

Abstract
CaV1.3 L-type calcium channels (LTCCs) have been a potential target for Parkinson’s disease since calcium ion influx through the channel was implicated in the generation of mitochondrial oxidative stress, causing cell death in the dopaminergic neurons. Selective inhibition of CaV1.3 over other LTCC isoforms, especially CaV1.2, is critical to minimize potential side effects. We recently identified pyrimidinetriones (PYTs) as a CaV1.3-selective scaffold; here we report the structure-activity relationship of PYTs with both CaV1.3 and CaV1.2 LTCCs. By variation of the substituents on the cyclopentyl and arylalkyl groups of PYT, SAR studies allowed characterization of the CaV1.3 and CaV1.2 LTCCs binding sites. The SAR also identified four important moieties that either retain selectivity or enhance binding affinity. Our study represents a significant enhancement of the SAR of PYTs at CaV1.3 and CaV1.2 LTCCs and highlights several advances in the lead optimization and diversification of this family of compounds for drug development.
Graphical abstract
INTRODUCTION
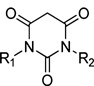
L-Type calcium channels1 (LTCCs) are cell membrane proteins activated upon membrane depolarization, selectively modulating calcium ion influx into the cells to initiate diverse intracellular events such as synaptic transmission, secretion, and gene expression. Although these channels are multimeric proteins of five subunits (α1, α2, β, γ, δ),2,3 the α1 subunit is the major pore-forming subunit, having four different subtypes (CaV1.1–1.4). CaV1.2 LTCCs are the major isoform (∼90%),4 expressed in cardiac myocites, smooth muscle, pancreas, and neurons, while CaV1.1 and CaV1.4 LTCCs are minor isoforms, restricted to skeletal muscle and retina, respectively.5 CaV1.3 LTCCs6 are expressed similarly as CaV1.2 LTCCs but are more neuron specific (cerebral cortex, hippocampus, basal ganglia, habenula, and thalamus); they are thought to serve predominantly as modulators in the neuronal system.7
Our recent studies8 showed that CaV13 LTCCs are used for rhythmic pacemaking in adult dopaminergic neurons of the substantia nigra pars compacta (SNc).9,10 The engagement of CaV1.3 LTCCs during autonomous pacemaking increases with age in animal models of Parkinson’s disease (PD),8 but CaV1.3 LTCCs seem to be unnecessary for normal functioning of SNc dopaminergic neurons. This reliance of CaV1.3 LTCCs elevates mitochondrial oxidant stress in SNc dopaminergic neurons, incurring continuous loss of SNc dopaminergic neurons, as demonstrated in a PD model.11 Antagonization of CaV1.3 LTCCs could provide a means to diminish cell loss in PD by protecting SNc dopaminergic neurons against toxins, thereby making CaV1.3 LTCCs an important therapeutic target for PD.12,13 However, drug candidates must selectively antagonize CaV1.3 over other ion channels, especially over CaV1.2 LTCCs, in order to avoid potential cardiovascular side effects.14
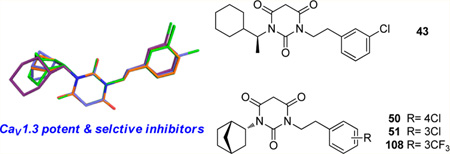
We have been interested in developing CaV1.3 selective inhibitors for treating PD; a calcium influx assay was developed using fluorometric imaging plate reader15 (FLIPR) with a Fluo-8 calcium dye16 and stably expressed CaV1.3 and CaV1.2 LTCCs in HEK293 cells. We tested diverse known CaV1.2 LTCCs blockers,17,18 such as isradipine (1), verapamil (2), and diltiazem (3), and synthesized over 100 modified DHPs19 and various hydropyrimidines20 (4) to obtain CaV1.3 selective antagonists; however, the best of these compounds have no greater than 2- to 3-fold selectivity for CaV1.3 over CaV1.2 LTCCs. More recently, high-throughput screening of molecular libraries identified pyrimidine-2,4,6-trione (PYT) 5 as a potential scaffold for the selective inhibition of CaV1.3 LTCCs.21 After SAR-based initial modification of the scaffold, we arrived at N-(3-chlorophenethyl)-N′-cyclopentylpyrimidine-2,4,6-trione (6) as a selective CaV1.3 LTCC antagonist and N-(4-chlorophenethyl)-N′-(±)-endo-norbornylpyrimidine-2,4,6-(1H,3H,5H)-trione (7) as a potent CaV1.3 LTCC antagonist among the series of compounds (Figure 1). PYT is a completely new scaffold as applied to the ion channels, and selective inhibition of CaV1.3 LTCCs with PYTs is the first approach for the treatment of PD as a potential cure. Herein, we report on our lead optimization efforts to obtain diverse potent and selective compounds.
Figure 1.

Example of CaV1.3 blockers. 1, 2, and 3 are known nonselective LTCC blockers used for the initial CaV1.3 study. 4 is a potent and slightly selective molecule. 5 is our initial hit from HTS. 6 is the most selective compound, and 7 is most potent compound in the previous study.
DESIGN
Our previous study showed that, starting from symmetric N,N′-disubstituted pyrimidine 2,4,6-trione (PYT) 5, changing a phenethyl group to a cyclopentyl ring led to the discovery of our most selective molecule, 6, and using an endo-norbornyl group led to most potent molecule (7) in this scaffold. We synthesized ∼100 PYT molecules utilizing electronically and/or sterically diverse arylalkyls, alkyls, and three- to seven-membered cycloalkyl rings. All selective compounds in this class share N-cycloalkyl and N-arylethyl moieties as the side chains. An overlapped visualization of compounds 6 and 7 was used as the starting point for further derivatization to obtain more potent and selective compounds. As shown in Figure 2, molecules 6 and 7 are only different at the cycloalkyl region and the substitution of the aromatic ring. The two extra carbons of the norbornyl ring in 7 or para-chloro substitution on the aromatic ring make 7 3-fold more potent than 6 toward CaV1.3 LTCC. It seems that a hydrophobic site is key for selective inhibition of CaV1.3 over CaV1.2 LTCCs. On the basis of this analysis, various 2- or 3-alkyl substituted cycloalkyl derivatives, as well as diverse bicycloalkyl derivatives, were prepared to probe the steric demands arising from this hydrophobic binding site. m-Chloro and p-chlorophenethyl groups were initially chosen as the other side chain to verify the SAR. Since racemic norbornyl derivatives showed good potency for CaV1.3 LTCCs, we synthesized each enantiomer of the endo-norbornyl derivatives to explore the effect of chirality. Diverse ethers and esters were incorporated at the 1, 2, and 3 positions of the cyclopentyl ring to investigate the electronic demands of the cyclopentyl ring region. To explore the steric demands on the ethylene linker of the N-phenethyl side chain while maintaining cyclopentyl or endo-norbornyl as the other side chain, diverse methyl, gem-dimethyl, phenyl, and carbonate moieties were employed on the α- or β-position of the ethylene linker. Similarly, diverse aromatic rings were employed for lead optimization. The moieties used for the construction of the PYT library are shown in Figure 3.
