

Summary
In T lymphocytes, polarization of the microtubule-organizing center (MTOC) to the immunological synapse enables the directional secretion of cytokines, cytolytic factors, and other soluble molecules toward the antigen-presenting cell. This is likely to be crucial for maintaining the specificity of T-cell effector responses. Here, we review recent advances in our understanding of MTOC reorientation in T cells, focusing first on the importance of diacylglycerol and protein kinase C isozymes and then on the molecular motor proteins that function downstream to drive MTOC movement.
Keywords: T cells, lipid mediators, protein kinases and phosphatases, T-cell receptors, signaling proteins, signal transduction
Introduction
Recognition by a T cell of cognate antigen on the surface of an antigen-presenting cell (APC) induces the formation of a stereotyped contact between the two cells known as an immunological synapse (IS). Although the IS was initially proposed to be a platform for T-cell antigen receptor (TCR) signaling (1, 2), subsequent studies demonstrated that TCR activation precedes and is required for IS formation (3, 4). This finding led to the now prevailing view that the IS does not function to trigger TCR signaling events but rather to modulate downstream T-cell effector responses (5). T cells operate in dense intercellular milieus containing numerous bystander cells that are often largely irrelevant to immune function. In such an environment, constraining the effects of a T-cell-mediated response to the APC alone or to a select group of target cells is an important issue. Indeed, multiple studies have suggested that the IS transforms the T cell-APC interface into a privileged zone designed to restrict the scope of intercellular communication. For example, certain cell surface signaling molecules, including Fas ligand (6) and CD40 ligand (7), are localized to the IS where they can exclusively engage the APC. IS formation also enables the directional release of soluble factors, such as cytokines and cytolytic molecules, into the synaptic space (8, 9). In addition, recent work has indicated that the IS can influence T-cell proliferation and differentiation. In a particularly intriguing set of studies, naive T cells were found to undergo asymmetric cell division while attached to dendritic cells, leading to the formation of distinct effector and memory T-cell pools (10, 11). IS formation plays an instrumental role in this context by establishing an anisotropic accumulation of proteins and organelles. Thus, it is becoming clear that the IS shapes not only the function but also the fate of activated T cells.
The cytoskeletal architecture of the IS reflects its role in specifying intercellular communication and creating a privileged environment at the T-cell-APC contact site. Within minutes of TCR stimulation, the T-cell microtubule-organizing center (MTOC) (also known as the centrosome) moves to a position just beneath the center of the IS (12–15). The MTOC serves as a focal point for vesicular trafficking and is closely associated with both the Golgi apparatus and the endosomal compartment. In cytotoxic T lymphocytes (CTLs), the MTOC also coordinates lytic granules containing perforin and granzyme (9). Upon TCR stimulation, these granules traffic toward the MTOC in a Ca2+-dependent manner, thereby hitching a ride to the IS (16). Concomitant with MTOC polarization, synaptic filamentous actin (F-actin) is reorganized into a ring-shaped configuration characterized by robust accumulation in the periphery and depletion from the center (17–19). Although some cortical F-actin remains in the central domain, its relative paucity provides vesicular compartments direct access to the synaptic membrane (20, 21). In this manner, MTOC reorientation, coupled to F-actin ring formation at the IS, enables the directional secretion of both nascent cytokines and cytolytic factors toward the APC (8, 9).
Although MTOC polarization has long been considered a hallmark of IS architecture, the molecular mechanisms that control it have remained elusive, largely because T cells (and lymphocytes in general) are a poor cell biology system. In short, they are too small, and they do things too fast! Recent improvements in single-cell imaging, however, combined with better loss-of-function strategies, have enabled some notable progress in this area. In this review, we discuss recent advances in the understanding of how signals emanating from the TCR control MTOC dynamics, focusing on (i) how the lipid second messenger diacylglycerol (DAG) guides polarization to the IS and (ii) how molecular motor proteins translate polarized signaling into forces that actually move the MTOC (Fig. 1).
Fig. 1. Mechanism of T-cell microtubule-organizing center (MTOC) reorientation to the immunological synapse (IS).
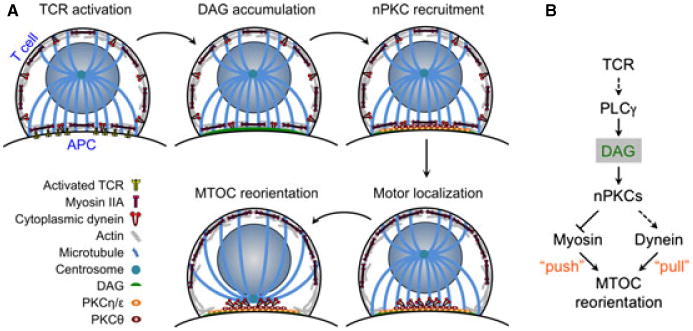
Localized diacylglycerol accumulation induces the ordered recruitment of protein kinase C (PKC) isozymes. This drives the recruitment of dynein to the IS and the redistribution of non-muscle myosin II (NMII) to the sides and rear of the cell. Dynein and NMII then collaborate to move the MTOC. The process is schematized to the left, with a pathway diagram to the right. In the pathway diagram, solid arrows denote direct connections, and dotted arrows denote either indirect or multistep connections.
A new imaging approach to study MTOC dynamics
Reorientation of the MTOC to the IS was originally documented over 30 years ago, well before the advent of the term ‘immunological synapse’. Berke, Kupfer, and their colleagues (12–14, 22) were the first to postulate a link between MTOC polarization and the targeted secretion of cytokines and cytotoxic molecules. It was subsequently shown that localized TCR activation is necessary and sufficient to drive the recruitment of the MTOC. In a particularly elegant set of experiments, Sedwick and colleagues mixed T cells with target cells expressing either cognate peptide-major histocompatibility complex (pMHC) or the leukocyte function-associated anti-gen-1 (LFA-1) ligand intercellular adhesion molecule-1 (ICAM-1) (23). In ternary conjugates containing one T cell and one of each kind of target cell, the MTOC consistently polarized toward the target expressing pMHC. Conversely, strong F-actin accumulation was observed at the interface with the cell expressing ICAM-1. Hence, TCR signaling controls MTOC positioning in the absence of and even in opposition to integrin-mediated adhesion. Consistent with this notion, a number of TCR-proximal signaling molecules, including Lck, linker for activation of T cells (LAT), ζ-associated protein of 70 kDa (Zap70), and SH2 domain-containing protein of 76 kDa (Slp76), were shown to be required for polarization responses (24, 25). Precisely how signals from the TCR and its associated proteins are coupled to cytoskeletal machinery controlling MTOC movement, however, remained unresolved.
Although a number of systems exist for investigating MTOC polarization, the most prevalent is the imaging of live or fixed T cell-APC conjugates. Because this approach makes use of bona fide target cells, it is considerably more physiologically relevant than alternative strategies in which T cells are stimulated with antibody-coated beads or glass slides. That being said, the spatial and temporal resolution of T cell-APC conjugate experiments is fundamentally limited, because the conjugates are small and dynamic and also because it is difficult to establish when initial TCR stimulation occurs under these conditions. Together, these issues complicate efforts to correlate cytoskeletal remodeling with intracellular signaling responses. This is particularly problematic when studying a process like MTOC reorientation, which occurs within minutes of T-cell activation. Further complicating matters, the MTOC polarization response in conjugates is actually a combination of two sequential processes: adhesion to the APC, followed by MTOC reorientation. This obfuscates the interpretation of perturbation experiments, as molecules involved solely in APC adhesion would also be expected to affect polarization secondarily.
