

Abstract
Major advances in carrier-mediated agents, which include nanoparticles, nanosomes and conjugates, have revolutionized drug delivery capabilities over the past decade. While providing numerous advantages, such as greater solubility, duration of exposure and delivery to the site of action over their small-molecule counterparts, there is substantial variability in systemic clearance and distribution, tumor delivery and pharmacologic effects (efficacy and toxicity) of these agents. This review provides an overview of factors that affect the pharmacokinetics and pharmacodynamics of carrier-mediated agents in preclinical models and patients.
Keywords: carrier-mediated agents, clearance, Doxil, nanoparticles, pharmacokinetics, S-CKD602
Nanoparticle formulations
The number of available nanoparticle (NP)-based drug systems has seen exponential growth in the past decade. In 2006 alone, nearly 130 nanotechnology-based products were estimated to be undergoing the drug development process worldwide [1]. While the number of agents used clinically is still limited, the plethora that are emerging as potential therapeutic agents warrants the need for detailed studies of their unique pharmacology and mechanisms of action in humans. Caron et al. summarize currently available and late-stage development of chemotherapeutic carrier-mediated agents (CMAs) in Supplementary Table 1 (see online at www.futuremedicine.com/doi/suppl/10.2217/nnm.14.179) [2]. In this review, we focus on factors that affect the pharmacokinetics (PK) and pharmacodynamics (PD) of anticancer CMAs in patients, and we hypothesize that many concepts presented within this review will be applicable to the vast field of agents to soon enter the clinic.
Pharmacokinetic characterization
The disposition of CMAs is dependent upon the carrier and not the therapeutic entity until the drug gets released [3]. The nomenclature used to describe CMA PK includes: encapsulated (the drug within or bound to the carrier), released (active drug that gets released from the carrier) and sum total (encapsulated drug plus released drug) [4,5]. After the drug is released from its carrier, it is pharmacologically active and subject to the same routes of metabolism and clearance as the noncarrier form of the drug [5]. In theory, the PK disposition of the drug after release from the carrier should be the same as after administration of the small molecule or standard formulations. Thus, the pharmacology and PK of CMAs are complex and comprehensive. Analytical methods must be performed in order to assess the disposition of encapsulated or released forms of the drug in plasma and tumor [6]. Considerable interpatient variability exists in the PK/PD of CMAs, and while the exact factors are unclear, it is hypothesized that the mononuclear phagocyte system (MPS; or reticuloendothelial system) plays a key role [7].
The PK of liposomal encapsulated drug and released drug is very different, and compared with conventional small-molecule anti-cancer agents, the PK variability in liposomal formulations is often much greater [5,8]. Inter-individual variability in drug exposure, represented by area under the concentration versus time curve (AUC), of encapsulated drug can be 20- to 100-fold. Factors with the potential to affect CMA PK include CMA-associated physical characteristics and host-associated characteristics [9]. Perhaps the greatest influence on the PK variability of CMA, however, is the MPS. Figure 1 illustrates the unique clearance mechanisms associated with CMA as compared with conventional small molecules [9,10].
Figure 1. Metabolism and elimination pathway for small molecule and carrier mediated agents.
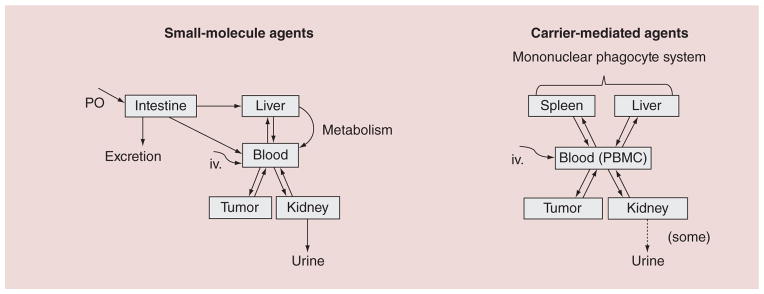
Small-molecule anticancer agents undergo a standard route of metabolism and elimination, including enterohepatic recycling and removal through the kidney. Carrier-mediated agents, however, which are engulfed by phagocytes, are contained primarily in compartments such as the spleen, liver and peripheral blood mononuclear cells.
IV: Intravenous administration; PBMC: Peripheral blood mononuclear cell; PO: Oral administration.
Reproduced with permission from [2].
For color figures, see online at www.futuremedicine.com/doi/full/10.2217/NNM.14.179
Mononuclear phagocyte system
Once an NP enters the bloodstream, it encounters plasma proteins and immune cells [11]. The interaction and subsequent effects of NP therapeutics on the immune system have not been fully elucidated, but presently, are generally placed into one of two categories: responses to NPs that are specifically modified to stimulate the immune system (e.g., vaccine carriers) and undesirable interactions and/or side effects [12]. In this review, we will focus on the latter, which may have profound clinical implications. Here, we describe the MPS and the mechanisms of CMA uptake. In the next section, we will describe how MPS function affects CMA PK/PD.
NP uptake by immune cells can occur in circulating monocytes, platelets, leukocytes and dendritic cells (DCs) of the bloodstream [11,12]. In addition, NPs can be taken up in tissues by phagocytes, such as Kupffer cells in the liver, DCs in the lymph nodes, macrophages and B cells in the spleen [11]. Figure 2 illustrates the interaction and clearance of CMA with these immune cells, collectively termed the MPS. Uptake mechanisms may occur through different pathways and are often facilitated by the adsorption of opsonins to the NP surface and subsequent phagocytosis [11].
Figure 2. Clearance of nanoparticles and carrier-mediated agents via the mononuclear phagocyte system.

When nonstabilized liposomal agents were first tested, they were found to only minimally increase the circulation time of the small-molecule agent encapsulated within the carrier (rapid clearance). However, stabilization with PEG has helped to reduce uptake and CL of CMA by MPS (slower clearance). While the clearance of PEGylated liposomes are slower than non-PEGylated liposomes, both are phagocytized by peripheral blood mononuclear cells, phagocytes of the liver and spleen. Greater tumor exposure is seen after administration of PEGylated liposomes, which in part due to the EPR effect and possibly, the MPS in tumors.
CL: Clearance; CMA: Carrier-mediated agent; EPR: Enhanced permeability and retention; MPS: Mononuclear phagocyte system.
Adapted with permission from [2].
Many NPs have been developed for the purpose of evading rapid clearance from the bloodstream and thereby extending systemic circulation time [11]. While this allows for a greater probability of NP delivery to a target site, an increase in circulation time results in a proportional increase of duration of contact with components of the immune system [11]. CMA PK/PD varies between human patients and can be attributed to many variables including duration of contact and overall activity of MPS components. In addition, we continue to explore the interspecies relationship of MPS function and CMA PK. We have seen a positive association between murine models and humans, which will be touched upon later in the article [13].
Delivery of CMA in tumor
While conventional drugs encounter numerous obstacles en route to their target, CMAs can take advantage of tumor’s leaky vasculature to extravasate into tissue via the enhanced permeability and retention effect (EPR) [14,15]. Furthermore, the poor lymphatic drainage in tumors leads to accumulation of the CMA for prolonged duration, allowing them to release the drug in tumor cells over time. Passive targeting exploits the classic features of tumor biology in order to increase exposure of CMA in the tumor.