Figure 2.

Overlay of two standard compounds, 6 (orange) and 7 (blue). The figure was generated using Sybyl-X and PyMOL.
Figure 3.

Diverse N-alkyl moieties used for side chains on PYTs.
RESULTS AND DISCUSSION
Chemistry
A majority of PYT analogues were synthesized from commercially available amines and isocyanates using the previously established one-pot synthesis of N,N-disubstituted PYTs, which involved the Wöhler22 urea synthesis and a Biltz and Wittek23 condensation with activated malonic acid (Scheme 1). Urea formation was accomplished by coupling isocyanates with amines in dichloromethane. If the starting amine was an acid salt, 1 equiv of triethylamine was added to the reaction mixture to initiate urea formation. The addition of malonyl chloride to the urea, performed under dilute conditions (0.02 M in dichloromethane) to avoid intermolecular acylation, provided the condensation products in moderate to high yields. The use of these synthetic approaches permitted the construction of the major library members in sufficient quantities and purity for assay against CaV1.3 and CaV1.2 LTCCs.
Scheme 1. General Synthetic Process for Assembling the PYT Ringa.
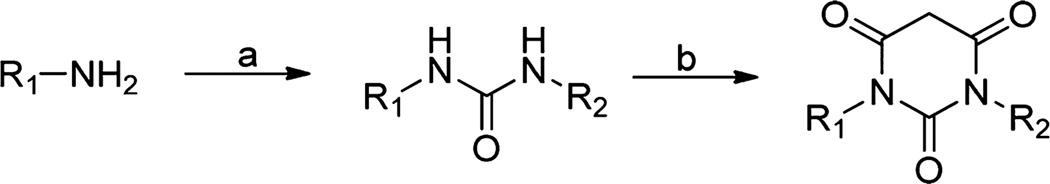
aReagents and conditions: (a) isocyanate, CH2Cl2 (0.1 M), room temp, 1–5 h; (b) malonyl chloride, CH2Cl2 (0.02 M), room temp, 1 h.
Commercially unavailable isocyanates and amines were prepared from related ketones, acetonitriles, carboxylic acids, and other amines. Enantiomerically pure (1S,2R,4R)-(−)-endo-2-norbornyl isocyanate (10) and (1R,2S,4S)-(+)-endo-2-nor-bornyl isocyanate (13) were prepared via a combination of the Poll24 and Fukaya25 procedures (Scheme 2); asymmetric Diels–Alder reactions of cyclopentadiene with an acrylate using (R)-pantolactone or (S)-2-hydroxy-N-methylsuccinimide chiral auxiliary provided chiral carboxylate intermediates. The resulting mixture underwent saponification and catalytic hydrogenation to give enantiomerically pure endo-norbornyl carboxylic acids 9 and 12. Carbonyl azide formation, followed by Curtius rearrangement, provided enantiomerically pure (1S,2R,4R)- or (1R,2S,4S)-endo-norbornyl isocyanates (10, 13), which were stored in the refrigerator and used for several months without further purification.
Scheme 2. Asymmetric Syntheses of endo-Nornornyl Isocyanates 10 and 13a.

aReagents and conditions: See Poll’s22 and Fukaya’s23 procedure. (a) cyclopentadiene, TiCl4; (b) LiOH; (c) 10% Pd/C, H2; (d) (i) ethyl chloroformate, triethylamine, 0 °C; (ii) NaN3, acetone-water, 2 h; (iii) hexane, reflux 2 h.
Commercially unavailable methyl-substituted phenethylamines (16a–d) required for the syntheses were prepared as shown in Scheme 3.26 Methylation of commercially available phenylacetonitriles (14a,b) with iodomethane, using sodium hydride as base, yielded a mixture of mono- and dimethylated acetonitriles (15a–d). Reduction of the nitriles with LiAlH4 provided the target phenethylamines (16a–d) in high yields.
Scheme 3. Synthesis of Methyl-Substituted Phenethylamines 16a–da.

aReagents and conditions: (a) NaH, MeI, DMF, room temp, overnight; (b) LiAlH4, THF, 0 °C, 2 h.
A majority of alkoxyl or carboxylate substituted cyclopentyl-amines used in these syntheses were easily prepared from the corresponding (N-Boc-amino)cyclopentanecarboxylic acids or (N-Boc-amino)cyclopentyl alcohols in a two-step process: esterification of the acids/alcohols with appropriate alcohols/ acids, and Boc deprotection with 50% TFA solution in dichloromethane. Alkyl-substituted cycloalkylamines were synthesized from the corresponding cyclopentanone by reductive amination with a benzylamine (Scheme 4). Especially, cis-2-methyl or 2,2-gem-dimethylcyclopentyl27 PYT derivatives were produced by asymmetric reductive amination of a cyclopentanone with a nonracemic α-methylbenzylamine,28 which occurs specifically from the face opposite29 that occupied by the adjacent alkyl substituent, to furnish the racemic cis-diastereomer in a low yield. This product was then coupled to the isocyanate as before, and the ethylbenzene was removed by treatment with 75% trifluoroacetic acid in dichloromethane. The urea was cyclized with malonyl chloride to furnish the final PYT ring. When the same procedure was applied for the synthesis of 3-alkyl substituted cyclopentyl derivatives, inseparable racemic mixtures were obtained.