To circumvent these issues, we developed methodology allowing us to control precisely where and when the T cell receives antigenic stimulus and then to monitor responses with high spatiotemporal resolution. Our system is built around a photoactivatable pMHC reagent that is non-stimulatory to T cells until it is irradiated with ultraviolet (UV) light (26, 27). CD4+ T cells expressing the 5C.C7 TCR, which recognizes the moth cytochrome c88–103 peptide presented by the class II MHC I-Ek, are plated on glass coverslips containing a photoactivatable version of the MCC-I-Ek complex together with an antibody against H2-Kk, a class I MHC expressed by the T cell. The anti-H2-Kk antibody serves to induce T-cell attachment and spreading without activating the TCR. Focused UV light is then used to decage the pMHC in a micrometer-scale region beneath the T cell, triggering localized TCR activation in the plasma membrane attached to the glass. Photostimulation of the TCR in this manner typically induces reorientation of the MTOC to the irradiated region in less than 3 min (27, 28). This polarization response and its associated intracellular signaling events can be monitored with genetically encoded fluorescent reporters (e.g. proteins linked to GFP or RFP) using either epifluorescence or total internal reflection fluorescence (TIRF) microscopy. TIRF illumination generates high-resolution images of the first 100 nm of the cell attached to the glass and is particularly well suited for the imaging of signaling dynamics at the membrane. Recently, we have extended our photoactivation and imaging approach to CTLs expressing the OT-1 TCR, enabling us to compare polarized signaling responses in CD4+ and CD8+ T cells.
As with any in vitro system, there are caveats that should be considered. Immobilization of pMHC on the glass surface would presumably hinder the trafficking of TCRs in the plasma membrane, which could alter the downregulation and attenuation of activated receptor complexes. It is also possible that photoactivation damages the T cell. We have no evidence, however, that the UV pulses we use adversely affect cellular physiology over the timescale of our experiments (typically less than 10 min). Indeed, the intracellular signaling responses we observe are completely dependent on the presence of photoactivatable pMHC (i.e. they are not elicited by UV alone) (27). Nevertheless, to the extent that it is possible, we use T cell-APC conjugate experiments to validate results obtained in photoactivation studies.
The importance of localized DAG signaling
TCR engagement induces the phosphorylation of its associated CD3 chains by the Src family kinase Lck, leading to the recruitment and activation of the Syk family kinase Zap70 (29, 30). Lck and Zap70 then phosphorylate multiple residues within the scaffolding proteins LAT and Slp76, which form a complex that serves as a platform to recruit a number of downstream effector enzymes. One of the most important of these enzymes is phospholipase C-γ (PLCγ), which hydrolyzes phosphatidylinositol 4,5 bisphosphate (PIP2), a plasma membrane phospholipid, to yield two second messengers: inositol trisphosphate (IP3), the soluble headgroup, and diacylglycerol (DAG), the lipid remnant. IP3 activates calcium (Ca2+) signaling by triggering the depletion of intracellular Ca2+ stores in the endoplasmic reticulum, which induces the influx of Ca2+ through CRAC channels at the cell surface. DAG, for its part, signals by recruiting proteins containing DAG-specific C1 domains to the plasma membrane. We became particularly interested in the role of DAG in T-cell polarity, because it had been shown using fluorescent biosensors that DAG accumulates in a polarized manner at the IS (31). Furthermore, a small molecule inhibitor of PLCγ completely blocked MTOC reorientation in our hands (28).
TCR photoactivation experiments revealed a striking spatiotemporal correlation between DAG and the MTOC (28) (Fig. 2). DAG consistently accumulated at the site of TCR stimulation 10–15 s before MTOC recruitment, strongly suggesting that the two events were causally related. Indeed, disrupting polarized DAG-dependent signaling with the phorbol ester PMA (phorbol myristate acetate), which engages DAG-binding proteins in a generalized manner, completely disrupted MTOC reorientation in both photoactivation experiments and T cell-APC conjugates. By contrast, blocking Ca2+ signaling with a combination of extracellular and intracellular chelators did not substantially alter polarization responses. Thus, PLCγ functioned through DAG, and not Ca2+, to control the MTOC. These results were somewhat surprising given previous reports that Ca2+ signaling was required for MTOC reorientation (24, 32). In these prior studies, however, polarization was scored using assays that required the T cells to first make close contact with an APC or a stimulatory surface, and then polarize toward it. If Ca2+ signaling were required for productive contact formation in these contexts, it may have affected MTOC polarization secondarily. Indeed, we have found that conjugate formation is suppressed by Ca2+ chelators (Fig. 3).
Fig. 2. Analysis of microtubule-organizing center (MTOC) polarization by T-cell antigen receptor photoactivation.
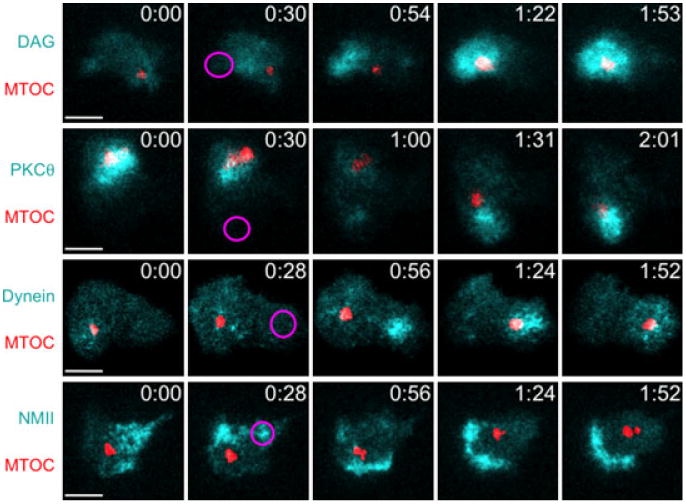
The 5C.C7 T-cell blasts expressing fluorescent reporters for the MTOC (red) and the indicated signaling molecules and motor proteins (cyan) were imaged and ultraviolet (UV)-irradiated on glass coverslips containing photoactivatable peptide-major histocompatibility complex. Representative time-lapse montages are shown (time in m:ss in the upper right corner of each image) with the time and region of UV irradiation indicated by a magenta oval. Scale bars = 5 μm.
Fig. 3. Ca2+ signaling is required for optimal T-cell-antigen-presenting cell conjugate formation.