In theory, EPR has been the primary route of CMA delivery to tumors, but heterogenicity of EPR between tumor types and the inability to ensure uniform delivery to all regions of the tumor is pressing researchers to dive deeper into EPR fundamentals and the effects of tumor vasculature [16–18]. Unevenly distributed blood flow, dense microenvironment and variations in vessel permeability play an important role in the distribution and penetration of CMAs to tumor [19]. Better and more effective CMAs that exploit EPR are needed while also employing methods to address structural hindrances in tumor microenvironment. Jain and colleagues, well-known for his efforts in the field of tumor vasculature, explains the EPR effect is attributed by physiologic contributions such as vascular pore dimensions, vascular structure, surrounding stroma and MPS function [16]. From a preclinical perspective, there is a need for tumor models that accurately represent the types of tumors seen in patients in order to conduct informative profiling and developmental studies of CMAs [16]. It is thought that metastatic, orthotopic and genetically engineered mouse models are better options for CMA studies than flank tumor xenografts [16]. However, systematic studies of several types of CMAs in each tumor model have not been reported and are desperately needed to advance the field of CMA in the treatment of cancer. Clinically, there is a need for diagnostic aids to determine EPR activity in patients and predict responders to individual CMAs [16]. Recent approaches to normalize both tumor vasculature and physical forces surrounding vessels have been explored [19]. Co-medications that effect stroma and blood pressure are known to influence EPR effect [16]. Mathematical models employed by Jain and colleagues have the potential to reveal an optimal vessel perfusion region that maximizes distribution of particular drug or CMA to tumor [18].
Active targeting of CMAs may further improve tumor delivery and activity by allowing the CMA to bind to specific cells in tumor using surface-attached ligands capable of recognizing and binding to cells of interest [20]. Relative to normal cells, tumor cells have certain overexpressed surface receptors or antigens that maximize the specificity of binding of targeted agents. One such strategy for the tumor-targeted drug delivery is the development of immunoliposomes. For example, anti-HER2-targeted liposomal doxorubicin was associated with higher efficacy compared with its nontargeted counterpart in a breast cancer model [21]. While antibody-mediated targeting has been the method of choice, other targeting strategies using nucleic acids, carbohydrates, peptides, aptamers and vitamins are also being evaluated [17].
MM-302, currently in early-phase clinical studies, incorporates anti-HER2 scFv-PEG-DSPE conjugates to the outer surface of PEGylated liposomal doxorubicin (PLD). In preclinical models, MM-302 has shown superior antitumor activity to free doxorubicin and PLD in HER2 overexpressing tumors with a pharmacokinetic profile similar to that of PLD. In addition, MM-302 expressed a favorable cardiosafety profile compared with free doxorubicin [22].
Several preclinical studies support extensive tumor delivery and prolonged exposure of PEGylated liposomes in tumors [5]. The development of PEGylated liposomes that contain lipid conjugated to PEG was based on a theory that their incorporation into nanosomes would evade the immune system and help prolong duration of exposure [9]. This is consistent with higher antitumor activity of Doxil in preclinical models compared with doxorubicin and with clinical activity in patients with refractory ovarian cancer and Kaposi sarcoma [23]. In studies comparing the disposition of PEGylated liposomal CKD-602 (S-CKD602) and nonliposomal CKD-602 in mice bearing A375 human melanoma xenografts, S-CKD602 provided pharmacokinetic advantages in plasma, tumor and tumor extracellular fluid (ECF) at 1/30th of the dose [24].
Tumor exposure and antitumor activity of liposomal anticancer agents was found to be related to the presence of the MPS in tumors [24]. This was demonstrated in mice bearing SKOV-3 human ovarian and A375 human melanoma xenografts [24]. The ratio of S-CKD602 in tumor to plasma was 1.7-fold higher in mice bearing SKOV-3 compared with A375 [24]. The ratio of released CKD-602 to S-CKD602 in tumor was twofold higher in mice bearing SKOV-3 compared with A375 [24]. The staining of MPS cells was 4.5-fold higher in SKOV-3 compared with A375 (p < 0.0001) [24]. In addition, SKOV-3 was fourfold more sensitive to S-CKD602 compared with A375 [24]. The increased tumor delivery and release of CKD-602 from S-CKD602 in SKOV-3 ovarian compared with A375 melanoma xenografts was consistent with the increased staining of MPS cells in SKOV-3 suggesting that variability in the MPS may affect the tumor disposition and activity of nanosomal anticancer agents [24]. MPS cell staining was utilized to check for consistency of results. Limitations such as complete cell specificity do exist. This suggests that tumor type may play a role in the PK of CMAs.
In another study using murine colon tumor xenografts, the sum total tumor platinum exposure was fourfold higher with PEGylated liposomal cisplatin (SPI-077) compared with nonliposomal cisplatin [25]. In spite of fourfold higher exposure associated with SPI-77 compared with cisplatin, this did not translate into antitumor activity in clinical trials. Consistent with that lack of antitumor effects in patients, a subsequent study showed that SPI-077 entered tumors but did not release platinum into tumor extracellular fluid and formed significantly less platinum (Pt)-DNA adducts than cisplatin [6]. Therefore, as with all drugs, it is important that the disposition of both encapsulated and released versions of liposomes be evaluated in tumors of patients for successful development of CMA. While concentrations of SPI-077 and cisplatin present in tumor ECF do represent an exposure in the vicinity of the malignancy, efficacy cannot be assumed based off of this, as cytotoxicity is dependent upon the formation of Pt-DNA adducts. An effort to measure direct adduct formation would better represent drug efficacy [6]. To address this, microdialysis has been used to study the relationship between unbound Pt in tumor ECF and total Pt in tumor homogenates, as well as formation of Pt-DNA adducts in a group of female C57BL/6 bearing B15 melanoma flank tumors [6]. Overall, CMAs have great utility for targeting many different tumor types and degree of efficacy is influenced by many variables including MPS activity and proper drug action.
Methods to target brain tumors
Treating primary or metastatic tumors within the CNS remains extremely challenging. Numerous approaches are being explored to enhance drug delivery and anti-cancer efficacy, and the use of NPs and CMAs provides a promising approach to enhance delivery to the brain and brain tumors. The mechanism of CMA delivery to the brain is not fully understood, but hypotheses such as NP altered distribution and longer blood circulation time may allow for enhanced permeation of drugs that have the ability to cross the blood–brain barrier (BBB). In addition, brain tumors may also employ similar EPR effect as solid tumors in other parts of the body which may be the foundation of enhanced delivery to the CNS when compared with small-molecule drug [26–28]. However, the magnitude of the EPR effect in intracranial tumors is relatively weak when compared with that of peripheral tumors. Explanations for this finding claim smaller pore size for solute passage in these tumor types compared with peripheral tumors [29,30]. Systematic studies evaluating these mechanisms are needed.
Anders and colleagues utilized an intracranial model of aggressive triple-negative breast cancer in athymic mice to evaluate the pharmacologic disposition and activity of PLD compared with small-molecule doxorubicin. Treatment with PLD resulted in approximately 1500-fold higher plasma and 20-fold higher intracranial tumor sum total doxorubicin AUC compared with small-molecule doxorubicin [31]. PLD, measured as sum total doxorubicin, was detected at 96 h in plasma and tumor, while small-molecule doxorubicin was undetectable after 24 h in plasma and tumor. Median survival of PLD-treated animals was 32 days ([CI]: 31–38), which was significantly longer than tumor grafts exposed to phosphate buffered saline alone (26 days; [CI: 25–28]; p = 0.0012) or mice treated with small-molecule doxorubicin (23.5 days; [CI: 18–28]; p = 0.0002) [31]. These data suggest that PLD is a treatment option for patients with intracranial metastatic breast cancer.