Scheme 4. Synthesis of Alkyl Substituted Cycloalkylaminea.

aReagents and conditions: (a) (i) (S)-α-methylbenzylamine, cat. TsOH, benzene, reflux, overnight; (ii) NaBH3CN, EtOH; *(R)-1-phenylethanamine was used under the same conditions; (b) 2-(3-Cl-phenyl)ethyl isocyanate, CH2Cl2, room temp; (c) 75% TFA in CH2Cl2; (d) malonyl chloride, CH2Cl2.
Biological Activity
The CaV1.3 and CaV1.2 LTCCs assays were carried out with the synthesized PYT molecules using the previously described high-throughput screen with a FLIPR system and Fluo-8 calcium assay kit.19 Each IC50 value, the concentration of test compounds required to inhibit 50% of calcium-dependent fluorescence response in the assay, was determined in triplicate (Tables 1–4). IC50 values and associated standard deviations were calculated by curve fitting of the percent inhibition data from the FLIPR assay to a sigmoidal model for a one-site compound target interaction using XLfit. The selectivity of antagonism was defined as the inverse of the ratio of the IC50 value with CaV1.2 LTCCs to that with CaV1.3 LTCCs. The Z′ score of the HTS was >0.6 on the basis of the values from at least a one-half of a plate of positive and negative controls, and standard deviations associated with the calcium channel FLIPR assay were usually <20%.
Table 1.
IC50 Values and Selectivities of Various Cycloalkyl Analogues
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | Compound # |
Cav1.3 IC50(µM) |
3-Cl Cav1.2 IC50(µM) |
Selectivitya | Compound # |
Cav1.3 IC50(µM) |
4-Cl Cav1.2 IC50(µM) |
Selectivitya |
| 6 | 6.3 | >100 | >100b | 7 | 5.6 ±1.1 | 50 ±13 | 8.9 | |
 |
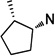
19 | 5.5 ±0.6 | 13 ±2.6 | 2.3 | 20 | 3.1 ±0.2 | 5.1 ±0.5 | 1.7 |
 |
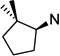
24 | 7.3 ±1.3 | 9.3 ±2.1 | 1.3 | 25 | 2.8 ±0.7 | 10 ±1.6 | 3.7 |
 |
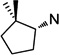
21 | 2.1 ±0.2 | 3.5 ±1.1 | 1.6 | 22 | 1.7 ±0.1 | 2.3 ±0.4 | 1.4 |
 |
26 | 2.5 ±0.4 | 4.7 ±0.5 | 1.9 | 27 | 4.7±0.6 | 4.8 ±0.8 | 1.0 |
| 30 | 3.7 ±0.1 | 38 ±3.4 | 10 | 31 | 9.8 ±0.8 | 20.4 ±1.1 | 2.1 | |
| 32 | 5.6 ±0.5 | 43 ±3.3 | 7.7 | 33 | 3.7 ±0.7 | 26 ±8.9 | 6.8 | |
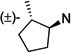
| 34 | 7.3 ±1.7 | 15 ±1.5 | 2.0 | 35 | 6.7 ±0.7 | 29 ±10 | 4.4 | |
 |
37 | 11 ±3.9 | 21 ±0.8 | 1.8 | 38 | 7.0 ±0.8 | 23 ±4.4 | 3.3 |
| 39 | 35 ±6.8 | 46 ±0.4 | 1.3 | 40 | 11 ±5.4 | 30 ±4.5 | 2.7 | |
| 41 | 3.7 ±0.8 | 3.7 ±0.1 | 1.0 | 42 | 6.1 ±1.7 | 8.3 ±0.5 | 1.4 | |
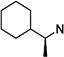 |
43 | 4.3 ±0.5 | >100 | >29 | 44 | 8.3 ±0.7 | >100 | >12 |
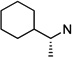 |
45 | 9.7 ±1.0 | >100 | >10 | 46 | 8.1 ±1.0 | >100 | >12 |
| 47 | >100 | 41 ±11 | 48 | >100 | >100 | |||
| 49 | 3.5 ±0.2 | >100 | >29 | 50 | 2.1 ±0.4 | 100 ±10 | 48 | |
| 51 | 2.0 ±0.1 | >100 | >50 | 52 | 1.9 ±0.3 | 54 ±5.8 | 11 | |
 |
53 | >100 | >100 | 54 | >100 | >100 | ||
| 55 | 40 ±2.9 | 47 ±1.9 | 1.2 | 56 | >100 | >100 | ||
 |
57 | >100 | >100 | 58 | 18 ±0.1 | >100 | >6 | |
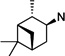 |
59 | >100 | >100 | 60 | 22 ±4.2 | 28 ±2.8 | 1.3 | |
Ratio of the IC50 values (CaV1.2/CaV1.3).
Selectivity was confirmed from the previous study.
Table 4.
