. Although there is an imbalance of the number of women in the early gestational age group across treatments, analyses indicated that the effect of retosiban vs. placebo was similar across gestational ages.
Table 1.
Summary of subject demographic and baseline characteristics
| Treatment group | Retosiban | Placebo |
|---|---|---|
| Number treated | 30 | 34 |
| Age (years) mean (range) | 25.2 (18–39) | 27.8 (19–41) |
| BMI (kg m–2) mean (SD) | 25.8 (4.4) | 26.9 (4.3) |
| Race, n (%) | ||
| African American | 4 (13) | 2 (6) |
| American Indian or Alaskan Native | 1 (3) | 2 (6) |
| Asian | 2 (7) | 6 (18) |
| White – Arabic/North African | 1 (3) | 0 |
| White – White/Caucasian/European | 22 (73) | 24 (71) |
| Ethnicity, n (%) | ||
| Hispanic or Latino | 10 (33) | 12 (35) |
| Non-Hispanic or Latino | 20 (67) | 22 (65) |
| Gestational age, n (%) | ||
| 306/7–326/7 | 9 (30) | 15 (44) |
| 330/7–356/7 | 21 (70) | 19 (56) |
| Cervical dilatation, median cm | 2 | 2 |
| Contraction frequency, mean (SD) | 12.5 (7.5) | 12.9 (6.0) |
| Prior tocolytic, n (%) | 6 (20) | 10 (29) |
BMI, body mass index.
Pharmacodynamic and efficacy outcomes
Uterine contractions
Uterine quiescence was achieved in 62% of women who received retosiban compared with 41% who received placebo. The RR was 1.53 (95% CrI 0.98, 2.48; NS). The mean baseline contraction rates were 12.5 and 12.9 h–1 in the retosiban and placebo groups, respectively. The rates at hour 6 were 3.7 and 5.3 h–1, respectively (Figure2).
Figure 2.
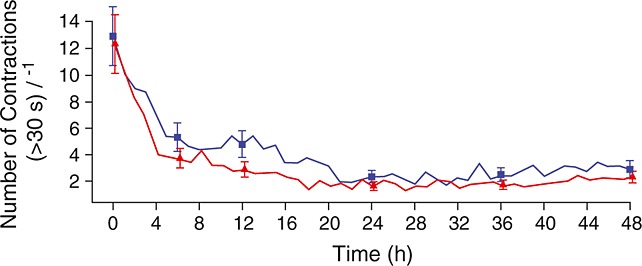
Mean uterine contraction frequency.  , retosiban, (n = 30);
, retosiban, (n = 30);  , placebo (n = 34)
, placebo (n = 34)
Intravenous infusion rates were increased in 37% (11/30) and 62% (21/34) of women assigned to retosiban and placebo, respectively. Ten women received rescue tocolysis, three (10%) in the retosiban group and seven (21%) in the placebo group. Rescue tocolytics included magnesium sulphate (n = 6), nifedipine (n = 3), fenoterol (n = 2), ritodrine (n = 1), atosiban (n = 1) and salbutamol (n = 1).
Time to delivery and preterm births
The time to delivery was longer in women treated with retosiban compared with placebo (mean difference 8.2 days [95% CrI 2.7, 13.74] and median time to delivery 34.5 and 25 days, respectively). There were no deliveries within 7 days in the retosiban group but six births (17.6%) in the placebo group. The time to delivery at each gestational age for women who received retosiban or placebo is shown in Figure3.
Figure 3.