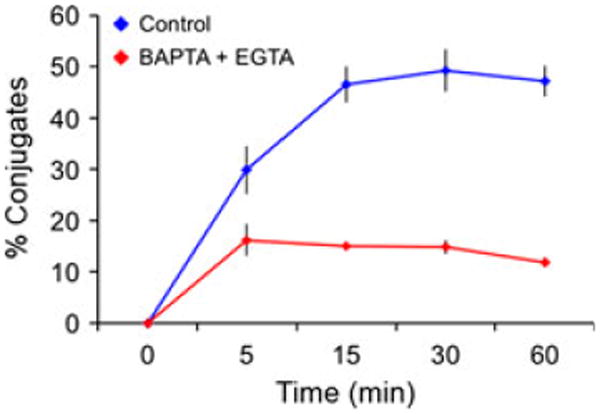
OT-1 T-cell blasts were stained with CFSE and mixed with antigen-loaded, PKH26-stained RMA-s cells in the presence or absence of Ca2+ chelators as indicated. After incubation at 37°C for the indicated times, samples were analyzed by flow cytometry and conjugates identified as GFP+ PKH26+ entities. Percent conjugates = [GFP+PKH26+/(GFP+PKH26−+ GFP+PKH26+)]. Error bars indicate standard error of the mean for triplicate measurements. Data are representative of two independent experiments.
To explore whether localized DAG accumulation was sufficient to drive recruitment of the MTOC, we made use of a photocaged form of DAG that could be activated by UV light (33). Localized photoactivation of this reagent in T cells did induce polarization of the MTOC, consistent with a central role for DAG in this process (28). This polarization response was only transient, however, raising the possibility that other pathways could be involved, particularly in the maintenance phase of the response. It is also possible, however, that the DAG accumulations generated by direct decaging were spatially suboptimal, lacking the properties of an effective polarizing gradient. Indeed, photoactivation of the TCR produced DAG accumulations that were much more stable and sharply defined.
In that regard, how might a sustained, polarizing DAG gradient be maintained over time at the IS? This presumably would require not only the localized production of DAG but also its rapid destruction so as to prevent broadening of the gradient by diffusion. In migrating Dictyostelium, which are guided by an accumulation of phosphatidylinositol 3,4,5 trisphosphate (PIP3) at their leading edge, this problem is solved by the coordinated action of phosphoinositide 3-kinase, which generates PIP3, and the lipid phosphate PTEN (phosphatase and tensin homologue), which hydrolyzes it (34). PTEN has been found to localize to the sides and rear of these cells, thereby constraining PIP3 accumulation to the front. In T cells, DAG-dependent signaling is attenuated by DAG kinases (DGKs), which convert DAG to phosphatidic acid (35). Consistent with a role for DGKs in DAG-induced polarity, we found that a small molecule DGK inhibitor impaired the maintenance of both TCR-induced DAG accumulation and MTOC polarization (28). T cells express a number of DGK isoforms, the most abundant being DGK-α and DGK-ζ. T cells lacking either protein display hyperactive TCR signaling (36, 37), suggesting that they function to curtail signals emanating from DAG. A possible role for these molecules in the induction of polarity, however, remains to be investigated.
Protein kinase C isozymes induce and maintain MTOC polarization
The protein kinase C (PKC) family of serine/threonine kinases plays a key role in coupling lipid- and protein-based signals at the plasma membrane to various downstream responses (38). PKCs can be divided into three subfamilies based on their regulatory properties: conventional isoforms (cPKCs) require both Ca2+ and DAG for activation, novel isoforms (nPKCs) are strongly dependent on DAG, but do not require Ca2+, and atypical isoforms (aPKCs) require neither Ca2+ nor DAG, and are instead regulated by protein-protein interactions. Because the activation requirements of the nPKCs (i.e. DAG but not Ca2+) most closely matched those we had established for MTOC polarization (28), we chose to focus on these isoforms.
TCR photoactivation experiments revealed that three out of the four nPKC isoforms, PKCε, PKCη, and PKCθ, were recruited to the region of TCR stimulation prior to the MTOC (PKCδ did not localize in this way) (39) (Fig. 2). In the case of PKCθ, this behavior was expected based on previous studies demonstrating that it accumulates at the IS and promotes TCR-induced activation of the nuclear factor κ-B (NF-κB) pathway (40, 41). The recruitment of PKCε and PKCη, however, was more surprising, because there were no indications at the time that either protein was involved in T-cell activation. Indeed, T cells derived from PKCε−/− mice displayed essentially no defects in TCR signaling and downstream effector responses (42). Closer examination of the recruitment dynamics of PKCε, PKCη, and PKCθ revealed that they moved to the IS in an ordered cascade (39). PKCs and PKCη arrived first (15–20 s before the MTOC), and occupied a broad swath of membrane encompassing the entire IS, whereas PKCθ arrived approximately 5 s later and was constrained to a more central zone (Fig. 1). We also performed loss-of-function studies to assess the importance of all three proteins for MTOC reorientation (39). Small interfering RNA (siRNA)-mediated suppression of either PKCε or PKCη alone had no effect on the response. Cells lacking both isozymes, however, displayed a significant polarization defect. By contrast, suppression of PKCθ alone was sufficient to inhibit MTOC reorientation. We also found that simultaneous knockdown of PKCε and PKCη inhibited recruitment of PKCθ. Hence, PKCε and PKCη play redundant roles in this pathway and operate upstream of PKCθ.
The functional redundancy we observed between PKCε and PKCη mirrored their essentially identical recruitment properties. It was also consistent with the phylogenetic relationships among PKC isoforms; PKCε and PKCη display higher levels of sequence homology with each other than with other members of the family (Fig. 4). In future studies, it will be interesting to investigate whether PKCε and PKCη play compensatory roles in other aspects of T-cell physiology and also in other cell types. PKCε−/− and PKCη−/− mice both reach adulthood and are visibly normal, but they display subtle phenotypes in specific organ systems. PKCε is highly expressed in the brain, and knockout mice are less prone to alcohol and nicotine addiction due to defects in dopamine-based reward pathways (43–45). PKCη, for its part, is highly expressed in epithelial tissues, and knockouts exhibit impaired wound healing and enhanced carcinogenesis in the skin (46). Recent studies have also documented a modest proliferative defect in PKCη−/− T cells (47). It is likely that these phenotypes will become more pronounced and perhaps give way to more dramatic developmental problems in mice lacking both proteins.
Fig. 4. Schematic diagram of novel protein kinase C (nPKC) domain structure.
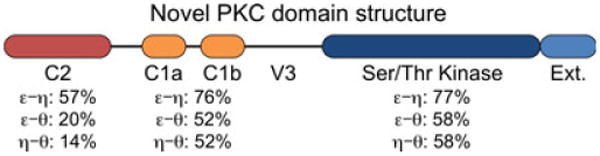
Percent amino acid sequence identities for the C2 domain, the tandem C1 domains, and the kinase domain were calculated from pairwise comparisons (LALIGN, Expasy server) of PKCε, PKCη, and PKCθ and are shown below the region of the protein to which they correspond. V3 = V3 linker, Ext. = Kinase extension.
