

Abstract
Transistor-based nanoelectronic sensors are capable of label-free real-time chemical and biological detection with high sensitivity and spatial resolution, although the short Debye screening length in high ionic strength solutions has made difficult applications relevant to physiological conditions. Here, we describe a new and general strategy to overcome this challenge for field-effect transistor (FET) sensors that involves incorporating a porous and biomolecule permeable polymer layer on the FET sensor. This polymer layer increases the effective screening length in the region immediately adjacent to the device surface, and thereby enables detection of biomolecules in high ionic strength solutions in real-time. Studies of silicon nanowire (SiNW) field-effect transistors (FETs) with additional polyethylene glycol (PEG) modification show that prostate specific antigen (PSA) can be readily detected in solutions with phosphate buffer (PB) concentrations as high as 150 mM, while similar devices without PEG modification only exhibit detectable signals for concentrations ≤ 10 mM. Concentration-dependent measurements exhibited real-time detection of PSA with a sensitivity of at least 10 nM in ~130 mM ionic strength PB with linear response up to the highest (1000 nM) PSA concentrations tested. The current work represents an important step toward general application of nanoelectronic detectors for biochemical sensing in physiological environments, and is expected to open up exciting opportunities for in-vitro and in-vivo biological sensing relevant to basic biology research through medicine.
Keywords: Semiconductor nanowires, field-effect-transistor, bioelectronics, Debye length, polymer-modified, polyethylene glycol
Since the first demonstration of biosensors based on nanoelectronic FETs in 2001,1 SiNW FET biosensors2 – 4 have been used for ultrasensitive, multiplexed real-time detection of a variety of biological species including protein disease biomarkers at a femtomolar level,5, 6 DNA and DNA mismatch identification at the tens of femtomolar level7 and even single viruses.8 Furthermore, SiNW FETs have demonstrated the capability to be used as a label-free sensing platform for investigating small molecule-protein interactions for drug screening5, 9 and for determining the kinetics of basic biomolecular association/dissociation processes and the rate and inhibition of enzymatic synthesis.5, 10 The robustness of these concepts introduced first with SiNW devices have been confirmed in subsequent studies of nanoscale FET biosensor devices configured from a variety of semiconductor nanomaterials, including other NW systems11 – 14, which have been used to detect proteins and DNA, carbon nanotube15 and subsequently graphene15, 16 FET devices were used for sensing proteins,17 - 19 glucose,20 DNA,21 and bacteria.22 Recently, similar results have also been repeated with layered MoS2 FET devices in pH and proteins sensing.23
The above results testify to the robustness and potential of nanoelectronic FET biosensors, although these sensors also have been limited to measurements in relatively low ionic strength solutions by the Debye-screening length.24 Specifically, in physiological solution environments, which are relevant to many important biological, medical and diagnostic applications, the short screening length, <1 nm, reduces the field produced by charged biomolecules at FET surface and thus makes real-time label-free detection difficult.
Several indirect and direct methods have been reported to overcome this intrinsic limitation of nanoelectronic FET biosensors. First, indirect methods have been reported5, 25 for detection of blood serum disease marker proteins that involve an initial ‘desalting’ step followed by detection in low ionic strength buffers. While effective, these indirect methods are not real-time and cannot be generally applied to many types of in-vitro and in-vivo detection. More recently, several groups have reported a direct approach based on smaller receptors26, 27 to reduce the distance between the FET surface and biomolecule analyte being detected. These studies are promising, although further studies are still needed to determine how general detection is under the limit of physiological conditions (Debye length < 1 nm) since the sizes of the aptamer26 and antibody fragment27 receptors are similar to or greater than this critical length scale. Last, Zhong and coworkers28 have also reported a direct high-frequency measurement strategy that can be applied to standard biological receptors, although the more complex device geometry required for these measurements may limit applicability, especially for cellular and in-vivo sensing applications.
Here we report a new direct strategy for real-time detection of biomolecules in physiological environment using nanoscale FET device and applicable to both in-vitro and in-vivo sensing. Our method (Figure 1a) involves linking a porous and biomolecule permeable polymer to the surface of the FET such that the effective Debye screening length is increased, making it possible to detect proteins and other biological analytes directly in real-time in physiology-relevant high ionic strength solutions. This approach is motivated by previous studies reporting that polymers, including PEG, can change substantially the dielectric properties in aqueous solutions,29 and thus serve to increase the effective Debye screening length for a given solution ionic strength. In the studies described below, we demonstrate this general approach for SiNW FET devices modified with PEG,30, 31 although we note that the basic concept will be general to FET biosensors configured from other materials.