Researchers are taking advantage of PLD with its inherent longer half-life and better brain penetration compared with small-molecule doxorubicin by adding other structural moieties that may enhance brain tumor delivery and efficacy. Preclinical studies of PLD with a glutathione coating (2B3-101) have also shown promising results in brain cancer models. Glutathione, an endogenous tripeptide, is actively transported across the BBB. This inherent property is thought to facilitate 2B3-101 in gaining access into the brain. Mouse models treated with 2B3-101 have shown greater inhibition of brain tumor growth with significant increase in median survival time when compared with PLD [32].
An alternative targeting strategy utilizing spherical nucleic acids (SNAs) NP conjugates has shown promise in targeting oncogenes within glioma tumor cells [33]. The ensuing dismantling of cancer-promoting signals is believed to cause tumor cell apoptosis. These SNAs consist of small interfering RNA (siRNA) coating a gold core. Researchers have created a prototypical SNA that has the ability to cross the BBB, where it can enter the tumor and exert its effect on a known oncogene (Bcl2L12). In murine models, systemic delivery of a particular siRNA-loaded SNA (siL12-2-SNA) rapidly accumulated in xenograft brain tumor tissue, where impaired tumor growth and increased survival was noted. Figure 3 shows differences in efficacy and survival in mice when treated with oncogene-targeted siL12-2-SNA, compared with control scrambled control sequence (siCo)-SNA [33]. Ultimately, CMAs are of high therapeutic interest for targeting difficult to treat organs like the brain due to its characteristic targeted approach of delivering active drug.
Figure 3. Intratumoral apoptosis in mice injected with siL12-2-SNA.

(A) The amount of aCasp-3 and terminal deoxynucleotidyl TUNEL. Data points are the number of stained cells per field. p-values were calculated with two-tailed Student’s t-test. (B) Kaplan–Meier survival curves of mice with TNS-derived xenografts treated with siL12-2-SNA (n = 6) or siCo-SNA (n = 7). p-value was calculated with the Mantel–Cox test.
aCasp-3: Activated caspase-3; HPF: High-power field; siCo: Scrambled control sequence; siL12-2-SNA: siRNA-loaded SNA; SNA: Spherical nucleic acid; TNS: Tumor neurosphere; TUNEL: Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling.
Adapted with permission from [32].
Physical characteristics
Particle size
Many groups have demonstrated that tumor uptake of NPs can depend on particle size. In one study of liposomes, particles that had a hydrodynamic diameter between 100 and 200 nm had a fourfold higher rate of uptake in tumors compared with particles less than 50 nm or greater than 300 nm [34]. These findings were replicated by Charrois, who found that liposomes ranging between 8 and 160 nm resulted in a significantly greater accumulation in tumor compared with liposomes greater than 240 nm [35]. It has been shown that particles with a hydrodynamic diameter smaller than 50 nm or greater than 300 nm have much shorter blood circulation times compared with particles with a hydrodynamic diameter between 100 and 200 nm, due to increased uptake by the MPS [34]. It is hypothesized that the shorter circulation times of particles smaller than 50 nm or greater than 300 nm do not allow the particles enough tumor exposure to take advantage of the EPR effect, leading to lower tumor accumulation [34]. However, the optimal size of NPs needed to increase blood circulation time and delivery to tumors remains unknown and may be influenced by the type of carrier and tumor. As particle size can affect what accumulates in tumor, it can also influence the concentrations that can be measured in plasma as well as eventual efficacy and toxicity measures.
Particle shape
Particle shape has also been studied to determine the effect on MPS particle recognition. Particle-Replication-In-Non-wetting-Templates (PRINT) technology is a soft-lithography process used to fabricate monodisperse populations of poly lactic-co-glycolic acid particles with high loadings of anticancer drugs [36]. The PRINT particle fabrication technique was used to fabricate two different monodisperse shape-specific poly lactic-co-glycolic acid particles loaded with the chemotherapeutic docetaxel [36]. The PK of two cylindrical-shaped particles, one with a diameter of 80 nm and a height of 320 nm (PRINT-Doc-80×320), and one with a diameter of 200 nm and a height of 200 nm (PRINT-Doc-200×200), were compared with small-molecule (SM) docetaxel in mice bearing SKOV-3 flank xenografts [36]. The concentration versus time profiles for plasma, tumor and tissues are shown in Figure 4 [36]. The docetaxel plasma exposure exceeded an increase by 20-fold for both particles compared with SM docetaxel [36]. Additionally, the volume of distribution of docetaxel in PRINT formulations was approximately 18-fold (PRINT-Doc-80×320) and approximately 33-fold (PRINT- Doc-200×200) lower than SM docetaxel [36]. The prolonged duration of docetaxel in plasma when dosed with PRINT formulations subsequently led to increased tumor exposure of docetaxel from 0 to 168 h (~53% higher than SM docetaxel for PRINT-Doc-80×320 and ~76% higher than SM docetaxel for PRINT-Doc-200×200 particles) [36]. PRINT-Doc-80×320 had lower exposures in the organs of the MPS compared with PRINT-Doc-200×200 [36]. Thus, particles shaped to have a large aspect ratio may be preferred to decrease clearance by organs of the MPS [36].
Figure 4. Plasma and tumor doectaxel vs time curves in mice bearing SKOV3 ovarian flank xenografts after administration of three different docetaxel formulations.

(A) Tumor (0–168 h), (B) tumor (0–24 h), (C) plasma, (D) lung, (E) spleen and (F) liver. Doc concentration values for each mouse are represented in the key. The lines are connected by the mean value for each time point.
Reproduced with permission from [37].
Surface modification
In an attempt to minimize opsonization and the subsequent rate of MPS uptake, the most commonly used strategy is to conjugate PEG onto the surface [38]. Sadzuka and colleagues conducted a PK study using irinotecan (CPT-11) loaded into bare liposomes or PEGylated liposomes [38]. They found that the overall exposure in plasma of CPT-11 contained within a PEGylated liposome was sixfold higher than that of non-PEGylated liposomes [38].
Surface charge
Drummond and colleagues have studied the effects of surface charge on particle clearance and found that uncharged liposomes have a lower clearance than either positively or negatively charged liposomes [39]. This is believed to be due to reduced opsonization on the uncharged liposomal surface, which leads to less MPS uptake [40]. Levchenko et al. conducted a study in which they prepared liposomes of 200 nm in size with different charges and studied tissue distribution over time [41]. They found that the rate of clearance from blood was significantly higher for negatively charged particles versus uncharged particles [41]. Additionally, the negative particles had a higher rate of MPS uptake in the liver compared with the uncharged particles, indicating that phagocytic cells resident to the liver preferentially take up negatively charged particles and increase the rate of clearance of particles from blood [41]. The most common surface modification, PEGylation, comes in to play here, as Levchenko has shown that PEGylation can shield the negative charge on liposomes, leading to a significantly reduced rate of liver uptake and prolonged blood circulation [41].