The differences in recruitment time and accumulation pattern observed between PKCε, PKCη, and PKCθ are quite striking given that the localization of all three isozymes depends on DAG. Biophysical studies have shown that PKCθ binds to DAG in membranes with lower affinity than PKCε (and presumably also PKCη) (48, 49), which could possibly explain the delayed and more limited accumulation of PKCθ at the IS. However, the fact that PKCδ, which also recognizes DAG with high affinity (50), is not recruited to the IS at all suggests that differential DAG binding is unlikely to be the only explanation. Indeed, it is becoming increasingly clear that nPKCs are designed to recognize DAG in a context-dependent manner, together with specific combinations of lipids and proteins. Certain C1 domains have been shown to recognize lipids other than DAG and also to mediate protein-protein interactions (51, 52). These alternative binding events could potentially refine the localization of tandem C1 modules in a combinatorial fashion. The atypical C2 domain, which is positioned just N-terminal to the C1 region in nPKCs (Fig. 4), could also contribute to PKC localization via protein-lipid or protein-protein interactions. Of particular relevance to this review, the C2 domains of PKCδ and PKCθ have been shown to bind specifically to phosphotyrosine (pTyr)-containing peptides (53, 54). In T cells, this interaction mediates association of PKCh with the phosphorylated adapter protein CDCP1 and is required for PKCθ-dependent activation of NF-κB (54). pTyr binding by the C2 domain also drives the recruitment of PKCθ into signaling microclusters at the natural killer cell IS, which play an important role in the amplification of downstream effector responses (55). Finally, there has been much recent interest in the V3 linker region between the tandem C1 domains and the kinase domain (Fig. 4). In PKCθ, the V3 promotes recruitment to the center of the IS, at least in part by interacting with Lck in a protein complex that includes the costimulatory receptor CD28 (56). Taken together, these results indicate that the N-terminal regulatory region of nPKCs controls their localization in a combinatorial fashion by engaging DAG together with other key protein and lipid determinants.
Although the nPKCs are certainly crucial for T-cell MTOC polarization, our results do not exclude a role for other polarity pathways that are not directly regulated by DAG. The aPKC isoform PKCζ, in particular, appears to play an important role in the process. PKCζ is a core component of the Partitioning defective (Par) polarity complex, which also contains the adapter proteins Par-3 and Par-6 (57–59). In more stable polarized cell types, such as neurons, fibroblasts, and epithelial cells, the Par complex functions in conjunction with other polarity inducers, including the Scribble/Dlg complex, to maintain polarity over long timescales (i.e. hours to days). Par and Scribble/Dlg complexes associate with distinct plasma membrane compartments and recruit distinct downstream effectors. In addition, they mutually antagonize each other's accumulation, thereby partitioning the membrane into separate domains. The first indications that polarity complexes were functionally important in T cells came from studies of Par-3 and Scribble/Dlg localization in T-cell-APC conjugates (60). Par-3 accumulated at the IS in these experiments, while Scribble and Dlg localized to the back of the cell. Subsequently, it was shown that phosphorylated PKCζ is also enriched at the IS and furthermore, that blocking PKCζ activity with a small molecule inhibitor impairs MTOC polarization (61). In addition, overexpression of Par1b, a kinase that antagonizes the Par complex, was shown to inhibit MTOC polarization in certain contexts (62). Together, these results imply an important role for PKCζ and Par proteins in T-cell polarity.
How might this aPKC-based system interface with the DAG-nPKC pathway during polarization responses? Although PKCζ is not regulated directly by DAG, a recent report showed that Par-3 interacts with phosphatidic acid (63), which is generated from DAG by DGK activity. Therefore, it is conceivable that both nPKCs and aPKCs function downstream of the same lipid mediator, providing overlapping layers of control that would make MTOC polarization more robust. It is also, possible, however, that the two systems operate sequentially. nPKC accumulation occurs within 2 min of TCR activation (39). By contrast, enrichment of Par-3 at the IS was only observed 30 min after conjugate formation with APCs (60). Phosphorylated PKCζ was also shown to accumulate at the IS over the same timescale (61). These data suggest a two-step model for MTOC polarization in which DAG-dependent nPKC signaling serves as an initial direction sensor to induce MTOC reorientation, at which point the PKCζ and the Par proteins are recruited to stabilize this polarized state (15). Although this model is intellectually satisfying, it has been challenged by recent data indicating that PKCζ can influence MTOC polarization as early as 5 min after TCR stimulation (64). Hence, precisely how nPKCs and aPKCs collaborate remains an open question. It is interesting, though, that these two distinct polarizing systems should both employ PKCs for signal transduction. Although nPKCs and aPKCs are activated by different upstream signals, they display similar substrate specificity at the level of primary sequence, and it is reasonable to expect that they might phosphorylate overlapping pools of downstream proteins. These proteins would likely serve as components of key regulatory modules required for polarized MTOC movement, and their identification remains an important goal for future research.
Collaborative reorientation of the MTOC by dynein and myosin II
It is generally thought that MTOC reorientation to the IS in T cells is mediated by dynein, a molecular motor that accounts for essentially all minus end-directed transport along microtubules in the eukaryotic cytoplasm (65). Dynein is a large, multisubunit protein containing homodimeric heavy chains and a variety of light chains. The heavy chains contain motor domains that hydrolyze ATP and move along microtubules from the plus to the minus end, while the light chains coordinate the assembly of the complex and mediate interactions with cofactors. The most important dynein cofactor is dynactin, an equally large multiprotein complex that functions to enhance the processivity of dynein and also link it to various cargos (65). The first indications that dynein is involved in T-cell MTOC polarization came from studies showing that it accumulates in the IS within minutes of TCR engagement (66, 67). Subsequent photoactivation experiments demonstrated that dynein is recruited to the site of TCR stimulation approximately 5 s before the MTOC (28) (Fig. 2), suggesting that it operates at a very late step in the polarization response that is well downstream of the nPKCs.
Observations like this led to the appealing hypothesis that dynein, once localized to the IS, moves the MTOC by pulling on the microtubules that radiate from it. These microtubules would be oriented with their minus ends in and their plus ends out, so movement of synaptically anchored dynein along them would necessarily drag the MTOC toward the IS. Consistent with this hypothesis, it was found that disrupting dynein function in Jurkat T cells, either by overexpression of the dynamitin subunit of dynactin or by siRNA-mediated suppression of dynein heavy chain, significantly inhibited MTOC reorientation (67). Other results, however, were less encouraging. In primary T cells, knockdown of dynein heavy chain induced only subtle polarization defects (68). Weak phenotypes were also observed after treatment with the small molecule dynein inhibitors ciliobrevin D and erythro-9-[3-(2-Hydroxynonyl)]adenine (68, 69). Taken together, these data suggested that mechanisms other than dynein-mediated pulling might also contribute to MTOC polarization.
In adherent cell types, dynein has been shown to collaborate with the actin-based motor non-muscle myosin II (NMII) to position certain key organelles, such as the MTOC and the nucleus, during polarity induction (70–72). NMII is a heterohexameric complex comprising two heavy chains, two essential light chains, and two regulatory light chains (RLCs). The heavy chain contains the motor domain and F-actin binding site, followed by a long coiled-coil region that mediates homodimerization and also clustering with other NMII complexes. The RLC, for its part, bears a number of key phosphorylation sites that can both positively and negatively regulate motor activity (72). NMII plays a well-established role in the migration of T cells, neutrophils, Dictyostelium, and other amoeboid cell types by driving contractility of the trailing uropod from the sides and rear of the cell (72–74). The possibility that it might contribute to MTOC polarization, however, had until recently not been extensively explored.