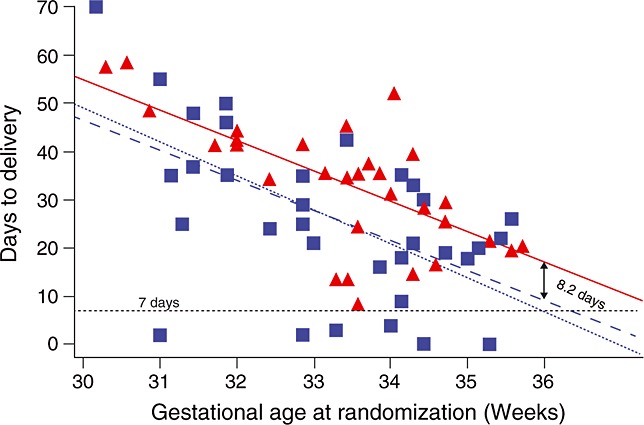
Scatter plot of days to delivery vs. gestational age at randomization. The solid line represents the time in days to achieve 37 weeks. Women falling below the solid line delivered before 37 weeks and those above the line delivered after 37 weeks. The dashed red and blue lines display the linear fit for time to delivery for the retosiban and placebo groups, respectively. The difference between the red and blue lines represents the difference between retosiban and placebo in mean days to delivery, which was consistent across gestational ages.  , retosiban;
, retosiban;  , placebo
, placebo
The proportion of preterm births in the retosiban and placebo groups was 18.7% (95% CrI 7.4%, 33.7%) and 47.2% (95% CrI 31.4%, 63.4%), respectively. The RR of preterm birth in women treated with retosiban was 0.38 (95% CrI 0.15, 0.81).
Statistical inference
Partially informative priors from the dose-ranging study (data from 14 subjects treated with retosiban i.v. and five treated with placebo i.v.) were included in the evaluation of uterine quiescence and time to delivery. The analyses were repeated using non-informative priors, which indicated that the use of these partially informative priors from the dose-ranging study had no impact on the statistical inferences, although there were minor changes to the point estimates and 95% CrI, as shown in Table2.
Table 2.
Effect of partially informative vs. non-informative prior
| Prior | Relative risk of uterine quiescence | Difference in time to delivery |
|---|---|---|
| Partially informative prior using data from phase 2 dose-ranging study | 1.53 (95% CrI 0.98, 2.48) | 8.2 (95% CrI 2.7, 13.74) |
| Non-informative prior (phase 2a proof of concept study only) | 1.54 (95% CrI 0.94, 2.63) | 7.3 (95% CrI 1.0, 13.5) |
CrI, credible interval.
Safety
Maternal assessments
Results from maternal ECGs, vital signs and clinical laboratory assessments were comparable for both groups. There were no significant changes in maternal blood pressure with treatment. Mean systolic and diastolic blood pressures following infusion of retosiban or placebo (0–48 h) are shown in Figure4. Maternal adverse events and serious adverse events were generally similar across treatment groups. There were 14/30 (47%) adverse events and 2/30 (7%) serious adverse events reported in the retosiban group, compared with 17/34 (50%) and 2/34 (6%) in the placebo group. Adverse events are displayed in Table3 and a summary of serious adverse events is shown in Table4. There was one report of post-partum haemorrhage that occurred more than 30 days after the completion of retosiban. The event was considered not related to treatment.
Figure 4.
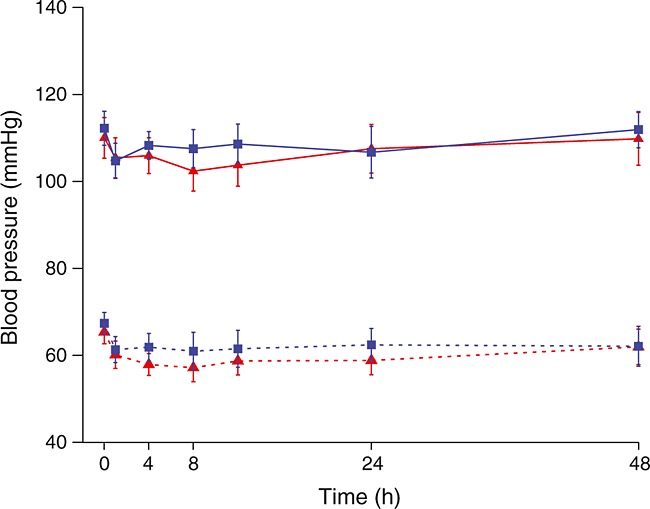
Maternal blood pressure (mean and 95% CrI) following administration of retosiban or placebo (0–48 h).  , retosiban, systolic;
, retosiban, systolic;  , placebo, systolic;
, placebo, systolic;  , retosiban, diastolic;
, retosiban, diastolic;  , placebo, diastolic
, placebo, diastolic
Table 3.