Particle dose
Chu et al. studied the effects of particle dose on particle clearance [42]. Docetaxel-loaded PRINT NPs with identical size, shape and surface chemistry, but with variable docetaxel loading were created [42]. The two different formulations of NPs had 9% (9%-NP) and 20% (20%-NP) docetaxel drug loading [42]. Both formulations were administered at identical docetaxel doses of 10 mg/kg that resulted in a total NP dose of 109 mg/kg in the 9%-NP group compared with only 50 mg/kg in the 20%-NP group [42]. The 9%-NP formulation, which is associated with administration of a higher number of NPs, was found to have a superior pharmacokinetic profile and enhanced efficacy when compared with that of the 20%-NPs [42]. Figure 5 shows the pharmacokinetic profiles of the two formulations in various tissues [42]. The 9%-NPs increased tumor docetaxel exposure and reduced liver, spleen and lung exposure when compared with that of the 20%-NPs [42]. Chu et al. theorize that the higher particle dose associated with the 9%-NP formulation may have saturated the MPS of the liver and spleen resulting in reduced docetaxel accumulation at those sites accounting for the increased tumor docetaxel exposure relative to the 20%-NP group [42]. While Chu et al. did not present any data on the MPS function or activity, there are ongoing studies to investigate the effect of particle dose on the MPS.
Figure 5. Plasma and tumor doectaxel vs time curves in mice after administration of two different shaped nanoparticle docetaxel formulations.

Pharmacokinetic profiles of (A) plasma, (B) tumor, (C) liver, (D) spleen and (E) lung. (F) In vitro release kinetics of 9%-NP (●) and 20%-NP (■). Each replicate is shown and the lines are connected by the mean of three replicates.
Reproduced with permission from [41].
The effect of MPS mediators on NP PK
A Phase I study looked at the relationship between the disposition of the CMA S-CKD602 (PEGylated liposomal CKD-602, a camptothecin analogue) and changes in monocytes and absolute neutrophil count (ANC) [43]. The control of this study was changes in monocytes and ANC after patients with refractory solid tumors received nonliposomal CKD-602 [43]. The percent decrease in ANC and monocytes from baseline to nadir from the blood of all patients administered S-CKD602 on cycle 1 was measured. The mean ± standard deviation% decrease in ANC and monocytes at the nadir was 42 ± 30 and 58 ± 34%, respectively (p = 0.001). The ratio of percent decrease in monocytes to ANC at the nadir was 2.1 ± 2.0. The percent decrease in ANC and monocytes at the nadir in the blood of patients was also assessed after administration of nonliposomal CKD-602. After administration, the percent decrease in ANC and monocytes were 86 ± 11 and 87 ± 12%, respectively (p > 0.05). The ratio of percent decrease in monocytes to ANC was 1.0 ± 0.2. The results suggest that monocytes are more sensitive to S-CKD602 compared with neutrophils. The increased sensitivity also appears to be related to the liposomal formulation and not the released drug from the liposome or the small-molecule formulation.
Some of the significant variability in the pharmacokinetic dispositions of CMA versus small molecules was found to be related to linear and nonlinear clearance of S-CKD602 in patients [44]. In the previously mentioned study, despite being treated with the same dose of 1.7 mg/m2, some patients were found to have linear clearance, while the majority portrayed nonlinear clearance [44]. Those with linear clearance had a plasma concentration versus time profile of CKD-602 with a distribution phase and an elimination phase [44]. For patients with nonlinear clearance, the plasma concentration versus time profile of CKD-602 yielded little change from 0 to 24 h [44]. The incidence of linear or nonlinear clearance was found to be associated with the dose of S-CKD602 administered [44]. At doses from 0.1 to 1.1 mg/m2, the S-CKD602 sum total and encapsulated CKD-602 plasma concentrations were best described using a model with linear clearance in all the patients (n = 33) [44]. At doses of 1.7–2.5 mg/m2, the S-CKD602 sum total and encapsulated CKD-602 plasma concentrations were best described using a model with linear (n = 2) and nonlinear (n = 10) clearances [44]. The observation of nonlinear PK at higher doses of CMA coincides with the hypothesis of MPS uptake in particle clearance and its subsequent saturation. However, there was significant interpatient variability in clearance with patients exhibiting linear and nonlinear clearance at the same dose.
The Pheno–GLO–high-throughput screening platform (HTSP) is an ex vivo flow cytometry-based system, used to measure the clearance of NPs by the MPS and bidirectional interaction between the MPS and NPs and conjugates [16]. Monocyte (MO) and DC function, as assessed by phagocytosis and oxidative burst can predict the PK of PEGylated liposomal NPs in several species as well as in patients [13]. Caron et al. measured MO/DC phagocytosis and reactive oxygen species production in mice, rats, dogs and patients with refractory solid tumors that had been administered a PEGylated liposomal drug formulations [13]. Preclinical pharmacokinetic studies of PLD, CKD-602 (S-CKD602) and cisplatin (SPI-077) were performed at the maximum tolerated dose [13]. Monocyte/DC function was also evaluated in patients with recurrent epithelial ovarian cancer (EOC) administered PLD [13]. Figure 6 shows that across species, a positive association was observed between monocyte/DC function and clearance of all three PEGylated liposomes. In patients with EOC, associations also were observed between PLD clearance and phagocytosis (coefficient of determination [R2] = 0.43, p = 0.04) and reactive oxygen species production (R2 = 0.61, p = 0.008) in blood monocytes/DCs (Figure 7) [13]. These findings suggest that probes of MPS function may help predict PEGylated liposome clearance across species, PLD clearance in patients with EOC and perhaps utilized to find the correct dose of PLD and other CMAs in the future [13].
Figure 6. Relationship between phagocytosis in monocytes/dendritic cells from blood and clearance of PEGylated liposomal agents in mice, rats, dogs and patients.
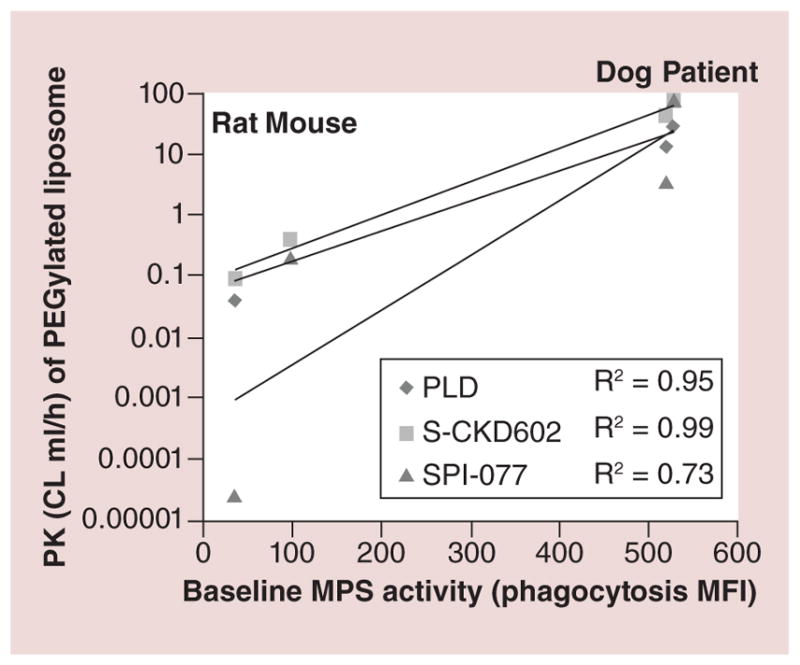
The mean values for three species are represented by individual symbols, with diamonds as PLD, squares as S-CKD602 and triangles as SPI-077. The species data are in vertical columns from left to right: rats, mice, dogs and patients. The best fit line for each group is represented by the solid lines. Across species, a positive association was observed between cell function and CL of PEGylated liposomes.
CL: Clearance; MFI: Mean fluorescent intensity; MPS: Mononuclear phagocyte system; PK: Pharmacokinetics; PLD: PEGylated liposomal doxorubicin; R2: Coefficient of determination.