Using both shRNA- and small molecule inhibitor-based approaches, we demonstrated that NMII is indeed required for optimal MTOC reorientation both in TCR photoactivation experiments and in T cell-APC conjugates (68). The polarization phenotypes we observed were subtle, similar to what had been seen in T cells lacking dynein. Simultaneous suppression of both dynein and NMII function, however, led to an essentially complete block in polarization, indicating that the two motor complexes operate in a partially redundant manner. Compensation between dynein and NMII explains why we and others have found that inhibition of either protein alone has such weak effects on MTOC polarization (68, 69, 73).
To explore how NMII and dynein collaborate to move the MTOC, we monitored the localization of both motors by TIRF microscopy in photoactivation experiments (68). These studies revealed a striking anti-correlation between NMII and dynein behavior during polarization; whereas dynein accumulated at the region of TCR stimulation, NMII localized to the opposite side of the cell, forming transient, fibrous clusters behind the MTOC as it moved (Fig. 2). The asymmetric distribution of NMII resulted not from enhanced recruitment to the rear but rather from suppression of clustering at the site of TCR stimulation. In this manner, the total distribution of NMII clusters was skewed toward plasma membrane regions distal to the zone of TCR activation. Taken together, these data suggested that dynein ‘pulls’ on the microtubule cytoskeleton from the front while NMII ‘pushes’ on it from behind (Fig. 1).
Precisely how NMII, an actin-based motor, might move the MTOC toward the synapse is not clear. Microtubule plus ends interface with cortical F-actin at the cell periphery, and contractile forces generated by NMII within this F-actin mesh could be propagated along microtubules to the MTOC. Such a mechanism would presumably require close coordination between the microtubule and F-actin cytoskeletons, placing very specific demands on proteins that regulate the growth and stability of both actin- and microtubule-based structures. It has been known for some time that ezrin-ra-dixin-moesin (ERM) family proteins stabilize cortical F-actin by cross-linking it to membrane-associated molecules (75). Interestingly, ERM proteins localize to the back of the T cell during IS formation (76–78), where they could potentially influence NMII cluster formation. Although their role in T-cell activation and MTOC reorientation is still a matter of debate (78, 79), it will be interesting to see if and how they interface with NMII during reorientation responses. Recent studies have also implicated two microtubule regulators, casein kinase 1 and stathmin, in MTOC reorientation (80, 81). T cells lacking either protein display polarization defects that are associated with reduced microtubule growth rates. These results suggest that the formation of extended microtubules is a prerequisite for polarity. Consistent with this interpretation, certain diaphanous formin proteins, including Dia, FMNL-1, and INF2, are known to be required for optimal polarization responses in both Jurkat and primary T cells (82, 83). Formins have a well-established role as actin bundling proteins that drive the formation of filopodia, stress fibers, and other unbranched structures (84). However, formins also promote the accumulation of long-lived microtubules that contain α-tubulin subunits lacking their C-terminal tyrosine (85, 86). Intriguingly, detyrosinated tubulin (also called Glu-tubulin) is rapidly and robustly induced by TCR stimulation, and this process has been shown to require formin activity (82). A pool of stabilized microtubules would presumably be ideal force transducers for both dynein-mediated pulling and NMII-mediated pushing, and it will be interesting in future studies to investigate whether the cellular distribution of Glu-microtubules matches that of either motor complex.
Coupling PKC activity to molecular motors
The reorganization of dynein and NMII that we observed in photoactivation experiments occurred concomitantly with the growth of TCR-induced PKC activity, implying a close link between PKCs and the reciprocal remodeling of both motors. Indeed, pharmacological inhibition of PKC activity blocked both the depletion of NMII and the accumulation of dynein at the region of TCR stimulation (68). A similar phenotype was observed in T cells lacking PKCε, PKCη, and PKCθ. Interestingly, loss of PKCθ alone or PKCε together with PKCη failed to disrupt motor localization, indicating that the three nPKCs control this process redundantly.
nPKCs appear to regulate cortical NMII distribution at least in part through direct phosphorylation of the RLC. It has been known for some time that the RLC contains three phosphorylation sites near its N-terminus that can be targeted by PKCs (Ser1, Ser2, and Thr9) (87–89). Phosphorylation of these residues has been reported to reduce myosin motor activity (88, 89), and more recently to alter its sub-cellular localization (90). Consistent with this latter data, we found that mutation of Ser1 and Ser2 to Ala blocked suppression of NMII clustering in the region of TCR photoactivation (68). We also demonstrated that acute stimulation of PKC activity (by addition of PMA) could dissolve cortical NMII clusters within seconds, independent of TCR stimulation. This effect was blocked by a small molecule PKC inhibitor, indicating that it was mediated by the PKCs and not some other PMA-activated protein. Hence, nPKCs control the localization of NMII in T cells via direct RLC phosphorylation. RLC also contains well-characterized activating phosphorylation sites (Thr18 and Ser19), which are targeted by the Ser/Thr kinases ROCK and MLCK (72). We found that mutation of these residues completely disrupted cortical NMII clustering both in the presence and the absence of TCR signaling (68). Furthermore, acute inhibition of ROCK with the small molecule Y27632 rapidly dissolved existing NMII clusters. ROCK inhibition also impaired MTOC reorientation, consistent with an active role for NMII clusters in polarization responses. Importantly, cells treated with both ROCK and PKC inhibitors also lacked cortical NMII, implying that the function of nPKCs in this context is to counteract the effects of ROCK-mediated phosphorylation. Taken together, these results are consistent with a model whereby NMII localization is established by polarized nPKC accumulation superimposed on a background of global ROCK activity.
It is important to note that MTOC reorientation does not appear to require complete depletion of NMII from the IS. Indeed, a pool of NMII remains that localizes to the inner edge of the peripheral F-actin ring (91, 92). The precise role of this synaptic pool is not clear. Some studies have indicated that it promotes the centripetal movement of signaling microclusters containing the TCR (92, 93), and it has been suggested that NMII activity is crucial for early TCR signaling events (93). Other work, however, has cast doubt on the importance of NMII for microcluster dynamics and TCR signaling, suggesting instead that it stabilizes synaptic architecture and symmetry (91). This controversy will not be considered further in this review as it has been discussed at length elsewhere (94). It seems fair to speculate, however, that there are multiple cellular pools of NMII, some that contribute to MTOC reorientation from the sides and rear of the cell, and others that may influence F-actin organization at the IS. The presence of multiple, functionally distinct cohorts of NMII is not surprising given the ubiquitous use of actomyosin contractility in a variety of cellular structures.
Although loss-of-function experiments clearly demonstrate that nPKCs control dynein organization, the precise mechanisms for this control have remained elusive. Indeed, we have been unable to link PKC activation directly to dynein dynamics. Acute application of PMA had no effect on the distribution of cortical dynein (68), and we have been unable to document PKC-mediated phosphorylation of any dynein component or associated cofactors. Taken together, our observations suggest that nPKC activity is necessary but not sufficient for dynein remodeling. Precisely which additional molecules might contribute to this process remain to be seen. One possible candidate is the scaffolding protein ADAP, which plays a well-established role in T-cell activation by promoting inside-out integrin signaling (95). ADAP interacts directly with dynein and is required for dynein recruitment to the IS in Jurkat cells (66). Furthermore, siRNA knockdown of ADAP in this system inhibits MTOC reorientation. Whether ADAP plays a similar role in primary T cells is not clear, however, and requires further study. Another intriguing possibility is that dynein recruitment does not respond to PKC activity per se but rather to the gradient of PKC activity that is centered at the IS. Various mechanisms for translating signaling asymmetries into the formation of compartmentalized complexes and cellular structures have been proposed (74, 96), and it will be interesting to see which, if any, apply in this system.