Maternal adverse events*
| Adverse event | Retosiban n = 30 n (%) | Placebo n = 34 n (%) |
|---|---|---|
| Headache | 4 (13) | 1 (3) |
| Dyspepsia | 3 (10) | 1 (3) |
| Back pain | 2 (7) | 0 |
| Nausea | 2 (7) | 1 (3) |
| Abdominal pain, upper | 1 (3) | 1 (3) |
| Amniotic fluid, decreased | 1 (3) | 1 (3) |
| Constipation | 1 (3) | 1 (3) |
| Premature rupture of membranes | 1 (3) | 2 (6) |
| Paraesthesia | 0 | 2 (6) |
| Polyhydramnios | 0 | 2 (6) |
| Post-partum depression | 0 | 2 (6) |
Listed are all adverse events, regardless of treatment group. Women with more than one occurrence of the same type of adverse event are listed only once.
Table 4.
Maternal serious adverse events*
| Treatment | Adverse event | Investigator assessment of drug relatedness | Comments |
|---|---|---|---|
| Retosiban | Post-partum haemorrhage | No | The event occurred >30 days post-discontinuation of retosiban |
| Retosiban | Musculoskeletal pain | No | The event was secondary to a motor vehicle accident and occurred >26 days post-discontinuation of retosiban |
| Placebo | Amniotic fluid volume decreased | No | AFI decreased to 5.8 cm 2 days after completion of placebo infusion. Event resolved as AFI subsequently increased to 6.6 cm and 7.1 cm |
| Placebo | Hypertension | No | Elevated systolic and diastolic blood pressure at 3-week post-partum visit, approximately 5 weeks after completion of placebo infusion. Admitted to hospital and blood pressure normalized with antihypertensives |
AFI, amniotic fluid index.
A serious adverse event is any untoward medical occurrence that results in death, is life threatening, results in hospitalization or prolongation of existing hospitalization, results in disability/incapacity, is a congenital anomaly/birth defect, drug-induced liver injury, or any other event deemed medically important.
Fetal assessments
There were no significant changes in the modified biophysical profile and values were similar across all treatment groups. Fetal heart rate parameters were not significantly different in women treated with retosiban or placebo. In one woman in the placebo group, there was a fetal heart rate deceleration that resolved spontaneously. Mean fetal and maternal heart rates following maternal administration of retosiban or placebo (0–48 h) are shown in Figure5.
Figure 5.