IC50 Values and Selectivities of Various Norbornyl Analogues
 | |||||
|---|---|---|---|---|---|
| Compound # |
R1 | R2 | Cav1.3 IC50(µM) |
Cav1.2 IC50(µM) |
Selectivity |
| 100 | 3.4 ±0.2 | 8.0 ±1.0 | 2.4 | ||
| 101 | 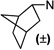 |
3.6 ±0.3 | 4.1 ±0.6 | 1.1 | |
| 102 | 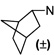 |
2.4 ±0.3 | 5.1 ±1.1 | 2.1 | |
| 103 | 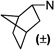 |
2.3 ±0.4 | 2.8 ±0.3 | 1.2 | |
| 104 | 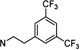 |
1.8 ±0.7 | 3.8 ±0.4 | 2.2 | |
| 105 | 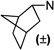 |
 |
2.7 ±0.2 | 4.6 ±1.7 | 1.7 |
| 106 |  |
2.4 ±0.2 | 100 ±6.3 | 40.8 | |
| 107 | 3.0 ±0.5 | >100 | >33.0 | ||
| 108 | 2.1 ±0.1 | 76.6 ±.8.0 | 36.0 | ||
| 109 | 2.7 ±0.2 | 3.1 ±0.2 | 1.2 | ||
To better understand the steric effect of the original N-cyclopentyl, N-3-chlorophenethyl PYT molecule (6) on potency and selectivity, our initial analogue efforts were focused on variations of steric bulkiness at the cyclopentyl moiety by alkyl substitutions on that ring. We used 3-chlorophenethyl and 4-chlorophenethyl moieties as the other side chain to confirm the substitution effect on the aromatic ring. As shown in Table 1, analogues with 1-cis-methyl (19, 20, 24, 25), 1,1-dimethyl (21, 22, 26, 27), 2-methyl (30, 31), and 2,2-dimethyl (32, 33) substitutions on the cyclopentyl ring had generally 1- to 4-fold increased binding affinity for both CaV1.3 and CaV1.2 LTCCs; 1,1-dimethyl-cyclopentyl compound 22 displayed an IC50 of 1.7 µM for CaV1.3 LTCCs. It seems that the binding site preferentially recognizes the stereochemistry of 1,2-cis substituted compounds. The favored 1,2-substitution pattern is also shown when utilizing cyclohexyl as the cycloalkyl chain (41). This steric preference was slightly increased with the combination of 4-chlorophenethyl as the other side chain (compounds 20 vs 19, 25 vs 24, and 22 vs 21). Alkyl substituted cyclopentyl derivatives had ∼3-fold lower IC50 for CaV1.3 LTCCs, but they also had increased CaV1.2 binding affinity; their CaV1.3 selectivities, therefore, were moderate to low. However, it is noteworthy that the 3-methyl- or 3,3-dimethylcyclopentyl derivatives still retained >7-fold selectivity for CaV1.3 LTCCs. Those compounds having (±)-trans-2-methylcyclopentyl (37, 38), 3-ethylcyclopentyl (31, 35), or 4-methylcyclohexyl (39–42) moieties were similar in potency to compound 6 but far less selective. In the bicyclic ring series, a significant potency drop was noted by increasing the bulkiness of the bicyclic rings. It seems the active site that interacts with the cyclopentyl moiety is not big enough to fit bornyl (53, 54), cis-myrtanyl (55, 56), or isopinocamphyl (57–60) groups; these bulky molecules have almost no binding to either CaV1.3 or CaV1.2 LTCCs. Racemic endo-norbornyl compound 7 was the most potent molecule in the previous study.19 To confirm the stereoactivity relationship, we asymmetrically synthesized each pair of enantiomers (49 and 50, 51 and 52, and 107 and 108) to isolate the more potent and selective enantiomer, as each enantiomer of known chiral LTCC blockers displays different efficacies.30 Although chiral compounds 50 and 51 displayed excellent selectivity (>48-fold) with good potency (∼2.0 µM) for CaV1.3 LTCCs, there was only a small stereorelated preference for CaV1.3 LTCCs with the (1S,2R,4R)-norbornyl moiety; (1S,2R,4R)-norbornyl analogues are 1.1- to 1.8-fold more potent than (1R,2S,4S)-norbornyl analogues for CaV1.3 LTCCs. Presumably, each endo-norbornyl group can freely rotate in the binding site; therefore, they are recognized similarly.
The other interesting molecule resulting from this study is cyclohexylethyl drivative (S)-N-(3-chlorophenethyl)-N-(1-cyclohexylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (43). This compound is slightly more potent (IC50 = 4.3 µM) than lead compound 6 and is a moderately selective antagonist for CaV1.3 LTCCs. In addition to 43, all three N-(1-cyclohexylethyl) derivatives (44–46) in this study displayed good selectivity over CaV1.2, with IC50 of 4.3–9.7 µM for CaV1.3 LTCCs. These compounds have additional carbons between the cycloalkyl and PYT rings that will allow further structural variations on the cyclopentyl side chain.
Another trend that emerges from the SAR of various PYT molecules is the unfavorable effect of polar functionality on the cyclopentyl ring. Previous SAR studies showed that an electron lone pair or charge (hydrophilicity) on the aryl ring limits its binding affinity for both calcium channels. As shown by compounds 65 and 67 in Table 2, charged molecules or even an alcohol derivative (69) has no binding affinity for either CaV1.3 or CaV1.2 LTCCs, even at 100 µM. A hydrophobic cycloalkyl chain also is favored, and bulky esters or ethers (63, 66, 68, 74) do not seriously reduce (1- to 2-fold reduction) binding affinity for both CaV1.2 and CaV1.3 LTCCs. It is thought that the hydrophilicity arising from the heteroatoms are counterbalanced by the nonpolar groups, and so the ether bond on the cycloalkyl chain can be utilized as a linker for further modification. The cis-1,2-substitution pattern is also favored when acetoxyl is substituted on the cyclopentyl chain (71 and 72), even though it results in 6-fold reduced binding affinity. The favored 3-substitution pattern for selectivity was not observed with the ether or ester substituted derivatives; presumably the binding site in CaV1.2 LTCCs recognizes hydrophobic functional groups of various inhibitors differently.
Table 2.