Adapted with permission from [44].
Figure 7. Relationship between monocytes/dendritic cells function and encapsulated doxorubicin clearance in patients presented in a linear regression model.

Measuring phagocytosis and production of ROS of MO/dendritic cell from patient blood samples at baseline (prior to the start of chemotherapy) was used as a phenotypic probe of MPS function and encapsulated doxorubicin CL. Each diamond represents an individual patient, and the solid line is the regression line. (A) Phagocytic activity (MFI) is significantly correlated with CL of encapsulated doxorubicin in ten patients receiving PLD alone or PLD + carboplatin (R2 = 0.43, p = 0.04). (B) Production of ROS: MFI is significantly correlated with CL of encapsulated doxorubicin in ten patients receiving PLD alone or PLD + carboplatin (R2 = 0.61, p = 0.008). (C) Phagocytic activity: MFI is significantly correlated with CL of encapsulated doxorubicin in six patients receiving PLD alone (R2 = 0.57, p = 0.03). (D) Production of ROS: MFI is significantly correlated with CL of encapsulated doxorubicin in six patients receiving PLD alone (R2 = 0.61, p = 0.001).
CL: Clearance; MFI: Mean fluorescent intensity; MO: Monocyte; MPS: Mononuclear phagocyte system; PLD: PEGylated liposomal doxorubicin; R2: Coefficient of determination; ROS: Reactive oxygen species.
Reproduced with permission from [44].
Age
Pharmacokinetic studies of Doxil administered in three Phase I and Phase II studies in patients with solid tumors and in patients with AIDS-related Kaposi’s sarcoma were performed. The mean ± standard deviation clearance of sum total Doxil in patients aged <60 and ≥60 years old were 54.6 ± 28.5 and 23.3 ± 10.8 ml/h/m2, respectively (p < 0.0001) [45]. A more recent study focusing on the effect of age on doxorubicin PK and toxicity after administration of PLD was published earlier this year [46]. A total of 35 patients (≥70 years old) were enrolled in the study. The mean t1/2 of the first three cycles of PLD in patients aged >77 versus ≤77 years old was 99 h versus 72 h (p = 0.02). A more profound 42% increase in mean t1/2 was seen when comparing octogenarians to patients in their 70s (p = 0.005) [46].
Age-related effects on PD of CMAs have also been reported. The relationship between neutropenia and monocytopenia after administration of S-CKD602 was tested by evaluating the percent decrease and rate of decrease of ANC and monocytes in patients aged <60 and ≥60 years of age [43]. Based on overall exposure to encapsulated S-CKD602, it appears that there is a greater reduction in monocytes at the nadir in patients aged <60 years of age versus ≥60 years of age [43]. This study concluded that monocytes are more sensitive than neutrophils to S-CKD602, increased sensitivity is related to the encapsulated liposomal formulation of CKD-602 and that age impacts this interaction [43]. This is consistent with the age-related effects on the clearance (CL) of PEGylated liposomes.
Age has also been reported to be associated with the toxicity and efficacy of CMA. Figure 8 from Gusella reports the relationship between age, PLD half-life and palmar-plantar erythrodysesthesia (PPE), a hallmark side effect of PLD. Both increasing age and PLD plasma half-life correlated with PPE severity [46]. In a multicenter Phase II trial of elderly patients (n = 60) with metastatic breast cancer who were treated with Doxil, severe nonhematological toxicities were correlated to patient aged >80 years old [47]. In terms of efficacy, an increase in age was found to be inversely proportional with progression-free survival; however, no correlation between age and overall survival was found [47]. Alterations in the PK and PD of CMA may involve age-related decline in immune system functioning, specifically the association of aging to the function of monocytes [48]. In theory, there is a loss of MPS activity or function in elderly patients which decreases the clearance of CMA by MPS and leads to increased drug exposures and toxicity in elderly patients.
Figure 8. The relationship between hand foot syndrome and PLD pharmacokinetics or age in patients receiving PLD treatment.

HFS (also called palmar-plantar erythrodysesthesia) association with PLD T1/2 (A) and age (B). Longer PLD T1/2 and advanced age resulted in an overall greater severity PPE grade.
ANOVA: Analysis of variance; HFS: Hand foot syndrome; PLD: PEGylated liposomal doxorubicin; PPE: Palmar-plantar erythrodysesthesia.
Reproduced with permission from [46].
Gender
Gender was also found to be one of the many factors contributing the PK/PD variability of CMAs. The effect of gender on clearance of Doxil (n = 70), IHL-305 (n = 39) and S-CKD602 (n = 45) was evaluated in PK studies conducted as part of Phase I and Phase II studies [49]. Female patients had a 2.4-fold lower overall clearance of Doxil (p < 0.001), a 1.4-fold lower overall clearance of IHL-305, and a 1.3-fold lower overall clearance of S-CKD602 compared with male patients [49]. In addition, an evaluation of the plasma PK disposition of TLI (liposomal topotecan) and S-CKD602 in male and female rats found the clearance of TLI and S-CKD602 to be 1.2-fold and 1.4-fold (p = 0.009) lower in female rats compared with male rats, respectively [50]. The basis for the differences in the PK and PD of CMA associated with gender is unclear with some ideas suggesting differences may be attributed to sex hormones on immune cell function [49].
Tissue & organ effects
Body habitus
Defined as the physical characteristics of an individual leading to a body type of underweight, normal weight or overweight, body habitus has been suggested to play an important role in PK of drugs cleared by the MPS [44,45]. Common terms used to describe an individual’s body habitus include BMI and body surface area (BSA).
Pharmacokinetic studies of Doxil were conducted as part of three Phase I and Phase II studies in patients with solid tumors (34 patients) and in patients with AIDS-related Kaposi’s sarcoma (36 patients) [45]. BSA and BMI were evaluated as potential factors that affect the clearance of Doxil [45]. The linear regression coefficient between BSA and Doxil clearance on cycle 1 was R2 = 0.25. BSA contributed to a relative reduction in variability of clearance by 8.6% [45]. There was no significant relationship found between Doxil clearance on cycle 1 and body composition as measured by actual body weight/ideal body weight (ABW/IBW) or BMI (R2 = 0.22 and 0.13, respectively) [45]. However, it was reported that patients with a lean body composition (ABW/IBW <1.35) had a higher plasma exposure of S-CKD602 as compared with patients with an ABW/IBW ≥ 1.35 (p = 0.02), perhaps due to a smaller volume of distribution [44]. While there was no relationship found between body composition and Doxil clearance in this study, this could be due to a lower number of patients enrolled [45]. Additional data collection through CMA clinical pharmacology studies is warranted in order to further explore the potential relationship between body composition and NP clearance.
Presence of liver tumors
In a Phase I pharmacokinetic study evaluating encapsulated and released CKD-602, 45 patients were enrolled, of which 26 individuals had tumor(s) of all types in their liver [51]. Linear and nonlinear pharmacokinetic models were constructed for encapsulated CKD-602, and it was determined that liver tumor was a significant covariate for maximum velocity. Including liver tumor as a covariate in the model decreased the variation in Vmax by 29%, meaning that liver tumors (primary or metastases) were outliers from the general population and influenced CMA PK [51]. The interpatient variability in S-CKD602 pharmacokinetic disposition could possibly be explained by the presence of metastatic liver tumors, as the Vmax of patients with these tumors is 1.5-fold higher than those patients without liver tumors [51]. These data suggest that patients with liver tumor may have 35% lower plasma exposure of encapsulated CKD-602, leaving them at risk for having a lower response potential. This finding is interesting, as historically, most studies show a decrease in clearance of small-molecule drugs when patients have tumors in the liver [52,53].