Figure 1. Polymer surface modification to increase the effective Debye length for FET biosensing.

(a) Schematic illustration of a NW FET device (top) without and (bottom) with a porous and biomolecule permeable polymer (green) surface modification. The magenta features on both NW surfaces represent APTES in these studies, or more generally, specific receptors. (b) Optical image of device chip (central light purple square, ca. 2 × 2 cm2) mounted on a PCB interface board that is plugged into the input/output interface connected (left side of image) to a computer controlled data acquisition system32 (not shown). The copper squares surrounding the device chip are connected to the chip by wire-bonding (not visible). A poly(dimethylsiloxane) (PDMS)-based microfluidic channel is mounted over the central SiNW device region of the chip with solution input/output via tubing (translucent, center to upper left/right of image) during real-time biosensing experiments. The inset shows a bright-field microscopy image of a portion of device chip containing 18 of 188 total FET devices on the chip; scale bar is 40 μm. Metal lines are visible in the image with common source (S) and one addressable drain (D) electrode labeled; the other addressable D electrodes are visible as the thin gold colored lines oriented upwards and downwards to right. The blue arrow highlights one SiNW FET as shown schematically in (a).
SiNWs (p-type, 30 nm diameter) synthesized by the nanocluster catalyzed vapor-liquid-solid method were used to fabricate FET sensor chip (Figure 1b) as described previously.32, 33, 34 The SiNW S-D contacts and metal interconnects were passivated with Si3N4.34 The SiNW device chips were modified with either a 4:1 mixture of (3-aminopropyl)triethoxysilane (APTES) and silane-PEG (10 kD) or pure APTES in manner similar to previous reports.33, 35 Following modification of the SiNW device chip, a PDMS microfluidic channel was mounted on the chip (Figure 1b) for delivery of buffer and protein/buffer solutions.33, 36 SiNW FET signals were recorded simultaneously from three devices, and signals were converted to absolute millivolt (mV) values using the device transconductance sensitivities determined from water gate measurements.36 Conductance versus liquid-gate voltage for typical APTES-modified and APTES/PEG-modified Si nanowire FET devices as well as a summary of transconductance values for modified devices (Figure S2) show that the transconductance is reduced by PEG modification as expected.. PSA37 was used as a protein model with all experiments carried out below the PSA isoelectric point38 at pH 6 PB.
Initial measurements made as a function PB concentration on APTES modified SiNW devices (Figure 2a) show that the PSA signal response is significantly diminished with increasing PB buffer ionic strength. First, as the PB concentration was increased from 1, 2, 5 to 10 mM, the signal response for 100 nM PSA dramatically decreased from ca. 112, 56, 23 to 8 mV, respectively. Second, when the PB concentration was further increased to 50 mM, no obvious response was observed. These results are consistent with previous studies27, 28, 39 showing a decrease in FET signal amplitudes with increasing solution ionic strength, and indicate that the APTES modification has little influence on the local ionic environment at the FET surface.
Figure 2. Real-time PSA detection using SiNW FETs sensors with and without PEG surface modifications.

(a) Signal amplitude vs time data recorded from an APTES modified SiNW FET following addition of 100 nM PSA (black arrow) and pure buffer (initial and following green arrow) for different pH 6 PB concentrations. (b) – (d) Comparison of signal response traces recorded simultaneously from three SiNW FET devices following addition of 100 nM PSA in different concentration pH 6 PBs; the devices were modified with 4 : 1 APTES/silane-PEG. The PB concentrations of the pure and PSA/PB solutions in (b), (c) and (d) were 50, 100 and 150 mM, respectively. Black and green arrows in figure correspond to the points where the solution flow was switched from pure buffers to protein solutions and from protein solutions to pure buffers; the delay from solution switch to signal change corresponds to the time for solution to flow from the entry point to the devices in our set-up.
Significantly, measurements made with polymer-modified SiNW FET devices (Figures 2a-d, Figure S3) exhibited well-defined PSA signal response for PB concentrations up to 150 mM and highlight several key points. First, measurements showed a weak PB concentration dependence with response of ca. 44, 37, 40 and 28 mV for 10, 50, 100 and 150 mM PB. We note that no obvious signals were detected for >10 mM PB under same experimental conditions using only APTES-modified devices. Second, the data recorded for each PB concentration is reproducible as evidenced by similar responses recorded simultaneously from three different modified SiNW devices on the sensor chip. Third, the ionic strengths of the higher concentration solutions are similar to physiological conditions, and thus show the potential for direct real-time sensing in this regime. Moreover, the binding/unbinding process on the APTES-modified devices was 2-4 times slower than that on the APTES/PEG-modified devices (Figure S4). This difference might reflect different interactions on the APTES versus APTES/PEG modified device, although future studies will be needed to determine unambiguously the origin of these interesting results.