IC50 Values and Selectivity of Various Cycloalkyl Analogues
 | ||||
|---|---|---|---|---|
| Compound # |
R1 | Cav1.3 IC50(µM) |
Cav1.2 IC50(µM) |
Selectivity |
| 61 | 32.7 ±11 | 33.4 ±1.8 | 1.1 | |
| 62 |  |
39.3 ±6.8 | 29.1 ±1.3 | 0.7 |
| 63 |  |
11.9 ±0.4 | 10.6 ±1.0 | 0.9 |
| 64 | 50.3 ±15 | 33.9 ±0.2 | 0.7 | |
| 65 | >100 | >100 | ||
| 66 | 10.4 ±1.3 | 8.5 ±0.9 | 0.8 | |
| 67 |  |
>100 | >100 | |
| 68 | 9.7 ±0.1 | 6.7 ±0.1 | 0.7 | |
| 69 | >100 | >100 | ||
| 70 | 30.3 ±1.8 | 34.6 ±4.3 | 1.1 | |
| 71 | >100 | 48.1 ±5.3 | ||
| 72 | 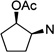 |
38.6 ±1.6 | 23.4 ±0.5 | 0.6 |
| 73 | 12.0 ±0.9 | 12.6 ±0.4 | 1.0 | |
| 74 | 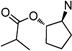 |
13.7 ±2.0 | 12.9 ±0.3 | 0.9 |
| 75 | 11.7 ±3.2 | 25.2 ±3.7 | 2.2 | |
| 36 | 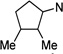 |
3.09 ±0.9 | 13.2 ±9.7 | 4.3 |
| 76 | 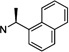 |
10.8 ±1.4 | 10.0 ±0.1 | 0.9 |
When the 3-chlorophenethyl substituent was changed to various fused aromatic rings, such as 1-tetrahydronaphthyl (77), 1-indanyl (78), and 2-indanyl (79), their binding affinity for CaV1.3 decreased 2-fold while their potency for CaV1.2 increased (Table 3). Although different lengths of alkyl linkers (81–84) were incorporated to adjust the space of this region, it was found that the ethyl linker was best for potency and selectivity. It is not clear whether this region disfavors hydrophilic functionalities, but carboxylate analogues 93–95 did not provide better potency or selectivity for CaV1.3 LTCCs. Another trend that emerged from the ethyl linker region is the steric preference; methyl- or dimethyl-substituted members or phenyl substituted members (89, 90, 96–99) have 1- to 3-fold improved binding affinity for CaV1.3 LTCCs. However, this modification also results in better interaction with CaV1.2 LTCCs; the overall selectivity for CaV1.3 LTCCs was 1- to 5-fold. 3,4- or 3,5-Dichloro and 3,5-bis(trifluoromethyl) substitution on the aromatic ring (85–87) led to ∼5-fold improved potency (IC50 = 1.2–1.4 µM) for CaV1.3 LTCCs, but selectivities were moderate.
Table 3.
IC50 Values and Selectivities of Various Arylalkyl Analogues
 | ||||
|---|---|---|---|---|
| Compound | R1 | Cav1.3 IC50(µM) |
Cav1.2 IC50(µM) |
Selectivity |
| 77 | 19.7 ±1.1 | 20.0 ±1.0 | 1.0 | |
| 78 | 19.9 ±4.9 | 24.8 ±0.6 | 1.2 | |
| 79 | 144 ±1.8 | 16.4 ±0.9 | 1.1 | |
| 80 | 4.6 ±0.4 | 6.15 ±1.5 | 1.3 | |
| 81 | >100 | >100 | ||
| 82 | 43.0 ±10 | 46.7 ±7.6 | 1.1 | |
| 83 | 17.9 ±1.9 | 51.7 ±0.7 | 2.9 | |
| 84 | 7.6 ±0.1 | 21.9 ±1.5 | 2.9 | |
| 85 | 1.4 ±0.4 | 3.3 ±0.1 | 2.3 | |
| 86 | 1.2 ±0.1 | 3.4 ±0.6 | 2.7 | |
| 87 | 1.4 ±0.3 | 2.6 ±0.5 | 1.8 | |
| 88 | 39.0 ±13 | 57.6 ±5.1 | 1.5 | |
| 89 | 5.2 ±1.1 | 8.1 ±1.0 | 1.6 | |
| 90 | 4.1 ±1.2 | 4.7 ±1.1 | 1.2 | |
| 91 | 4.4 ±0.3 | 5.4 ±0.4 | 1.2 | |
| 92 | 34.1 ±4.9 | 18.7 ±1.1 | 0.5 | |
| 93 | >100 | >100 | ||
| 94 | 55.0 ±1.0 | 44.3 ±1.7 | 0.8 | |
| 95 | 7.5 ±0.2 | 13.4 ±1.4 | 1.8 | |
| 96 | 7.3 ±1.0 | >100 | >26 | |
| 97 | 4.2 ±0.3 | 20.7 ±7.2 | 5.0 | |
| 98 | 2.4 ±0.2 | 12.6 ±2.6 | 4.9 | |
| 99 | 2.2 ±0.4 | 4.2 ±0.7 | 1.9 | |
Previously, CaV1.3 inhibitory activity was improved with substitution of the cyclopentyl group by the endo-norbornyl group; thus, a small series of norbornyl analogues (100–106) were prepared to determine optimal arylalkyl moiety (Table 4). The combination of endo- norbornyl with β-methyl-4- chlorophenethyl (100), α-methyl-4-chlorophenethyl (101), β-methyl-3-chlorophenethyl (102), β,β-dimethyl-3-chlorophe-nethyl (103), 2-(bis-3-(trifluoromethyl)phenyl)ethyl (104), 2-(3,5-dichlorophenyl)ethyl (105), or 2-(3-(trifluoromethyl)-phenyl)ethyl (106–108) led to good CaV1.3 LTCCs inhibitory activity, with IC50 values of 1.8–3.6 µM. Compounds 106–108, as racemic mixtures or individual enantiomers, gave IC50 values of 2–3 µM for CaV1.3 and ∼40-fold selectivity over CaV1.2 LTCCs. The (1S,2R,4R)-endo analogue (108) is slightly more potent than the (1R,2S,4S)-endo analogue (107), but they are almost the same in their CaV1.3 LTCCs inhibitory activity and selectivity.