Drug–drug interactions
Drug–drug interactions have been reported to influence the PK and PD of CMAs. In a drug interaction study, the PK of Doxil when given as a single agent and in combination with a taxane, either paclitaxel (n = 10) or docetaxel (n = 9), was evaluated. Both paclitaxel and docetaxel increased the Doxil AUC (p = 0.002 and 0.039, respectively) and decreased the clearance of Doxil (p = 0.013 and 0.16, respectively). Administering paclitaxel produced a more significant increase in systemic exposure of Doxil than coadministration with docetaxel. These findings may explain a high incidence of toxicity observed at relatively low doses of Doxil when coadministered with paclitaxel compared with docetaxel [54]. In addition, a Phase I study of patients with solid tumors (n = 26) showed that treatment with cisplatin followed by Doxil increased the clearance of Doxil compared with patients receiving single-agent Doxil. A possible mechanism for this accelerated clearance of Doxil is transient macrophage activation induced by cisplatin [55].
In a Phase I study of factors influencing the inter-patient variability in the pharmacokinetic disposition of S-CKD602, the effect of prior chemotherapy on the pharmacokinetic disposition of S-CKD602 was evaluated [44]. Patients with advanced malignancies received S-CKD602 intravenously (iv.) every 3 weeks. Patients who received prior therapy with Doxil (n = 5) had a 2.2-fold higher exposure of sum total AUC/dose S-CKD602 compared with patients who did not receive prior Doxil (n = 39; p = 0.045) [44]. In addition, the encapsulated AUC/dose of S-CKD602 in patients previously treated with Doxil (n = 5) was 1.8-fold higher compared with patients who did not receive Doxil (n = 34; p = 0.11). However, prior Doxil therapy did not alter the disposition of released CKD-602 [44]. There was no relationship found between the disposition of S-CKD602 and the number of PLD cycles or the time interval between stopping PLD and starting S-CKD602 [44]. It is unclear, if changes in S-CKD602 PK were related to Doxil or patient-related factors as all patients that received Doxil were patients with refractory ovarian cancer already exposed to chemotherapy.
Bevacizumab is a recombinant humanized mono-clonal VEGF-A antibody that inhibits the binding of human VEGF-A to its receptors. Combination treatment of locally recurrent or metastatic breast cancer patients (n = 39) with bevacizumab at 10 mg/kg iv. and Doxil at 20 mg/m2 iv. every 2 weeks resulted in the premature discontinuation of a single-arm Phase II trial due to higher than anticipated toxicity [56]. The most significant toxicity noted was grade 3 PPE that occurred in an unusually high proportion of patients (41%), suggesting an additive toxic effect of combination bevacizumab and Doxil treatment [56]. Possible suggested mechanisms of this synergistic toxicity include: direct pharmacological interaction between Doxil and bevacizumab; effects of bevacizumab on the vasculature of soles, palms and possibly the oral mucosa, resulting in increased accumulation of Doxil in these areas; and bevacizumab impairing wound healing of dermal and mucosal injuries [56].
Prior treatment
In addition to the high interpatient variability in the PK of NPs, there may also be high intrapatient variability in PK of NPs. Gabizon and colleagues reported that the clearance of sum total PLD decreased by approximately 25–50% from cycle 1 to 3 in patients with ovarian cancer (Figure 9) [57]. La-Beck and colleagues reported that this reduction in clearance of Doxil from cycle 1 to 3 was associated with a reduction in precycle monocyte count. These studies suggest that there is a reduction in the clearance of liposomes over time that is associated with a reduction in MPS function. Thus, dose reductions may be needed in subsequent cycles to minimize the risk of toxicity [57]. However, how the reductions in the dose of PLD should be performed in each patient is unclear due to high inter- and intra-patient variability in changes in MPS function. By phenotypically probing patient MPS function prior to administering each cycle of PLD is of interest to guide patient-specific dosing [13]. Studies are ongoing to evaluate the use of phenotypic probes of MPS as a method to individualize the dose of PLD.
Figure 9. Clearance of PEGylated liposomal doxorubicin after chemotherapy cycle in human subjects.
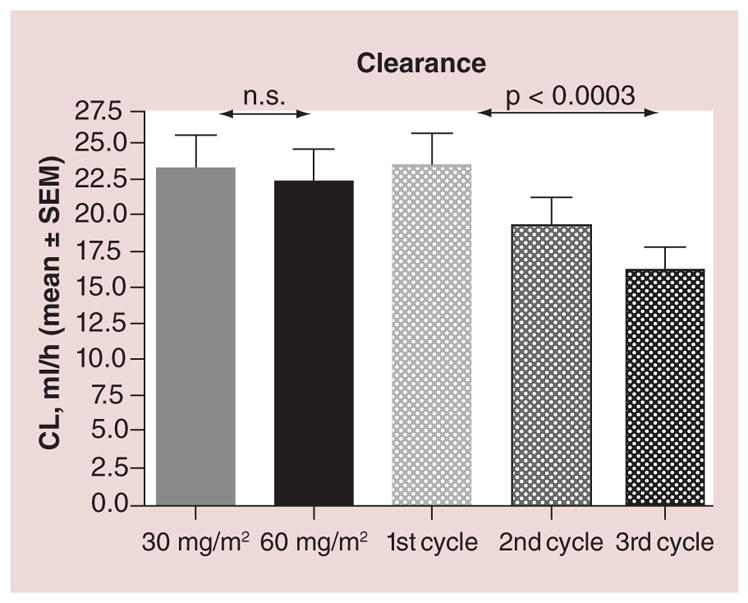
Bars represent mean values and SEM of clearance of PLD by dose and cycle. p values shown were calculated by repeated measures analysis of variance.
CL: Clearance; n.s: Not significant; PLD: PEGylated liposomal doxorubicin; SEM: Standard error of the mean.
Adapted with permission from [57].
Conclusion
There are many properties that make CMAs unique from the active small-molecule drug that is contained within the nanocarrier. These differences lead to significant variability in the PK and PD of CMAs. It has been shown that physical properties, the MPS, presence of tumors in the liver, EPR effect, drug–drug interactions, age, gender, body habitus and prior treatment all contribute in varying degrees to the pharmacokinetic disposition and pharmacodynamic endpoints of CMAs in patients.
Future perspective
Due to the unique and highly variable clearance mechanisms of CMAs, it is important to create, validate and utilize assays and models that represent EPR effect and MPS function, and use them to evaluate factors that alter PK/PD of CMAs during all phases of development. Patient factors that affect MPS function and NP PK/PD variability include age, gender and body habitus among others. However, these factors do not account for all of the PK/PD variability and thus the development of phenotypic probes of the MPS that account for all encompassing factors may be a more appropriate approach to predicting the PK/PD of NPs in animal models and patients (Figure 10). These probes could also be used to individualize the dose of NPs throughout therapy, as there is high inter- and intrapatient variability in MPS function and NP PK. Studies in animal models and patients using phenotypic probes of MPS function to predict the clearance of PEGylated liposomal agents support this plan. Additional areas of research that can aid in our understanding of how these agents are handled and how we may predict their actions in patients include: pharmacogenomics, more sensitive and accurate analytical PK methods and identification of the optimal preclinical models.
Figure 10. Summary of the process to use a phenotypic probe of mononuclear phagocyte system function in blood to measure mononuclear phagocyte system function in patients, which would predict nanoparticle pharmacokinetics, efficacy and toxicity.