We investigated further the reproducibility of PSA sensing at different PB concentrations with polymer-modified sensors by comparing results obtained from independent SiNW sensor chips. Notably, the signals recorded from three independent devices on each of two different sensor chips (Figure 3a) exhibit good consistency at all PB concentrations (i.e., 10, 50, 100 and 150 mM). These results indicate that the APTES/silane-PEG surface modification is relatively uniform, although there is a ca. 14 % variation of the PSA signal between the two different sensor chips. We believe that this chip-to-chip variation, which may reflect variations in the PEG density/porosity obtain using our modification approach, could be reduced in the future through optimization of the post-fabrication cleaning and modification strategies.
Figure 3. Reproducible PSA detection by FET sensors in high ionic strength solutions.
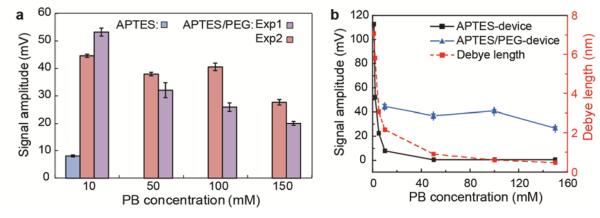
(a) Comparison of PSA response signals from independent sensor chips as labeled in the figure, where APTES/PEG corresponds to modification with 4 : 1 APTES/silane-PEG as described in the text. The error bars for each experiment correspond to ± 1 standard deviation from data acquired simultaneously from three independent devices. (b) The dependence of PSA signal amplitudes and Debye length on the PB concentrations. The SiNW FET functionalization is the same as in (a).
Comparison of these PB concentration-dependent experimental sensing data and the calculated Debye length as a function of PB concentration (Figure 3b) brings out several key points. For APTES-modified devices, the PSA (100 nM) response in 1 mM PB with a Debye length of ~7 nm was 112 mV, and decreased rapidly to ca. 8 mV in 10 mM PB where the Debye length is ~ 2.2 nm. Moreover, no obvious response was observed in 50 mM PB where the Debye length is ~1 nm. This rapid decrease and ultimate loss of signal response is consistent with the thickness of the APTES monolayer, 0.8 nm,40 and the diameter of PSA, ca. 2 nm,41 since charge screening at the FET surface will become increasingly effective as the Debye length becomes smaller than the sum of the APTES layer thickness and protein diameter. In contrast, for the APTES/PEG-modified devices, the PSA (100 nM) response in 10 mM PB was 44 mV (vs. 8 mV for APTES alone), and moreover, decreased only slowly to ca. 40 mV for 100 mM PB (Debye length ~ 0.67 nm) and 28 mV for 150 mM PB where the Debye length is ~ 0.54 nm. The significant difference between the APTES and APTES/PEG modified devices reveals that the polymer plays an important role in modulating the local ionic environment at the SiNW FET surfaces, and thus overcoming the Debye screening issue for FET biosensors under physiologically-relevant conditions.
In addition, the PSA concentration dependent sensor response was investigated under high ionic strength physiological conditions. Conductance versus time data recorded in 100 mM PB (Figure 4a), which has a Debye length ~ 0.67 nm comparable with physiological solution, showed clear sensor response for PSA concentrations from 10 – 1000 nM. A standard plot of this data versus log[PSA] (Figure 4b) yields a relatively linear detection regime for PSA concentrations ≥ 50 nM. Last, we investigated the PSA concentration-dependent sensor response in terms of a Langmuir adsorption.42 We find that the data can be well-fit by the expression for Langmuir adsorption isotherm (equation 1)42 :
Figure 4. Concentration-depended PSA detection in high ionic strength solutions.