CONCLUSION
Our SAR study of N-(3-chlorophenethyl)-N’-cyclopentylpyr-imidine-2,4,6-(1H,3H,5H)-trione (6) highlights several advancements in lead optimization of the PYT scaffold. One of the trends found from these analogues is that the endo-norbornyl moiety provides high selectivity for CaV1.3 LTCCs with good binding affinity if an appropriate arylalkyl moiety is used as the other side chain. Second, both CaV1.3 and CaV1.2 LTCCs have limited space to interact with various steric requirements at the cycloalkyl ring binding site; however, insertion of a one methylene linker between the cycloalkyl and PYT rings retains selectivity. Third, inactive compounds at both CaV1.3 and CaV1.2 LTCCs have a charged ion or electron lone pair near the cycloalkyl ring, implying that strong polarity should be avoided for antagonism of CaV1.3 LTCCs. Fourth, both CaV1.3 and CaV1.2 Ca + channels preferentially recognize the regiochemistry of 1,2/1,3-substituted cycloalkyl derivatives. The overall SAR of the PYT analogues identified four important series of molecules that either retained selectivity or enhanced binding affinity: The chiral N-endo-norbornyl analogues 50 (CaV1.3 IC50 = 2.1 ± 0.4 µM, selectivity ≥ 48), 51 (CaV1.3 IC50 = 2.0 ±0.1 µM, selectivity ≥ 50), and 108 (CaV1.3 IC50 = 2.1 ± 0.1 µM, selectivity = 36) have low micromolar activity against CaV1.3 LTCCs while retaining >36 selectivity. Dimethyl substitution on the cyclopentyl ring (22, CaV1.3 IC50 = 1.7 ± 0.1 µM) or on the arylethyl side chain (98, CaV1.3 IC50 = 2.4 ± 0.2 µM) leads to improved potency for CaV1.3. Among the studied PYT molecules, 3,4- or 3,5-disubstituted phenethyl derivatives (85, CaV1.3 IC50 = 1.4 ± 0.1 µM; 86, CaV1.3 IC50 = 1.2 ± 0.1 µM) are the most potent inhibitors for CaV1.3 LTCC. N-1-Cyclohexylethyl analogue 43 (CaV1.3 IC50 = 4.3 ± 0.5 µM, selectivity ≥ 29) is one of the potent and selective molecules; this side chain could be further modified. Our study highlights several advancements in the lead optimization and diversification toward the drug development of PYTs. The superimposition of the energy-minimized structures of selective or potent molecules highlights the overlapping and variable regions (Figure 4); these hotspots are located at or near the cycloalkyl and aromatic rings and should be important for future modifications.
Figure 4.

Overlay of the most interesting molecules: (A) selective compounds 43 (green), 50 (blue), and 51 (puple); (B) potent compounds 22 (skyblue), 85 (pink), 86 (white), and 98 (blue). The picture was generated using Sybyl-X and PyMOL.
EXPERIMENTAL SECTION
General Methods for Biology
Experimental procedures for the construction and stable transfection of HEK293 cells with CaV1.3 and CaV1.2 and procedure for high-throughput screening were previously reported.17,19 Rat CaV1.3α 1D (GenBank code AF370010) containing all alternative splice sites, rat CaV β3 (GenBank code M88751), rat CaV α2δ-1 (GenBank code 286488), and rabbit CaV1.2α 1C (GenBank code P15381) cDNAs were used. Screen Quest Fluo-8 no wash calcium assay kit (ABD Bioquest Inc., Sunnyvale, CA, U.S.) on a FLIPR tetra (Molecular Devices LLC, Sunnyvale, CA, U.S.) were used for the high-throughput assay. HEK293 cell density was 4 × 104 cells/ well and cultured in DMEM with 10% FBS for 4 days.
General Methods for Synthesis and Structure Characterization
Reagents were purchased from Sigma-Aldrich, Alfa-Aesar, TCI America, and Chem-Impex and were used without further purification. Solvents were purified by passage through a solvent column composed of activated alumina and a supported copper redox catalyst. Normal phase flash column chromatography was performed using SuperFlash Si 50 prepacked silica cartridges with an Agilent 971-FP flash purification system. Reaction progress was monitored by thin-layer chromatography (TLC) carried out on SiliCycle silica gel plates (2.5 cm × 7.5 cm, 250 µm thick, 60 F254), visualized by using UV (254 nm) and ninhydrin stain. 1H NMR and 13C NMR spectra were recorded in the indicated solvent on a Bruker Avance-III (500 and 126 MHz for 1H and 13C, respectively) spectrometer. Chemical shifts are given in ppm (δ) relative to the internal standard, and coupling constants (J) are in hertz (Hz). MS was performed on a system consisting of an electrospray ionization (ESI) source in a Thermo Finnigan LCQ mass spectrometer. High resolution mass spectra were obtained using an Agilent 6210 LC-TOF spectrometer. Melting points were measured in open capillaries on a Büchi B-540 melting point analyzer. The purity of the compounds was evaluated on an Agilent 1260 reverse phase analytical HPLC system using an Agilent Zorbax Eclipse XDB-C18 (4.6 mm × 50 mm, 5 µm) reverse phase column with UV absorbance and evaporative light scattering detection. Optical rotations were measured using an AA-100 polarimeter. The chiral purity of the compounds was evaluated on a Beckman Gold HPLC system using Daicel CHIRALPAK AD-RH or OD-RH (4.6 mm × 150 mm, 5 µm) reverse phase chiral columns.
General Procedure for PYT Synthesis Using Amine and Isocyanate. Method A
To a solution of an amine (1 mmol) in dry dichloromethane (10 mL), was added an isocyanate (1 mmol), and the mixture was stirred at room temperature for 1–5 h. After dilution with dry dichloromethane (40 mL), malonyl chloride (1.1 mmol) was added dropwise under vigorous stirring at room temperature for 5 min. The resulting pale yellow solution was stirred for an additional 1 h and washed with brine (50 mL). The organic layer was dried over anhydrous MgSO4 and concentrated at reduced pressure to a small volume. The resultant reaction mixture was purified using a silica gel cartridge (24 g) with an Agilent 971-FP purification system to give analytically pure compounds (20–90% yield).
(1S,2R,4R)-Bicyclo[2.2.1]heptane-2-isocyanate (10)
Precursor 9 was prepared according to Poll24 and Fukaya.25 To a solution of ethyl chloroformate (2.04 mL, 21.4 mmol) in hexane (20 mL) was added dropwise a solution of 9 (3.0 g, 21.4 mmol) and triethylamine (3.07 mL, 22 mmol) in hexane (20 mL) at 0 °C. After being stirred for 30 min at the same temperature, the mixture was quickly filtered. The filtrate was dried under vacuum and redissolved in acetone (20 mL). To this solution, a solution of sodium azide (2.2 g, 32 mmol) in water (20 mL) was added dropwise at 0 °C, and stirring continued for 2 h at the same temperature. The mixture was poured into ice–water (50 mL), and the organic materials were extracted with hexane (50 mL). The organic layer was dried over anhydrous MgSO4, filtered, and then heated to reflux for 5 h. After the mixture was cooled to room temperature, the solvent was removed by vacuum evaporation to give a crude product of 10 (2.26g, 75%). The product was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 3.87 (m, 1H), 2.35 (t, J = 4.3 Hz, 1H), 2.25 (t, J = 4.6 Hz, 1H), 2.12–1.98 (m, 1H), 1.79 (m, 1H), 1.67–1.54 (m, 1H), 1.47 (m, 1H), 1.38–1.29 (m, 3H), 1.05 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 121.20, 55.10, 42.28, 39.32, 37.36, 36.79, 29.54, 21.88. [α]25D (c 1, CHCl3) = −1.3.