his type of probe could be used as a test that could retrospectively be used to explain patients with highly variable PK and PD. This type of probe could also be used as a method to individualize the dose of nanoparticles as needed.
MPS: Mononuclear phagocyte system; PD: Pharmacodynamics; PK: Pharmacokinetics; PPE: Palmar-plantar erythrodysesthesia.
Supplementary Material
Executive summary.
Nanoparticle formulations
Currently hundreds of nanoparticle (NP)-based drug products are undergoing development.
Clinically available carrier-mediated agents (CMAs) are still limited but expected to grow greatly in the near future.
Pharmacokinetic characterization
The kinetics of CMAs are dependent upon the carrier until the parent drug is released.
Analytical studies must be performed to assess the disposition of CMAs.
Mononuclear phagocyte system
NPs can be taken up by cells of the mononuclear phagocyte system (MPS; Kupffer cells in the liver, dendritic cells in the lymph nodes, macrophages and B cells in the spleen).
Variations within the MPS greatly influence the pharmacokinetics and pharmacodynamics of a CMA.
Delivery of CMA in tumor
CMAs have the ability to extravasate into tissue with help of tumor’s leaky vasculature as a result of the enhanced permeability and retention effect.
New therapeutics are utilizing NPs as delivery systems in both preclinical and early phase studies in hard to target areas like the brain.
Physical characteristics
-
Particle size and shape:
Particles between 100 and 200 nm have been most efficient for CMA uptake in tumor;
Particle-Replication-In-Non-wetting-Templates (PRINT) technology can easily alter particle shape.
-
Surface modification and charge:
Conjugation of PEG to the surface of NPs increases circulation time and encapsulated drug plasma area under the concentration versus time curve;
Uncharged particles have less MPS uptake and longer circulation time.
-
Number of NPs administered:
Greater number of particles per dose increases the plasma and tumor drug exposure.
Monocytes & the MPS
-
There is a high degree of interpatient variability with regards to the clearance of NPs:
Both linear and nonlinear clearances (CLs) of NPs are seen in the clinic;
Nonlinear CL may be due to saturation of the MPS.
There is an apparent relationship between NP CL and age, gender, body habitus, presence of liver disease, drug–drug interactions and prior treatment.
Monocyte and dendritic cells function can predict the pharmacokinetics of PEGylated liposomal NPs in mice, rats and dogs.
Footnotes
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Financial & competing interests disclosure
Research reported in this publication was supported by the National Cancer Institute of the NIH under Award Number U54CA151652, and the University of North Carolina University Cancer Research Fund. The authors have no other relevant affiliations or financial involvement with any organization or entity with a interest in or financial conflict with the subject matter or materials discussed in the manuscript a part from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Ge Y, Tiwari A, Li S. Nanomedicine – bridging the gap between nanotechnology and medicine. Adv Mat Lett. 2011;2(1):1–2. [Google Scholar]
- 2.Caron WP, Song G, Kumar P, Rawal S, Zamboni WC. Interpatient pharmacokinetic and pharmacodynamic variability of carrier-mediated anticancer agents. Clin Pharmacol Ther. 2012;91(5):802–812. doi: 10.1038/clpt.2012.12. [DOI] [PubMed] [Google Scholar]
- 3.Laginha K, Mumbengegwi D, Allen T. Liposomes targeted via two different antibodies: assay, B-cell binding and cytotoxicity. Biochim Biophys Acta. 2005;1711(1):25–32. doi: 10.1016/j.bbamem.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Yurkovetskiy AV, Hiller A, Syed S, et al. Synthesis of a macromolecular camptothecin conjugate with dual phase drug release. Mol Pharm. 2004;1(5):375–382. doi: 10.1021/mp0499306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zamboni WC. Liposomal, nanoparticle, and conjugated formulations of anticancer agents. Clin Cancer Res. 2005;11(23):8230–8234. doi: 10.1158/1078-0432.CCR-05-1895. [DOI] [PubMed] [Google Scholar]
- 6.Zamboni WC, Gervais AC, Egorin MJ, et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53(4):329–336. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- 7.Laverman P, Boerman OC, Oyen WJG, Corstens FHM, Storm G. In vivo applications of PEG liposomes: unexpected observations. Crit Rev Ther Drug Carrier Syst. 2001;18(6):551–566. [PubMed] [Google Scholar]
- 8.Schell RF, Sidone BJ, Caron WP, et al. Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomedicine. 2014;10(1):109–117. doi: 10.1016/j.nano.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamboni WC. Concept and clinical evaluation of carrier-mediated anticancer agents. Oncologist. 2008;13(3):248–260. doi: 10.1634/theoncologist.2007-0180. [DOI] [PubMed] [Google Scholar]
- 10.Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol. 2002;72(4):621–627. [PubMed] [Google Scholar]
- 11.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5(4):487–495. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2(8):469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 13.Caron WP, Lay JC, Fong AM, et al. Translational studies of phenotypic probes for the mononuclear phagocyte system and liposomal pharmacology. J Pharmacol Exp Ther. 2013;347(3):599–606. doi: 10.1124/jpet.113.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonso MJ. Nanomedicines for overcoming biological barriers. Biomed Pharmacother. 2004;58(3):168–172. doi: 10.1016/j.biopha.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]
- 16.Prabhakar U, Maeda H, Jain RK, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73(8):2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 18.Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci USA. 2013;110(46):18632–18637. doi: 10.1073/pnas.1318415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12(11):958–962. doi: 10.1038/nmat3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 21.Park JW, Hong K, Kirpotin DB, et al. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8(4):1172–1181. [PubMed] [Google Scholar]
- 22.Wickham T, Reynolds J, Drummond DC, et al. Preclinical safety and activity of MM-302, a HER2-targeted liposomal doxorubicin designed to have an improved safety and efficacy profile over approved anthracyclines. www.merrimackpharma.com/sites/default/files/documents.