(a) Time-dependent signal response traces at different PSA concentrations for a PEG-modified SiNW FET sensor in pH6 100 mM PB. (b) Plot of the sensor response vs PSA concentration. The red line is fit of the data with Langmuir adsorption isotherm with k = 6.4 ×106 M−1. The inset shows sensor response (conductance change) versus logarithm of the PSA concentration.
| (1) |
where S and Smax represent the signal and saturation signal, respectively, in response to PSA concentration C, and k is a constant. Significantly, the value of k determined from our fit, 6.4 × 106 M−1, agrees well with the reported results for protein adsorption.43, 44
In conclusion, we describe a new and general strategy to overcome the limitation of Debye screening for FET biomolecule sensors. FET devices functionalized with a porous and biomolecule permeable polymer increase the effective screening length in the region immediately adjacent to the device surface, thus enabling detection of biomolecules in high ionic strength solutions in real-time. Measurements made with SiNW FETs with additional PEG modification show that PSA can be readily detected in solutions with PB concentrations as high as 150 mM, while similar devices without PEG modification only exhibit detectable signals for concentrations ≤ 10 mM. Concentration-dependent measurements exhibited real-time detection of PSA to at least 10 nM in ~130 mM ionic strength PB with linear response up to the highest (1000 nM) PSA concentrations tested. The current work is general and could also be applied to other nanowires, carbon nanotube, graphene and 2D semiconductors,11-23 which have shown similar results as previous SiNW FET biosensor studies.1-10 While additional work will be needed, for example, to incorporate specific receptors such as antibodies within the polymer modification layer to allow for selective protein detection, we believe this work represents an critical step toward general application of nanoelectronic detectors in many areas, including (i) real-time point-of-care diagnostics, (ii) intracellular and subcellular real-time monitoring of proteins and nucleic acids using 3D kinked SiNW FET devices previously used for real-time intracellular recording of action potential from single cells,45, 46 and (3) in-vitro/in-vivo monitoring of important biomolecules in engineered and natural tissues using our 3D free-standing nanoelectronic scaffolds/networks.47
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jinlin Huang for helpful discussion and assistance with sensor measurements. C.M.L. acknowledges support of this work by Hoffmann-La Roche, a NIH Director’s Pioneer Award, and a National Security Science and Engineering Faculty Fellow award.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Cui Y, Wei Q, Park H, Lieber CM. Science. 2001;293:1289–92. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- (2).Patolsky F, Lieber CM. Materials Today. 2005;8:20–28. [Google Scholar]
- (3).Patolsky F, Zheng G, Lieber CM. Nanomedicine. 2006;1:51–65. doi: 10.2217/17435889.1.1.51. [DOI] [PubMed] [Google Scholar]
- (4).Chen K-I, Li B-R, Chen Y-T. NanoToday. 2011;6:131–154. [Google Scholar]
- (5).Zheng G, Patolsky F, Cui Y, Wang WU, Lieber CM. Nat. Biotechnol. 2005;23:1294–1301. doi: 10.1038/nbt1138. [DOI] [PubMed] [Google Scholar]
- (6).Stern E, Klemic JF, Routenberg DA, Wyrembak PN, Turner-Evans DB, Hamilton AD, LaVan DA, Fahmy TM, Reed MA. Nature. 2007;445:519–522. doi: 10.1038/nature05498. [DOI] [PubMed] [Google Scholar]
- (7).Hahm J, Lieber CM. Nano Lett. 2004;4:51–54. [Google Scholar]
- (8).Patolsky F, Zheng G, Hayden O, Lakadamyali M, Zhuang X, Lieber CM. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14017–14022. doi: 10.1073/pnas.0406159101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang WU, Chen C, Lin KH, Fang Y, Lieber CM. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3208–3212. doi: 10.1073/pnas.0406368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Duan X, Li Y, Rajan NK, Routenberg DA, Modis Y, Reed MA. Nat. Biotechnol. 2012;7:401–407. doi: 10.1038/nnano.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ramgir NS, Yang Y, Zacharias M. Small. 2010;6:1705–1722. doi: 10.1002/smll.201000972. [DOI] [PubMed] [Google Scholar]