Concluding remarks
The work described in this review has provided a conceptual framework for understanding MTOC polarization at a mechanistic level. That being said, we have really only scratched the surface of this complex and functionally important process. The relationship between synaptic F-actin and the MTOC, for instance, remains largely unexplored. The marked correlation between MTOC recruitment and F-actin ring formation suggests that the two processes are causally linked (19). Indeed, microtubules have been observed to dock within the F-actin ring at the periphery of the IS (97). Hence, it is conceivable that the growth of this ring during IS formation might exert force on microtubules to pull the MTOC close to the synaptic membrane. Future studies will no doubt address this hypothesis directly.
In almost all eukaryotic cells, recruitment of the centriole-bearing MTOC (the centrosome) to the plasma membrane is required for growth of the primary cilium, a signaling structure that has been called a ‘cellular antenna’ (98). Lymphocytes lack obvious primary cilia, and it has been proposed that MTOC polarization to the IS represents a ‘frustrated’ attempt by the T cell to generate such a structure (9). Although there is, as yet, little evidence to support this idea, it is intriguing that intraflagellar transport proteins, which mediate polarized transport into and out of the primary cilium, also play a role in trafficking internalized TCR to the IS (99). Hence, at least some of the molecular machinery involved in the structure and function of the primary cilium may also operate at the IS. Studies aimed at defining the roles of other ciliary components during T-cell activation are currently underway.
Now that we can induce MTOC polarization in a controlled manner, the speed and reproducibility of the response make it an excellent model system for investigating polarized signaling. The synaptic DAG gradient induced by TCR activation is remarkably sharp, and we as yet do not understand how it is formed and maintained. Destruction of DAG by DGKs and other lipid modifying enzymes almost certainly plays a role, but the precise mechanism is likely to be more complex. Indeed, analogous gradients in other systems are sustained by robust positive feedback (96, 100, 101). Consistent with this notion, we have observed that localized decaging of DAG drives the polarized generation of more DAG by the T cell (28). We have also found that the T cell can discriminate between competing zones of TCR stimulation on the cell surface, polarizing preferentially toward the one containing more agonist pMHC (27). These results are consistent with data showing that T cells polarize selectively toward APCs bearing higher levels of antigenic peptide (102). Hence, T cells can locally integrate the amount of TCR stimulation, and use that information to inform a binary cytoskeletal remodeling response. In migratory leukocytes and Dictyostelium, the presence of a single leading edge is enforced by active inhibition of protrusive F-actin growth in the sides and rear of the cell (74). It will be interesting to see if analogous suppressive mechanisms enhance the fidelity and persistence of MTOC polarization in T cells.
Finally, we must explore in greater detail the role of synaptic polarity in T-cell effector function. In the past, the only known perturbations capable of disrupting MTOC polarization also blocked TCR signaling, making it impossible to focus on the specific contribution of the former. Intensive study of the pathways controlling TCR-induced cytoskeletal dynamics has now begun to generate better tools for modulating synaptic polarity in a selective manner in more complex experimental settings. In a recent study, for example, Bertrand and colleagues (64) used targeted inhibition of PKCf to demonstrate that CTLs can release cytolytic factors without MTOC reorientation to the IS. These results imply that the synaptic targeting of lytic granules is not required for killing, but rather functions to constrain the scope of cytolytic secretion, thereby limiting damage to innocent bystander cells. A better understanding of how polarity guides effector responses will enhance and refine our understanding of T-cell function. This should, in turn, aid in the development of strategies to modulate key aspects of lymphocyte function selectively in immunotherapeutic contexts.
Acknowledgments
Support provided by the U.S. National Institutes of Health (R01-AI087644) is acknowledged.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 2.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 3.Lee KH, et al. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302:1218–1222. doi: 10.1126/science.1086507. [DOI] [PubMed] [Google Scholar]
- 4.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 5.Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol. 2010;2:a002311. doi: 10.1101/cshperspect.a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lettau M, et al. The adaptor protein Nck interacts with Fas ligand: guiding the death factor to the cytotoxic immunological synapse. Proc Natl Acad Sci USA. 2006;103:5911–5916. doi: 10.1073/pnas.0508562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boisvert J, Edmondson S, Krummel MF. Immunological synapse formation licenses CD40-CD40L accumulations at T-APC contact sites. J Immunol. 2004;173:3647–3652. doi: 10.4049/jimmunol.173.6.3647. [DOI] [PubMed] [Google Scholar]
- 8.Huse M, Quann EJ, Davis MM. Shouts, whispers, and the kiss of death: directional secretion in T cells. Nat Immunol. 2008;9:1105–1111. doi: 10.1038/ni.f.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 10.Chang JT, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 11.Oliaro J, et al. Asymmetric cell division of T cells upon antigen presentation uses multiple conserved mechanisms. J Immunol. 2010;185:367–375. doi: 10.4049/jimmunol.0903627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kupfer A, Dennert G. Reorientation of the microtubule-organizing center and the Golgi apparatus in cloned cytotoxic lymphocytes triggered by binding to lysable target cells. J Immunol. 1984;133:2762–2766. [PubMed] [Google Scholar]
- 13.Kupfer A, Dennert G, Singer SJ. Polarization of the Golgi apparatus and the microtubule-organizing center within cloned natural killer cells bound to their targets. Proc Natl Acad Sci USA. 1983;80:7224–7228. doi: 10.1073/pnas.80.23.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiger B, Rosen D, Berke G. Spatial relationships of microtubule-organizing centers and the contact area of cytotoxic T lymphocytes and target cells. J Cell Biol. 1982;95:137–143. doi: 10.1083/jcb.95.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huse M. Microtubule-organizing center polarity and the immunological synapse: protein kinase C and beyond. Front Immunol. 2012;3 doi: 10.3389/fimmu.2012.00235. Article 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beal AM, et al. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity. 2009;31:632–642. doi: 10.1016/j.immuni.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- 18.Sims TN, et al. Opposing effects of PKCtheta and WASp on symmetry breaking and relocation of the immunological synapse. Cell. 2007;129:773–785. doi: 10.1016/j.cell.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 19.Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- 20.Brown AC, et al. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLoS Biol. 2011;9:e1001152. doi: 10.1371/journal.pbio.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 2011;9:e1001151. doi: 10.1371/journal.pbio.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kupfer A, Mosmann TR, Kupfer H. Polarized expression of cytokines in cell conjugates of helper T cells and splenic B cells. Proc Natl Acad Sci USA. 1991;88:775–779. doi: 10.1073/pnas.88.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sedwick CE, Morgan MM, Jusino L, Cannon JL, Miller J, Burkhardt JK. TCR, LFA-1, and CD28 play unique and complementary roles in signaling T cell cytoskeletal reorganization. J Immunol. 1999;162:1367–1375. [PubMed] [Google Scholar]