- 23.Vaage J, Barbera-Guillem E, Abra R, Huang A, Working P. Tissue distribution and therapeutic effect of intravenous free or encapsulated liposomal doxorubicin on human prostate carcinoma xenografts. Cancer. 1994;73(5):1478–1484. doi: 10.1002/1097-0142(19940301)73:5<1478::aid-cncr2820730526>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 24.Zamboni WC, Strychor S, Joseph E, et al. Plasma, tumor, and tissue disposition of STEALTH liposomal CKD-602 (S-CKD602) and nonliposomal CKD-602 in mice bearing A375 human melanoma xenografts. Clin Cancer Res. 2007;13(23):7217–7223. doi: 10.1158/1078-0432.CCR-07-1035. [DOI] [PubMed] [Google Scholar]
- 25.Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, PEGylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother Pharmacol. 1999;43(1):1–7. doi: 10.1007/s002800050855. [DOI] [PubMed] [Google Scholar]
- 26.Siegal T, Horowitz A, Gabizon A. Doxorubicin encapsulated in sterically stabilized liposomes for the treatment of a brain tumor model: biodistribution and therapeutic efficacy. J Neurosurg. 1995;83(6):1029–1037. doi: 10.3171/jns.1995.83.6.1029. [DOI] [PubMed] [Google Scholar]
- 27.Koukourakis MI, Koukouraki S, Giatromanolaki A, et al. Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer. J Clin Oncol. 1999;17(11):3512–3521. doi: 10.1200/JCO.1999.17.11.3512. [DOI] [PubMed] [Google Scholar]
- 28.Sharma US, Sharma A, Chau RI, Straubinger RM. Liposome-mediated therapy of intracranial brain tumors in a rat model. Pharm Res. 1997;14(8):992–998. doi: 10.1023/a:1012136925030. [DOI] [PubMed] [Google Scholar]
- 29.Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95(8):4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Lu W. Recent advances in brain tumor-targeted nano-drug delivery systems. Expert Opin Drug Deliv. 2012;9(6):671–686. doi: 10.1517/17425247.2012.682726. [DOI] [PubMed] [Google Scholar]
- 31.Anders CK, Adamo B, Karginova O, et al. Pharmacokinetics and efficacy of PEGylated liposomal doxorubicin in an intracranial model of breast cancer. PLoS ONE. 2013;8(5):e61359. doi: 10.1371/journal.pone.0061359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaillard PJ, Appeldoorn CC, Dorland R, et al. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3–101) PLoS ONE. 2014;9(1):e82331. doi: 10.1371/journal.pone.0082331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen SA, Day ES, Ko CH, et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med. 2013;5(209):209ra152. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu D, Mori A, Huang L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim Biophys Acta. 1992;1104(1):95–101. doi: 10.1016/0005-2736(92)90136-a. [DOI] [PubMed] [Google Scholar]
- 35.Charrois GJ, Allen TM. Rate of biodistribution of STEALTH liposomes to tumor and skin: influence of liposome diameter and implications for toxicity and therapeutic activity. Biochim Biophys Acta. 2003;1609(1):102–108. doi: 10.1016/s0005-2736(02)00661-2. [DOI] [PubMed] [Google Scholar]
- 36.Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 37.Chu KS, Hasan W, Rawal S, et al. Plasma, tumor and tissue pharmacokinetics of docetaxel delivered via nanoparticles of different sizes and shapes in mice bearing SKOV-3 human ovarian carcinoma xenograft. Nanomedicine. 2013;9(5):686–693. doi: 10.1016/j.nano.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci. 2008;97(11):4696–4740. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 39.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51(4):691–743. [PubMed] [Google Scholar]
- 40.Levchenko TS, Rammohan R, Lukyanov AN, Whiteman KR, Torchilin VP. Liposome clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. Int J Pharm. 2002;240(1–2):95–102. doi: 10.1016/s0378-5173(02)00129-1. [DOI] [PubMed] [Google Scholar]
- 41.Chu KS, Schorzman AN, Finniss MC, et al. Nanoparticle drug loading as a design parameter to improve docetaxel pharmacokinetics and efficacy. Biomaterials. 2013;34(33):8424–8429. doi: 10.1016/j.biomaterials.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zamboni WC, Maruca LJ, Strychor S, et al. Bidirectional pharmacodynamic interaction between pegylated liposomal CKD-602 (S-CKD602) and monocytes in patients with refractory solid tumors. J Liposome Res. 2011;21(2):158–165. doi: 10.3109/08982104.2010.496085. [DOI] [PubMed] [Google Scholar]
- 43.Zamboni WC, Strychor S, Maruca L, et al. Pharmacokinetic study of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. Clin Pharmacol Ther. 2009;86(5):519–526. doi: 10.1038/clpt.2009.141. [DOI] [PubMed] [Google Scholar]
- 44.La-Beck NM, Zamboni BA, Gabizon A, et al. Factors affecting the pharmacokinetics of pegylated liposomal doxorubicin in patients. Cancer Chemother Pharmacol. 2012;69(1):43–50. doi: 10.1007/s00280-011-1664-2. [DOI] [PubMed] [Google Scholar]
- 45.Gusella M, Bononi A, Modena Y, et al. Age affects pegylated liposomal doxorubicin elimination and tolerability in patients over 70 years old. Cancer Chemother Pharmacol. 2014;73(3):517–524. doi: 10.1007/s00280-014-2378-z. [DOI] [PubMed] [Google Scholar]
- 46.Falandry C, Brain E, Bonnefoy M, et al. Impact of geriatric vulnerability parameters on pegylated liposomal doxorubicin (PLD) tolerance and outcome in elderly patients with metastatic breast cancer: results of the DOGMES multicenter phase II GINECO trial. J Clin Oncol. 2011;29:Abstract 9122. [Google Scholar]
- 47.Lloberas J, Celada A. Effect of aging on macrophage function. Exp Gerontol. 2002;37(12):1325–1331. doi: 10.1016/s0531-5565(02)00125-0. [DOI] [PubMed] [Google Scholar]
- 48.La-Beck N, Wu H, Infante J, et al. The evaluation of gender on the pharmacokinetics (PK) of PEGylated liposomal anticancer agents. J Clin Oncol. 2010;28:Abstract e13003. [Google Scholar]
- 49.Song G, Wu H, La-Beck N, et al. Effect of gender on pharmacokinetic disposition of PEGylated liposomal CKD-602 (S-CKD602) and Optisomal topotecan (TLI) in rats. Presented at: AACR 101st Annual Meeting; Washington, DC, USA. 17–21 April 2010; 2010. p. Abstract 3700. [Google Scholar]
- 50.Wu H, Ramanathan RK, Zamboni BA, et al. Population pharmacokinetics of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. J Clin Pharmacol. 2012;52(2):180–194. doi: 10.1177/0091270010394851. [DOI] [PubMed] [Google Scholar]
- 51.Robieux I, Sorio R, Borsatti E, et al. Pharmacokinetics of vinorelbine in patients with liver metastases. Clin Pharmacol Ther. 1996;59(1):32–40. doi: 10.1016/S0009-9236(96)90021-1. [DOI] [PubMed] [Google Scholar]
- 52.Twelves CJ, O’Reilly SM, Coleman RE, Richards MA, Rubens RD. Weekly epirubicin for breast cancer with liver metastases and abnormal liver biochemistry. Br J Cancer. 1989;60(6):938–941. doi: 10.1038/bjc.1989.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyass O, Hubert A, Gabizon AA. Phase I study of doxil-cisplatin combination chemotherapy in patients with advanced malignancies. Clin Cancer Res. 2001;7(10):3040–3046. [PubMed] [Google Scholar]
- 54.Volk LD, Flister MJ, Chihade D, Desai N, Trieu V, Ran S. Synergy of nab-paclitaxel and bevacizumab in eradicating large orthotopic breast tumors and preexisting metastases. Neoplasia. 2011;13(4):327–338. doi: 10.1593/neo.101490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rochlitz C, Ruhstaller T, Lerch S, et al. Combination of bevacizumab and 2-weekly PEGylated liposomal doxorubicin as first-line therapy for locally recurrent or metastatic breast cancer. A multicenter, single-arm phase II trial (SAKK 24/06) Ann Oncol. 2011;22(1):80–85. doi: 10.1093/annonc/mdq319. [DOI] [PubMed] [Google Scholar]
- 56.Gabizon A, Isacson R, Rosengarten O, Tzemach D, Shmeeda H, Sapir R. An open-label study to evaluate dose and cycle dependence of the pharmacokinetics of pegylated liposomal doxorubicin. Cancer Chemother Pharmacol. 2008;61(4):695–702. doi: 10.1007/s00280-007-0525-5. [DOI] [PubMed] [Google Scholar]
- 57.La-Beck N, Tzemach D, Schmeeda H, Sapir R, Gabizon A, Zamboni W. Evaluation of the relationship between patient factors and the reduction in clearance of pegylated liposomal doxorubicin. ASCO Annual Meeting. J Clin Oncol. 2009;27(Suppl):Abstract 2548. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.