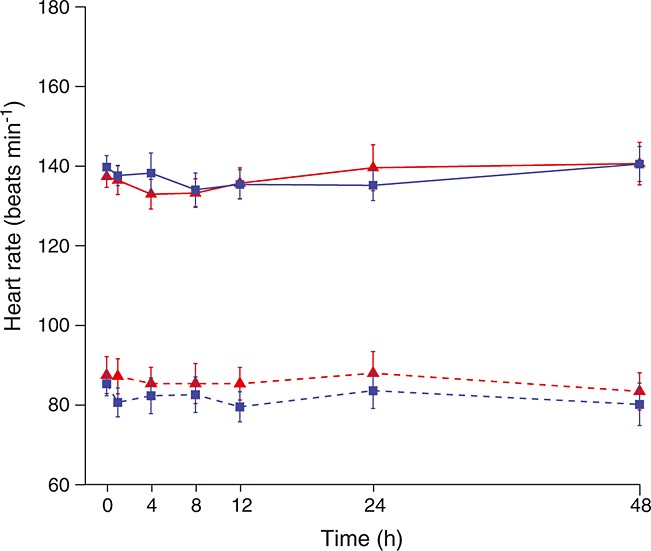
Maternal and fetal heart rate (mean and 95% CrI) following maternal treatment with retosiban or placebo (0–48 h).  , retosiban, fetal;
, retosiban, fetal;  , placebo, fetal;
, placebo, fetal;  , retosiban, mother;
, retosiban, mother;  , placebo, mother
, placebo, mother
Neonatal assessments
Apgar scores and growth parameters were consistent with those expected for the estimated gestational age at birth and were similar across groups. Neonatal endpoints and gross developmental follow-up at approximately 1 month are summarized in Table5A, B. Adverse events were reported in 4/30 (13%) and 7/34 (21%) of retosiban- and placebo-exposed neonates, respectively. Adverse events in newborns are summarized in Table6. Two of 30 (7%) neonates whose mothers received retosiban had a serious adverse event reported compared with 3/34 (9%) of placebo-exposed neonates (Table7). Neonatal adverse events and serious adverse events were generally associated with preterm birth complications or had confounding risk factors.
Table 5.
A,B. Outcomes
| Treatment | Time point | n | n | Mean (SD) | |
|---|---|---|---|---|---|
| A. Neonatal endpoints | |||||
| Apgar score at 1 min | Retosiban | Birth | 30 | 30 | 8.3 (0.88) |
| PBO | Birth | 34 | 34 | 8.2 (0.77) | |
| Apgar score at 5 min | Retosiban | Birth | 30 | 30 | 9 (0.53) |
| PBO | Birth | 34 | 34 | 8.9 (0.34) | |
| Length (cm) | Retosiban | Birth | 30 | 28 | 47.8 (2.78) |
| PBO | Birth | 34 | 34 | 47.8 (3.39) | |
| Retosiban | Follow-up | 30 | 22 | 50.6 (2.89) | |
| PBO | Follow-up | 34 | 29 | 51.3 (3.3) | |
| Neonatal weight (g) | Retosiban | Birth | 30 | 30 | 3099 (512.6) |
| PBO | Birth | 34 | 34 | 2940 (585.0) | |
| Retosiban | Follow-up | 30 | 22 | 3427 (635.5) | |
| PBO | Follow-up | 34 | 30 | 3602 (659.5) | |
| Neonatal head circumference (cm) | Retosiban | Birth | 30 | 27 | 33.3 (1.78) |
| PBO | Birth | 34 | 34 | 33.1 (1.63) | |
| Retosiban | Follow-up | 30 | 21 | 35.1 (1.75) | |
| PBO | Follow-up | 34 | 27 | 35.2 (1.77) | |
| Time to follow-up (days) | Retosiban | Follow-up | 30 | 22 | 18.4 (15.67) |
| PBO | Follow-up | 34 | 31 | 25.1 (20.45) | |
| B. Gross development reported at follow-up | |||||
| Treatment | n | Normal | Abnormal | Not reported | |
| Gross development | Retosiban | 30 | 22 | 0 | 8 |
| PBO | 34 | 30 | 1 | 3 | |
Table 6.
Adverse events occurring in neonates*
| Adverse event | Retosiban n = 30 (%) | Placebo n = 34 (%) |
|---|---|---|
| Hypoglycaemia | 2 (7) | 1 (3) |
| Hyperbilirubinaemia | 1 (3) | 5 (15) |
| Jaundice | 1 (3) | 1 (3) |
| Anaemia | 0 | 2 (6) |
| Hypercalcaemia | 0 | 2 (6) |
| Malnutrition† | 0 | 2 (6) |
Listed are all adverse events, regardless of treatment group. Neonates with more than one occurrence of the same type of adverse event are listed only once.