(1R,2S,4S)-Bicyclo[2.2.1]heptane-2-isocyanate (13)
13 was prepared from 12 following a similar reaction procedure described for 10, and spectral data were identical to those of 10. [α]25D (c 1, CHCl3) = +1.5.
2-(3-Chlorophenyl)propanenitrile (15a) and 2-(3-Chloro-phenyl)-2-methylpropanenitrile (15b)
To a solution of (3-chlorophenyl)acetonitrile (3.0 g, 20 mmol) in DMF (40 mL) was added NaH (1.6 g, 40 mmol, 60% in oil) portionwise at room temperature. After the mixture was stirred for 15 min, MeI (4.56 g, 32 mmol) was added. The mixture was stirred overnight. The reaction mixture was quenched with saturated NH4Cl (80 mL), extracted with ethyl acetate (200 mL), dried over MgSO4, and concentrated at reduced pressure. The organic residue was purified by flash column chromatography to give 15a (0.49 g, 15%) and 15b (1.61 g, 45%). 15a: 1H NMR (500 MHz, CDCl3) δ 7.46–7.13 (m, 4H), 3.91 (q, J = 7.3 Hz, 1H), 1.68 (d, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 138.90, 135.03, 130.49, 128.40, 127.03, 124.98, 120.97, 30.97, 21.33. MS (ESI): calculated for C9H8Cl [M + H]+, 166.04; found, 166.19. 15b: 1H NMR (500 MHz, CDCl3) δ 7.45–7.29 (m, 4H), 1.73 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 143.41, 130.28, 128.12, 126.13, 125.45, 123.95, 123.49, 37.05, 29.04. MS (ESI): calculated for C10H12F3N [M + H]+, 204.10; found, 204.24.
2-(3-(Trifluoromethyl)phenyl)propanenitrile (15c) and 2-Methyl-2-(3-(trifluoromethyl)phenyl)propanenitrile (15d)
15c and 15d were prepared (15c = 5%, 15d = 59% yield) from (3-(trifluoromethyl)phenyl)acetonitrile following a similar reaction procedure described for 15a,b. 15c: 1H NMR (500 MHz, CDCl3) δ 7.68–7.48 (m, 4H), 4.01 (q, J = 7.3 Hz, 1H), 1.71 (d, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 138.01, 131.65 (q, J = 32.6 Hz), 130.20, 129.83, 125.12 (d, J = 4.0 Hz), 123.72 (q, J = 272.5 Hz), 123.64 (q, J = 3.9 Hz), 120.81, 31.17, 21.37. MS (ESI): calculated for C10H8F3N [M + H]+, 200.07; found, 200.23. 15d: 1H NMR (500 MHz, CDCl3) δ 7.81–7.43 (m, 4H), 1.79 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 142.48, 131.56 (q, J = 32.2 Hz), 129.62, 128.82, 124.87 (d, J = 4.3 Hz), 123.79, 123.69 (q, J = 277 Hz), 121.82 (d, J = 3.8 Hz), 37.20, 29.06. MS (ESI): calculated for C10H8F3N [M + H]+, 214.20; found, 214.23.
2-(3-Chlorophenyl)propan-1-amine (16a)
To a solution of 15a (0.40 g, 2.45 mmol) in THF (25 mL) at 0 °C was added 1.0 M solution of LiAH4 (7.35 mL, 7.35 mmol). After being stirred for 2 h at the same temperature, the mixture was warmed to room temperature, quenched with saturated NH4Cl (30 mL), and partitioned with ethyl acetate (30 mL). The organic layer was washed with brine, dried over MgSO4, and concentrated to give 16a (0.253g, 61%). 1H NMR (500 MHz, CDCl3) δ 7.44–7.15 (m, 4H), 3.65 (m, 1H), 2.77 (m, 2H), 1.23 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 147.09, 134.97, 130.50, 128.36, 127.00, 124.99, 49.12, 30.92, 21.28. MS (ESI): calculated for C9H12ClN [M + H]+, 170.07; found, 169.85.
2-(3-Chlorophenyl)-2-methylpropan-1-amine (16b)
16b was prepared (yield 73%) from 15b following a similar reaction procedure described for 16a. Pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.43–7.10 (m, 4H), 2.79 (s, 2H), 1.30 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 149.42, 134.34, 129.64, 126.63, 126.15, 124.43, 54.47, 39.97, 26.22. MS (ESI): calculated for C10H15ClN [M + H]+, 184.09; found, 183.96.
2-(3-(Trifluoromethyl)phenyl)propan-1-amine (16c)
16c was prepared (yield 61%) from 15c following a similar reaction procedure described for 16a. Pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.66–7.45 (m, 4H), 3.62 (m, 1H), 2.81 (m, 2H), 1.50 (d, J = 6.6 Hz, 3H). MS (ESI): calculated for C10H12F3N [M + H]+, 204.09; found, 204.43.
2-Methyl-2-(3-(trifluoromethyl)phenyl)propan-1-amine (16d)
16d was prepared (yield 68%) from 15d following a similar reaction procedure described for 16a. 1H NMR (500 MHz, CDCl3) δ 7.71–7.30 (m, 4H), 2.90 (s, 2H), 1.40 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 147.04, 130.77 (q, J = 32.3 Hz), 129.59, 129.08, 128.85, 123.40 (d, J = 3.9 Hz), 123.15 (m), 122.80 (d, J = 3.7 Hz), 52.91, 38.96, 26.14. MS (ESI): calculated for C11H14F3N [M + H]+, 218.12; found, 218.23.