- (12).Li C, Curreli M, Lin H, Lei B, Ishikawa FN, Datar R, Cote RJ, Thompson ME, Zhou CW. J. Am. Chem. Soc. 2005;127:12484–12485. doi: 10.1021/ja053761g. [DOI] [PubMed] [Google Scholar]
- (13).Choi A, Kim K, Jung HI, Lee SY. Sens. Actuators, B. 2010;148:577–582. [Google Scholar]
- (14).Chen CP, Ganguly A, Lu CY, Chen TY, Kuo CC, Chen RS, Tu WH, Fischer WB, Chen KH, Chen LC. Anal. Chem. 2011;83:1938–1943. doi: 10.1021/ac102489y. [DOI] [PubMed] [Google Scholar]
- (15).Balasubramanian K, Kern K. Adv. Mater. 2014;26:1154–1175. doi: 10.1002/adma.201304912. [DOI] [PubMed] [Google Scholar]
- (16).Zhan B, Li C, Yang J, Jenkins G, Huang W, Dong X. Small. 2014;10:4042–4065. doi: 10.1002/smll.201400463. [DOI] [PubMed] [Google Scholar]
- (17).Vedala H, Chen YA, Cecioni S, Imberty A, Vidal S, Star A. Nano Lett. 2011;11:170–175. doi: 10.1021/nl103286k. [DOI] [PubMed] [Google Scholar]
- (18).Chen RJ, Bangsaruntip S, Drouvalakis KA, Kam NWS, Shim M, Li YM, Kim W, Utz PJ, Dai HJ. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4984–4989. doi: 10.1073/pnas.0837064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ohno Y, Maehashi K, Matsumoto K. J. Am. Chem. Soc. 2010;132:18012–18013. doi: 10.1021/ja108127r. [DOI] [PubMed] [Google Scholar]
- (20).Besteman K, Lee JO, Wiertz FGM, Heering HA, Dekker C. Nano Lett. 2003;3:727–730. [Google Scholar]
- (21).Star A, Tu E, Niemann J, Gabriel JCP, Joiner CS, Valcke C. Proc. Natl. Acad. Sci. U.S.A. 2006;103:921–926. doi: 10.1073/pnas.0504146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mohanty N, Berry V. Nano Lett. 2008;8:4469–4476. doi: 10.1021/nl802412n. [DOI] [PubMed] [Google Scholar]
- (23).Sarkar D, Liu W, Xie XJ, Anselmo AC, Mitragotri S, Banerjee K. ACS Nano. 2014;8:3992–4003. doi: 10.1021/nn5009148. [DOI] [PubMed] [Google Scholar]
- (24).Israelachvili, J. Intermolecular & Surface Forces. 2nd. Academic Press; London: 1991. In electrolytes at room temperature, the Debye length is given by , where λB is the Bjerrum length = 0.7 nm, ∑i is the sum over all ion species, and ρi and Zi are the density and valence, respectively, of ion species i. [Google Scholar]
- (25).Stern E, Vacic A, Rajan NK, Criscione JM, Park J, Ilic BR, Mooney DJ, Reed MA, Fahmy TM. Nat. Biotechnol. 2010;5:138–142. doi: 10.1038/nnano.2009.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Maehashi K, Katsura T, Kerman K, Takamura Y, Matsumoto K, Tamiya E. Anal. Chem. 2007;79:782–787. doi: 10.1021/ac060830g. [DOI] [PubMed] [Google Scholar]
- (27).Elnathan R, Kwiat M, Pevzner A, Engel Y, Burstein L, Khatchtourints A, Lichtenstein A, Kantaev R, Patolsky F. Nano Lett. 2012;12:5245–5254. doi: 10.1021/nl302434w. [DOI] [PubMed] [Google Scholar]
- (28).Kulkarni GS, Zhong Z. Nano Lett. 2012;12:719–723. doi: 10.1021/nl203666a. [DOI] [PubMed] [Google Scholar]
- (29).Arnold K, Herrmann A, Pratsch L, Gawrisch K. Biochim. Biophys. Acta. 1985;815:515–518. doi: 10.1016/0005-2736(85)90381-5. [DOI] [PubMed] [Google Scholar]
- (30).Harris JM. Poly(ethylene glycol) Chemistry. Plenum Press; 1992. [Google Scholar]
- (31).Knop K, Hoogenboom R, Fischer D, Schubert US. Angew. Chem. Int. Ed. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- (32). SiNW synthesis: The Si nanowires were grown by a gold nanoparticle-catalyzed vapor−liquid−solid growth method. Briefly, the growth substrate (600 nm SiO2/Si) was cleaned by oxygen plasma (100 W, 3 min), treated with poly-L-lysine solution (0.1%, Ted Pella) for 5 min, and then rinsed thoroughly with deionized water. For silicon nanowire synthesis, 30 nm gold nanoparticles (Ted Pella) were dispersed on growth substrates, and then nanowire growth was carried out at 450°C under a constant pressure of 40 Torr with SiH4 (2.5 SCCM), diluted B2H6 (100 ppm in He, 3 SCCM), and H2 (60 SCCM) as reactant, doping, and carrier gases, respectively. The growth time was 25 min to produce an average nanowire length of 25 μm. [Google Scholar]