Medical Dictionary for Regulatory Activities (MedDRA) preferred term for event reported as nutrition imbalance (infant).
Table 7.
Neonatal serious adverse events*
| Serious adverse event | Retosiban n = 30 (%) (events occurred in two neonates) | Placebo n = 34 (%) (events occurred in three neonates) |
|---|---|---|
| Hyperbilirubinaemia | 1† (3) | 2‡,§ (6%) |
| Hypoglycaemia | 1¶ (3) | 0 |
| Jaundice | 1¶ (3) | 0 |
| Meconium in amniotic fluid | 1¶ (3) | 0 |
| Apnoea | 0 | 1§ (3) |
| Malnutrition** | 0 | 2ठ(6) |
| Respiratory distress | 0 | 1§ (3) |
| Tachypnoea | 0 | 1†† (3) |
A serious adverse event is any untoward medical occurrence that results in death, is life threatening, results in hospitalization or prolongation of existing hospitalization, results in disability/incapacity, is a congenital anomaly/birth defect, drug-induced liver injury, or any other event deemed medically important.
37 week infant. Investigator noted a maternal/fetal blood group incompatibility (mother A+; infant O+).
Delivered at 33 weeks’ gestation.
Delivered at 31 weeks’ gestation.
Infant born to a mother with gestational diabetes (risk of neonatal hypoglycaemia) following post-date induction (risk of meconium). Investigator attributed neonatal jaundice to maternal-fetal blood group incompatibility.
Medical Dictionary for Regulatory Activities (MedDRA) preferred term for event reported as nutrition imbalance (infant).
Delivered at 346/7 weeks’ gestation.
Discussion
The results of this study demonstrate that short term treatment with retosiban significantly prolongs pregnancy and reduces the incidence of preterm birth. Few, if any, placebo-controlled studies have demonstrated an effect of this magnitude 7,15,16. This is encouraging, as data on the efficacy of current tocolytics are contradictory and adverse effects have been reported in mothers or offspring 17. To date, no tocolytic has been demonstrated effectively to delay delivery and improve outcome, although some agents, such as the β-sympathomimetics, have been demonstrated to delay delivery 18. This represents a dilemma, since it is known that babies born at later gestational ages have lower morbidity and mortality, yet delaying delivery has not been shown to improve outcome 9. There are many possible explanations, such as methodological issues in the clinical trials or failure to use the time to perform procedures that improve outcome. More worrying is the possibility that administration of a tocolytic could theoretically maintain the fetus in an adverse intrauterine environment. In light of this, it is important that patients are carefully selected for tocolysis, with exclusion of those having aetiological factors, such as abruption or intrauterine infection, which could adversely influence the outcome. The results of this study demonstrate a significant prolongation in time to delivery in women administered retosiban for the treatment of preterm labour and form the basis for phase 3 trials that will determine whether the prolongation of the pregnancy is associated with improved outcomes in the offspring.
Additional studies are in progress to elucidate further the mechanisms of retosiban action on the myometrium and other tissues, such as the amnion. Interestingly, in the present study there was a marked increase in the duration of pregnancy following a single, time-limited infusion. This observation raises questions about the pathophysiology of preterm labour, and suggests that in this patient population the initiating stimulus may be discrete, self-limited and non-recurrent.
The retosiban infusion was well tolerated, and there was no indication of a safety issue for mother, fetus or newborn. A theoretical safety concern with retosiban is an increased risk of post-partum haemorrhage if delivery occurs within a few hours of infusion. A case of post-partum haemorrhage occurred more than 30 days after retosiban and was not considered related to treatment. Because of the mechanism of action of retosiban and the role of oxytocin in promoting haemostasis after delivery, it will be important to monitor similar events in subsequent trials.