- 24.Kuhne MR, et al. Linker for activation of T cells, zeta-associated protein-70, and Src homology 2 domain-containing leukocyte protein-76 are required for TCR-induced microtubule-organizing center polarization. J Immunol. 2003;171:860–866. doi: 10.4049/jimmunol.171.2.860. [DOI] [PubMed] [Google Scholar]
- 25.Lowin-Kropf B, Shapiro VS, Weiss A. Cytoskeletal polarization of T cells is regulated by an immunoreceptor tyrosine-based activation motif-dependent mechanism. J Cell Biol. 1998;140:861–871. doi: 10.1083/jcb.140.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeMond AL, Starr T, Dustin ML, Groves JT. Control of antigen presentation with a photoreleasable agonist peptide. J Am Chem Soc. 2006;128:15354–15355. doi: 10.1021/ja065304l. [DOI] [PubMed] [Google Scholar]
- 27.Huse M, et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity. 2007;27:76–88. doi: 10.1016/j.immuni.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 28.Quann EJ, Merino E, Furuta T, Huse M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat Immunol. 2009;10:627–635. doi: 10.1038/ni.1734. [DOI] [PubMed] [Google Scholar]
- 29.Huse M. The T-cell-receptor signaling network. J Cell Sci. 2009;122:1269–1273. doi: 10.1242/jcs.042762. [DOI] [PubMed] [Google Scholar]
- 30.Lin J, Weiss A. T cell receptor signalling. J Cell Sci. 2001;114:243–244. doi: 10.1242/jcs.114.2.243. [DOI] [PubMed] [Google Scholar]
- 31.Spitaler M, Emslie E, Wood CD, Cantrell D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity. 2006;24:535–546. doi: 10.1016/j.immuni.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Kupfer A, Dennert G, Singer SJ. The reorientation of the Golgi apparatus and the microtubule-organizing center in the cytotoxic effector cell is a prerequisite in the lysis of bound target cells. J Mol Cell Immunol. 1985;2:37–49. [PubMed] [Google Scholar]
- 33.Suzuki AZ, et al. Coumarin-4-ylmethoxycarbonyls as phototriggers for alcohols and phenols. Organic Lett. 2003;5:4867–4870. doi: 10.1021/ol0359362. [DOI] [PubMed] [Google Scholar]
- 34.Swaney KF, Huang CH, Devreotes PN. Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu Rev Biophys. 2010;39:265–289. doi: 10.1146/annurev.biophys.093008.131228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong XP, Guo R, Zhou H, Liu C, Wan CK. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol Rev. 2008;224:249–264. doi: 10.1111/j.1600-065X.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olenchock BA, et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 37.Zhong XP, et al. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003;4:882–890. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 38.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quann EJ, Liu X, Altan-Bonnet G, Huse M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat Immunol. 2011;12:647–654. doi: 10.1038/ni.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manicassamy S, Gupta S, Sun Z. Selective function of PKC-theta in T cells. Cell Mol Immunol. 2006;3:263–270. [PubMed] [Google Scholar]
- 41.Monks CR, Kupfer H, Tamir I, Barlow A, Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- 42.Gruber T, Thuille N, Hermann-Kleiter N, Leitges M, Baier G. Protein kinase Cepsilon is dispensable for TCR/CD3-signaling. Mol Immunol. 2005;42:305–310. doi: 10.1016/j.molimm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Hodge CW, et al. Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- 44.Lee AM, Messing RO. Protein kinase C epsilon modulates nicotine consumption and dopamine reward signals in the nucleus accumbens. Proc Natl Acad Sci USA. 2011;108:16080–16085. doi: 10.1073/pnas.1106277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olive MF, Mehmert KK, Nannini MA, Camarini R, Messing RO, Hodge CW. Reduced ethanol withdrawal severity and altered withdrawal-induced c-fos expression in various brain regions of mice lacking protein kinase C-epsilon. Neuroscience. 2001;103:171–179. doi: 10.1016/s0306-4522(00)00566-2. [DOI] [PubMed] [Google Scholar]
- 46.Chida K, et al. Disruption of protein kinase Ceta results in impairment of wound healing and enhancement of tumor formation in mouse skin carcinogenesis. Cancer Res. 2003;63:2404–2408. [PubMed] [Google Scholar]
- 47.Fu G, et al. Protein kinase C eta is required for T cell activation and homeostatic proliferation. Sci Signal. 2011;4:ra84. doi: 10.1126/scisignal.2002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melowic HR, et al. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Ctheta. J Biol Chem. 2007;282:21467–21476. doi: 10.1074/jbc.M700119200. [DOI] [PubMed] [Google Scholar]
- 49.Stahelin RV, et al. Diacylglycerol-induced membrane targeting and activation of protein kinase Cepsilon: mechanistic differences between protein kinases Cdelta and Cepsilon. J Biol Chem. 2005;280:19784–19793. doi: 10.1074/jbc.M411285200. [DOI] [PubMed] [Google Scholar]
- 50.Stahelin RV, et al. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cdelta. J Biol Chem. 2004;279:29501–29512. doi: 10.1074/jbc.M403191200. [DOI] [PubMed] [Google Scholar]
- 51.Colon-Gonzalez F, Kazanietz MG. C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Kashiwagi K, Shirai Y, Kuriyama M, Sakai N, Saito N. Importance of C1B domain for lipid messenger-induced targeting of protein kinase C. J Biol Chem. 2002;277:18037–18045. doi: 10.1074/jbc.M111761200. [DOI] [PubMed] [Google Scholar]
- 53.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. 2005;121:271–280. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 54.Stahelin RV, et al. Protein kinase Ctheta C2 domain is a phosphotyrosine binding module that plays a key role in its activation. J Biol Chem. 2012;287:30518–30528. doi: 10.1074/jbc.M112.391557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merino E, et al. Protein kinase C-theta clustering at immunological synapses amplifies effector responses in NK cells. J Immunol. 2012;189:4859–4869. doi: 10.4049/jimmunol.1200825. [DOI] [PubMed] [Google Scholar]
- 56.Kong KF, Yokosuka T, Canonigo-Balancio AJ, Isakov N, Saito T, Altman A. A motif in the V3 domain of the kinase PKC-theta determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat Immunol. 2011;12:1105–1112. doi: 10.1038/ni.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Etienne-Manneville S, Hall A. Cell polarity: Par6, aPKC and cytoskeletal crosstalk. Curr Opin Cell Biol. 2003;15:67–72. doi: 10.1016/s0955-0674(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 58.Li R, Gundersen GG. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 2008;9:860–873. doi: 10.1038/nrm2522. [DOI] [PubMed] [Google Scholar]
- 59.Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- 60.Ludford-Menting MJ, et al. A network of PDZ-containing proteins regulates T cell polarity and morphology during migration and immunological synapse formation. Immunity. 2005;22:737–748. doi: 10.1016/j.immuni.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 61.Bertrand F, et al. Activation of the ancestral polarity regulator protein kinase C zeta at the immunological synapse drives polarization of Th cell secretory machinery toward APCs. J Immunol. 2010;185:2887–2894. doi: 10.4049/jimmunol.1000739. [DOI] [PubMed] [Google Scholar]