- (33).Patolsky F, Zheng G, Lieber CM. Nat. Protoc. 2006;1:1711–1724. doi: 10.1038/nprot.2006.227. [DOI] [PubMed] [Google Scholar]
- (34). Arrays of Si-NW devices were defined using photolithography with Ti/Pd/Ti (1.5 nm/55 nm/7 nm) contacts deposited by thermal evaporation on silicon substrates with a 600 nm-thick oxide layer. The metal contacts and interconnects were passivated with a ~ 60 nm thick Si3N4 layer deposited by magnetron sputtering. [Google Scholar]
- (35). The surface modification process for 4 : 1 APTES : silane-PEG and pure APTES (Fig. S1) was carried out as follows. APTES (741442, Sigma-Aldrich, St Louis, MO) and silane-PEG (Methoxyl silane PEG, No. PG1-SL-10k, Nanocs, Inc., Boston, MA) were first dissolved in the mixture of 95% ethanol and 5% DI water with a final concentration of APTES and silane-PEG of 4.8 and 1.2 mM, respectively; pure APTES solutions were made in a similar manner with only APTES.29 Precleaned SiNW device chips were reacted with theethanol solutions of APTES/silane-PEG or APTES for 45 mins, and then washed gently with ethanol and allowed to dry. [Google Scholar]
- (36). SiNW devices were initially characterized using a probe station, and then wire-bonded to the output pads of the chip carrier. A PDMS microfluidic channel was mounted on the sensor chip, with the channel aligned and overlapped to the central region of the device chip where the SiNW FETs are located. Polyethylene tubing was attached to the inlet and the outlet holes on the PDMS microfluidic channel, and PB or PSA/PB solution was drawn through the channel using a syringe pump at 0.2 ml/h. Measurements were carried out using up to 3 independent lock-in amplifiers with 30 mV modulation amplitudes and modulation frequencies of 79, 97 and 103 Hz to record in parallel 3 different SiNW FET elements selected from 188 elements on the chip. A Ag/AgCl reference electrode was used to fix the solution potential at a constant value of 0 V in all recording experiments. The conductance versus time data were digitized and recorded on computer using custom software. The watergate responses of devices were characterized before PSA detection experiments. Device transconductance values were determined from watergate measurements, and used to convert recorded conductance - time data to mV values. [Google Scholar]
- (37). Preservative free PSA (MBS537240, Mybiosource, Inc., San Diego, CA) was used without further purification and directly diluted in different ionic strength pH 6 PB prior to sensing measurements. [Google Scholar]
- (38).Wang MC, Papsidero LD, Kuriyama M, Valenzuela LA, Murphy GP, Chu TM. Prostate. 1981;2:89–96. doi: 10.1002/pros.2990020109. [DOI] [PubMed] [Google Scholar]
- (39).Stern E, Wagner R, Sigworth FJ, Breaker R, Fahmy TM, Reed MA. Nano Lett. 2007;7:3405–3409. doi: 10.1021/nl071792z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ouyang H, Striemer CC, Fauchet PM. Appl. Phys. Lett. 2006;88:163108. [Google Scholar]
- (41).Wegner KD, Jin ZW, Linden S, Jennings TL, Hildebrandt N. ACS Nano. 2013;7:7411–7419. doi: 10.1021/nn403253y. [DOI] [PubMed] [Google Scholar]
- (42).Krishnamurthy N, Vallinayagram P, Madhavan D. Engineering chemistry. PHI learning Pvt. Ltd.; Oct. 2014. [Google Scholar]
- (43).Latour RA. J. Biomed. Mater. Res. A. 2015;103:949–958. doi: 10.1002/jbm.a.35235. [DOI] [PubMed] [Google Scholar]
- (44).Gettens RTT, Bai ZJ, Gilbert JL. J Biomed Mater Res A. 2005;72A:246–257. doi: 10.1002/jbm.a.30218. [DOI] [PubMed] [Google Scholar]
- (45).Tian BZ, Cohen-Karni T, Qing Q, Duan XJ, Xie P, Lieber CM. Science. 2010;329:830–834. doi: 10.1126/science.1192033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jiang Z, Qing Q, Xie P, Gao RX, Lieber CM. Nano Lett. 2012;12:1711–1716. doi: 10.1021/nl300256r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tian B, Liu J, Dvir T, Jin L, Tsui JH, Qing Q, Suo Z, Langer R, Kohane DS, Lieber CM. Nat. Mater. 2012;11:986–994. doi: 10.1038/nmat3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.