Treatment discontinuations and dose escalations provide evidence that the initial retosiban dosing regimen, consisting of a 6 mg loading dose and 6 mg h–1 infusion, is the lowest effective dose. More women discontinued study drug in the placebo group than in the retosiban group. In addition, more women taking placebo had their infusion rates increased compared with women taking retosiban (65% vs. 37%). As almost 40% of women on retosiban required a dose increase, it is unlikely that a dose lower than 6 mg h–1 would provide adequate effect. Taken together, the data from this study support the initial 6 mg h–1 infusion rate as the lowest effective dose for the majority of women, while recognizing that a considerable number of women may require higher doses to attain a satisfactory response.
Strengths and limitations
The strength of this study is that it provides evidence for the efficacy, safety and tolerability of a specific oxytocin receptor antagonist for the treatment of spontaneous preterm labour, and thus represents a proof-of-concept study in this population. It is perhaps surprising that the effects on time to delivery, preterm birth and use of rescue tocolysis were so marked, given the heterogeneous population typical of such studies. The difference in the effect on uterine quiescence between active treatment and placebo groups was not statistically significant, although there was a markedly higher rate of quiescence in women who received retosiban (62% vs. 41%). A potential limitation of this study is that it was not, nor was it intended to be, a definitive trial to demonstrate the effectiveness of retosiban in clinical practice, nor was it designed to demonstrate improved neonatal morbidity and mortality. There was no long term neonatal follow-up. The inclusion criteria, similar to many prior trials investigating tocolytic agents, did not include fetal fibronectin or cervical ultrasound, which may be used in clinical practice. Furthermore, women at early gestational ages (<30 weeks) were not recruited.
The advent of a therapeutic intervention that could significantly prolong pregnancy in patients with spontaneous preterm labour would be invaluable. While the mode of action of retosiban in preterm labour is not fully understood, this placebo-controlled study found that the short term administration of retosiban halted preterm labour and prolonged pregnancy to a degree that could have a positive impact on perinatal outcomes.
In conclusion, this phase 2 study provides proof-of-concept evidence for the efficacy and safety of retosiban, a prerequisite for investment in phase 3 clinical trials. Whether the tocolytic effect of retosiban results in improved neonatal and infant outcomes following preterm labour at early and late gestational ages remains to be determined.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare ST received consultancy fees, travel reimbursement and/or honoraria payments with contract research and commercial organizations including GlaxoSmithKline, HM received grant funding for study administration, travel reimbursement and consultancy fees from GlaxoSmithKline and GV received grant funding for study administration and consultancy fees from GlaxoSmithKline. JS, BS, MF, TM, MP, and KB are employees of GlaxoSmithKline and own shares of GlaxoSmithKline company stock.
The authors would like to acknowledge the contribution of the investigators (funded by GlaxoSmithKline) and their patients who participated in this study. The authors also wish to thank the following individuals for their contributions: Sergio Forero-Swanhaeuser, MD, Trish McBride, MS, of GlaxoSmithKline and Annette Ferrell, formerly of GlaxoSmithKline, for clinical study oversight and management; Kay S. Tatsuoka for statistical design input and analysis, Douglas Wicks, MPH, CMPP, from GlaxoSmithKline, for management of manuscript development and Rosemary Perkins of Caudex Inc. for editorial assistance in preparation of the manuscript (funded by GlaxoSmithKline). This study was funded by GlaxoSmithKline, Research Triangle Park, NC, USA (NCT00404768).
Contribution to authorship
All authors met the International Committee for Medical Journal Editors criteria for authorship, were fully involved in manuscript development, and assume responsibility for the direction and content. Steve Thornton had a major role in the study design, implementation, data review and interpretation, Hugh Miller, Guillermo Valenzuela, Jerry Snidow and Brendt Stier were involved in study design, data analysis and data interpretation, Michael Fossler was involved in the pharmacological aspects of study design, data analysis and data interpretation, Timothy Montague was involved in statistical aspects of study design, data analysis, and data interpretation and Marcy Powell and Kathleen Beach were involved in data analysis and data interpretation.