- 62.Lin J, Hou KK, Piwnica-Worms H, Shaw AS. The polarity protein Par1b/EMK/MARK2 regulates T cell receptor-induced microtubule-organizing center polarization. J Immunol. 2009;183:1215–1221. doi: 10.4049/jimmunol.0803887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu CG, Harris TJ. Interactions between the PDZ domains of Bazooka (Par-3) and phosphatidic acid: in vitro characterization and role in epithelial development. Mol Biol Cell. 2012;23:3743–3753. doi: 10.1091/mbc.E12-03-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertrand F, Muller S, Roh KH, Laurent C, Dupre L, Valitutti S. An initial and rapid step of lytic granule secretion precedes microtubule organizing center polarization at the cytotoxic T lymphocyte/target cell synapse. Proc Natl Acad Sci USA. 2013;110:6073–6078. doi: 10.1073/pnas.1218640110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kardon JR, Vale RD. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol. 2009;10:854–865. doi: 10.1038/nrm2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Combs J, et al. Recruitment of dynein to the Jurkat immunological synapse. Proc Natl Acad Sci USA. 2006;103:14883–14888. doi: 10.1073/pnas.0600914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin-Cofreces NB, et al. MTOC translocation modulates IS formation and controls sustained T cell signaling. J Cell Biol. 2008;182:951–962. doi: 10.1083/jcb.200801014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu X, Kapoor TM, Chen JK, Huse M. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin II. Proc Natl Acad Sci USA. 2013;110:11976–11981. doi: 10.1073/pnas.1306180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hashimoto-Tane A, et al. Dynein-driven transport of T cell receptor microclusters regulates immune synapse formation and T cell activation. Immunity. 2011;34:919–931. doi: 10.1016/j.immuni.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 70.Gomes ER, Jani S, Gundersen GG. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell. 2005;121:451–463. doi: 10.1016/j.cell.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 71.Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10:970–979. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 72.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobelli J, Chmura SA, Buxton DB, Davis MM, Krummel MF. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat Immunol. 2004;5:531–538. doi: 10.1038/ni1065. [DOI] [PubMed] [Google Scholar]
- 74.Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol. 2004;5:626–634. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- 75.Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allenspach EJ, et al. ERM-dependent movement of CD43 defines a novel protein complex distal to the immunological synapse. Immunity. 2001;15:739–750. doi: 10.1016/s1074-7613(01)00224-2. [DOI] [PubMed] [Google Scholar]
- 77.Delon J, Kaibuchi K, Germain RN. Exclusion of CD43 from the immunological synapse is mediated by phosphorylation-regulated relocation of the cytoskeletal adaptor moesin. Immunity. 2001;15:691–701. doi: 10.1016/s1074-7613(01)00231-x. [DOI] [PubMed] [Google Scholar]
- 78.Shaffer MH, et al. Ezrin and moesin function together to promote T cell activation. J Immunol. 2009;182:1021–1032. doi: 10.4049/jimmunol.182.2.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ilani T, Khanna C, Zhou M, Veenstra TD, Bretscher A. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol. 2007;179:733–746. doi: 10.1083/jcb.200707199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Filbert EL, Le Borgne M, Lin J, Heuser JE, Shaw AS. Stathmin regulates microtubule dynamics and microtubule organizing center polarization in activated T cells. J Immunol. 2012;188:5421–5427. doi: 10.4049/jimmunol.1200242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zyss D, Ebrahimi H, Gergely F. Casein kinase I delta controls centrosome positioning during T cell activation. J Cell Biol. 2011;195:781–797. doi: 10.1083/jcb.201106025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Andres-Delgado L, et al. INF2 promotes the formation of detyrosinated microtubules necessary for centrosome reorientation in T cells. J Cell Biol. 2012;198:1025–1037. doi: 10.1083/jcb.201202137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gomez TS, Kumar K, Medeiros RB, Shimizu Y, Leibson PJ, Billadeau DD. Formins regulate the actin-related protein 2/3 complex-independent polarization of the centrosome to the immunological synapse. Immunity. 2007;26:177–190. doi: 10.1016/j.immuni.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- 85.Eng CH, Huckaba TM, Gundersen GG. The formin mDia regulates GSK3beta through novel PKCs to promote microtubule stabilization but not MTOC reorientation in migrating fibroblasts. Mol Biol Cell. 2006;17:5004–5016. doi: 10.1091/mbc.E05-10-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wen Y, et al. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 2004;6:820–830. doi: 10.1038/ncb1160. [DOI] [PubMed] [Google Scholar]
- 87.Bengur AR, Robinson EA, Appella E, Sellers JR. Sequence of the sites phosphorylated by protein kinase C in the smooth muscle myosin light chain. J Biol Chem. 1987;262:7613–7617. [PubMed] [Google Scholar]
- 88.Ikebe M, Hartshorne DJ, Elzinga M. Phosphorylation of the 20,000-dalton light chain of smooth muscle myosin by the calcium-activated, phospholipid-dependent protein kinase. Phosphorylation sites and effects of phosphorylation. J Biol Chem. 1987;262:9569–9573. [PubMed] [Google Scholar]
- 89.Ikebe M, Reardon S. Phosphorylation of bovine platelet myosin by protein kinase C. Biochemistry. 1990;29:2713–2720. doi: 10.1021/bi00463a014. [DOI] [PubMed] [Google Scholar]
- 90.Komatsu S, Ikebe M. The phosphorylation of myosin II at the Ser1 and Ser2 is critical for normal platelet-derived growth factor induced reorganization of myosin filaments. Mol Biol Cell. 2007;18:5081–5090. doi: 10.1091/mbc.E06-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Babich A, Li S, O'Connor RS, Milone MC, Freedman BD, Burkhardt JK. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J Cell Biol. 2012;197:775–787. doi: 10.1083/jcb.201201018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yi J, Wu XS, Crites T, Hammer JA., 3rd Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell. 2012;23:834–852. doi: 10.1091/mbc.E11-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hammer JA, 3rd, Burkhardt JK. Controversy and consensus regarding myosin II function at the immunological synapse. Curr Opin Immunol. 2013;25:300–306. doi: 10.1016/j.coi.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang H, Rudd CE. SKAP-55, SKAP-55-related and ADAP adaptors modulate integrin-mediated immune-cell adhesion. Trends Cell Biol. 2008;18:486–493. doi: 10.1016/j.tcb.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chau AH, Walter JM, Gerardin J, Tang C, Lim WA. Designing synthetic regulatory networks capable of self-organizing cell polarization. Cell. 2012;151:320–332. doi: 10.1016/j.cell.2012.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kuhn JR, Poenie M. Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity. 2002;16:111–121. doi: 10.1016/s1074-7613(02)00262-5. [DOI] [PubMed] [Google Scholar]
- 98.Singla V, Reiter JF. The primary cilium as the cell's antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 99.Finetti F, et al. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol. 2009;11:1332–1339. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wedlich-Soldner R, Altschuler S, Wu L, Li R. Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science. 2003;299:1231–1235. doi: 10.1126/science.1080944. [DOI] [PubMed] [Google Scholar]
- 101.Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Depoil D, et al. Immunological synapses are versatile structures enabling selective T cell polarization. Immunity. 2005;22:185–194. doi: 10.1016/j.immuni.2004.12.010. [DOI] [PubMed] [Google Scholar]