Investigators
Jean-Marc Ayoubi, Hôpital Foch, Suresnes, France; James Balducci, St. Joseph’s Hospital and Medical Center, Phoenix, AZ, USA; John Byron, Southern Pines Women’s Health Center, Southern Pines, NC, USA; Bobby Howard, Regional Obstetrical Consultants, Knoxville, TN, USA; Ahm Kim, Asan Medical Center, Seoul, Republic of Korea; Yun Chiang Kwek, KK Women’s and Children’s Hospital, Singapore; David Lewis, Greater Cincinnati Ob/Gyn Associates, Cincinnati, OH, USA; Hugh Miller, Tucson Medical Center, Tucson, AZ, USA; Javier Ardila Montealegre, Hospital Universitario Clinica San Rafael, Bogotá, Colombia; Assen Nikolov, SHAT ObGyn, Sofia, Bulgaria; Yong Won Park, Severance Hospital, Seoul, Republic of Korea; Luis Cabero Roura, Hospital Vall d’Hebrón, Barcelona, Spain; Patrick Rozenberg, CHI Poissy-Saint Germain, Poissy, France; Melvin Seid, Lyndhurst Gynecologic Associates, Winston-Salem, NC, USA; Alfonso Correa Uribe, Clinica del Country, Bogotá, Colombia; Guillermo Valenzuela, Arrowhead Medical Center, Colton, CA, USA; Roumen Velev, SHAT ObGyn, Sofia, Bulgaria; Tony Wen, The University of Texas Medical Branch at Galveston, TX, USA.
References
- American College of Obstetrics and Gynecologists, Committee on Practice Bulletins–Obstetrics. ACOG practice bulletin no. 127: Management of preterm labour. Obstet Gynecol. 2012;119:1308–17. doi: 10.1097/AOG.0b013e31825af2f0. [DOI] [PubMed] [Google Scholar]
- Engle WA, Tomashek KM, Wallman C. ‘Late-preterm’ infants: a population at risk. Pediatrics. 2007;120:1390–401. doi: 10.1542/peds.2007-2952. [DOI] [PubMed] [Google Scholar]
- Tucker J, McGuire W. Epidemiology of preterm birth. BMJ. 2004;329:675–8. doi: 10.1136/bmj.329.7467.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigal S, Doyle LW. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet. 2008;371:261–9. doi: 10.1016/S0140-6736(08)60136-1. [DOI] [PubMed] [Google Scholar]
- Royal College of Obstetricians and Gynaecologists. 2011. Tocolytic drugs for women in preterm labour. Green-top Guideline No 1B, February [online]. Available at http://www.rcog.org.uk/womens-health/clinical-guidance/tocolytic-drugs-women-preterm-labour-green-top-1b (last accessed 24 July 2014)
- Roos C, Spaanderman ME, Schuit E, Bioemenkam KW, Bolte AC, Cornette J, Duvekot JJ, van Eyck J, Franssen MT, de Groot CJ, Kok JH, Kwee A, Merién A, Nij Bijvank B, Opmeer BC, Oudijk MA, van Pampus MG, Papatsonis DN, Porath MM, Scheepers HC, Scherjon SA, Sollie KM, Vijgen SM, Willekes C, Mol BW, van der Post JA, Lotgering FK APOSTEL-II Study Group. Effect of maintenance tocolysis with nifedipine in threatened preterm labour on perinatal outcomes: a randomized controlled trial. JAMA. 2013;309:41–7. doi: 10.1001/jama.2012.153817. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos L, Kaunitz AM, Gaudier FL, Delke I. Efficacy of maintenance therapy after acute tocolysis: A meta-analysis. Am J Obstet Gynecol. 1999;181:484–90. doi: 10.1016/s0002-9378(99)70582-x. [DOI] [PubMed] [Google Scholar]
- Goodwin TM, Paul R, Silver H, Spellacy W, Parsons M, Chez R, Hayashi R, Valenzuela G, Creasy GW, Merriman R. The effect of the oxytocin antagonist atosiban on preterm uterine activity in the human. Am J Obstet Gynecol. 1994;170:474–8. doi: 10.1016/s0002-9378(94)70214-4. [DOI] [PubMed] [Google Scholar]
- Haas DM, Caldwell DM, Kirkpatrick P, McIntosh JJ, Welton NJ. Tocolytic therapy for preterm delivery: systematic review and network meta-analysis. BMJ. 2012;345 doi: 10.1136/bmj.e6226. : e6226. doi: 10.1136/bmj.e6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddle J, Allen MJ, Borthwick AD, Brooks DP, Davies DE, Edwards RM, Exall AM, Hamlett C, Irving WR, Mason AM, McCafferty GP, Nerozzi F, Peace S, Philp J, Pollard D, Pullen MA, Shabbir SS, Sollis SL, Westfall TD, Woollard PM, Wu C, Hickey DM. The discovery of GSK221149A: A potent and selective oxytocin antagonist. Bioorg Med Chem Lett. 2008;18:90–4. doi: 10.1016/j.bmcl.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th ed. Br J Pharmacol. 2011;164(Suppl. 1) doi: 10.1111/j.1476-5381.2011.01649_1.x. ): S1S324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty GP, Pullen MA, Wu C, Edwards RM, Allen MJ, Wollard PM, Borthwick AD, Liddle J, Hickey DM, Brooks DP, Westfall TD. Use of a novel and highly selective oxytocin receptor antagonist to characterize uterine contractions in the rat. Am J Physiol Regul Integr Comp Physiol. 2007;293:R299–305. doi: 10.1152/ajpregu.00057.2007. [DOI] [PubMed] [Google Scholar]
- Snidow J, Miller H, Valenzuela G, Thornton S, Stier B, Clayton L, Fossler M, Montague T, Beach K, Williams P. A multicenter, randomized, double-blind, placebo-controlled phase 2 trial of retosiban, a selective oxytocin receptor antagonist, for the management of preterm labor. Am J Obstet Gynecol. 2013;208 : S155:350 (abstract) [Google Scholar]
- Carlin BP, Louis TA, editors. Bayesian Methods for Data Analysis, Third Edition. Chapman & Hall/CRC, Taylor and Francis Group: Boca Raton, FL; 2009. [Google Scholar]
- Gyetvai K, Hannah ME, Hodnett ED, Ohlsson A. Tocolytics for preterm labour: a systematic review. Obstet Gynecol. 1999;94:869–77. doi: 10.1016/s0029-7844(99)00329-4. [DOI] [PubMed] [Google Scholar]
- Haas DM, Imperiale TF, Kirkpatrick PR, Klein RW, Zollinger TW, Golichowski AM. Tocolytic therapy: a meta-analysis and decision analysis. Obstet Gynecol. 2009;113:585–94. doi: 10.1097/AOG.0b013e318199924a. [DOI] [PubMed] [Google Scholar]
- Thornton S, Goodwin TM, Greisen G, Hedegaard M, Arce JC. The effect of barusiban, a selective oxytocin antagonist, in threatened preterm labor at late gestational age: a randomized, double-blind, placebo-controlled trial. Am J Obstet Gynecol. 2009;200:627–9. doi: 10.1016/j.ajog.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Neilson JP, West HM, Dowswell T. Beta-sympathomimetics for inhibiting preterm labour. Cochrane Pregnancy and Childbirth Group. Cochrane Database Syst Rev. 2014;2 doi: 10.1002/14651858.CD004352.pub3. : CD004352. doi: 10.1002/14651858.CD004352. [DOI] [PMC free article] [PubMed] [Google Scholar]