(1S,2R)-2-Methyl-N-((S)-1-phenylethyl)cyclopentanamine (18a)
A mixture of 2-methylcyclopentanone (0.294 g, 3.0 mmol), (S)-1-phenylethanamine (0.774 mL, 6.0 mmol), and a catalytic amount of TsOH in benzene (25 mL) was refluxed overnight. After being cooled to room temperature, the reaction mixture was diluted with 10 mL of absolute EtOH, and sodium cyanoborohydride (569 mg, 9 mmol) was then added to the reaction mixture in three portions over a ∼5 h period. The reaction mixture was quenched with brine (50 mL), partitioned with ethyl acetate (50 mL), dried over MgSO4, and concentrated on reduced pressure. The resulting residue was subjected to silica gel chromatography to purify the major component as 18a (0.238 g, 36%). 1H NMR (500 MHz, CDCl3) δ 7.49–7.04 (m, 5H), 3.78 (q, J = 6.6 Hz, 1H), 2.82 (m, 1H), 2.10 (m, 1H), 1.74–1.56 (m, 3H), 1.46–1.35 (m, 1H), 1.33 (d, J = 6.5 Hz, 3H), 1.28 (m, 2H), 0.87 (d, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 146.29, 128.30, 126.72, 126.67, 59.51, 56.26, 34.02, 31.51, 30.18, 24.99, 20.25, 13.56. MS (ESI): calculated for C14H21N [M + H]+, 204.18; found, 204.14.
(1R,2S)-2-Methyl-N-((R)-1-phenylethyl)cyclopentanamine (23a)
23a was prepared (yield 33%) with (R)-1-phenylethanamine following a similar reaction procedure described for 18a. Spectral data were identical to those of 18a.
(S)-2,2-Dimethyl-N-((S)-1-phenylethyl)cyclopentanamine (18b)
18b was prepared (yield 63%) from (S)-1-phenylethanamine and 2,2-dimethylcyclopentanone following a similar reaction procedure described for 18a. 1H NMR (500 MHz, CDCl3) δ 7.49–7.06 (m, 5H), 3.91 (q, J = 6.6 Hz, 1H), 2.56 (dd, J = 9.6, 7.7 Hz, 1H), 2.44–2.30 (m, 1H), 1.82–1.70 (m, 1H), 1.61–1.54 (m, 1H), 1.48–1.36 (m, 3H), 1.35 (d, J = 6.6 Hz, 3H), 1.30–1.19 (m, 1H), 1.05 (s, 3H), 0.89 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 146.72, 128.29, 126.76, 126.24, 65.58, 57.30, 40.90, 39.96, 31.87, 28.15, 24.36, 21.14, 19.71. MS (ESI): calculated for C15H24ClN [M + H]+, 218.19; found, 218.18.
(R)-2,2-Dimethyl-N-((R)-1-phenylethyl)cyclopentanamine (23b)
23b was prepared (yield 60%) from 2,2-dimethylcyclopenta-none and (R)-1-phenylethanamine following a similar reaction procedure described for 18a. Spectral data were identical to those of 18b.
3-Methyl-N-((S)-1-phenylethyl)cyclopentanamine (29a)
Diastereomeric mixture 29a was prepared (yield 71%) from 3-methylcyclopentanone and (S)-1-phenylethanamine following a similar reaction procedure described for 18a. 1H NMR (500 MHz, CDCl3) δ 7.48–7.31 (m, 12H), 7.26–7.12 (m, 4H), 4.07–3.94 (m, 1H), 3.14–2.89 (m, 1H), 2.21–2.05 (m, 1H), 1.91–1.75 (m, 2H), 1.74–1.64 (m, 1H), 1.55 (dd, J = 6.8, 2.1 Hz, 3H), 1.41–1.13 (m, 2H), 1.01–0.91 (m, 3H); 13C NMR (126 MHz, CDCl3) δ 141.56, 128.79, 127.86, 127.24, 56.97, 56.87, 56.63, 56.05, 41.09, 39.99, 33.34, 32.99, 32.50, 32.25, 32.13, 30.96, 29.62, 24.13, 22.64, 20.44. MS (ESI): calculated for C14H21N [M + H]+, 204.18; found, 204.12.
3,3-Dimethyl-N-((S)-1-phenylethyl)cyclopentanamine (29b)
Diastereomeric mixture 29b was prepared (yield 75%) from 3,3-dimethylcyclopentanone and (S)-1-phenylethanamine following a similar reaction procedure described for 18a. Major isomer: 1H NMR (500 MHz, CDCl3) δ 7.36–7.20 (m, 5H), 3.98–3.77 (m, 1H), 2.93–2.78 (m, 1H), 1.95–1.87 (m, 1H), 1.75–1.67 (m, 1H), 1.57–1.54 (m, 3H), 1.47–1.39 (m, 4H), 0.96 (s, 3H), 0.93 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 146.52, 128.31, 126.76,126.31, 57.28, 46.85, 38.86, 35.29, 29.36, 29.24, 20.72. MS (ESI): calculated for C15H24ClN [M + H]+, 218.19; found, 218.13.
3-Ethyl-N-((S)-1-phenylethyl)cyclopentanamine (29c)
Diastereomeric mixture 29c was prepared (yield 74%) from 3-ethyl-cyclopentanone and (S)-1-phenylethanamine following a similar reaction procedure described for 18a. Isomeric mixture: 1H NMR (500 MHz, CDCl3) δ 7.40–7.09 (m, 5H), 3.81 (p, J = 6.8 Hz, 1H), 3.01–2.82 (m, 1H), 2.20–2.04 (m, 1H), 1.98–1.75 (m, 2H), 1.69–1.57 (m, 1H), 1.52–1.35 (m, 3H), 1.34 (d, J = 6.8 Hz, 3H), 1.28–1.19 (m, 2H), 0.89–0.80 (m, 3H); 13C NMR (126 MHz, CDCl3) δ 146.03, 128.35, 126.77, 126.75, 126.63, 126.59, 56.98, 56.85, 56.57, 56.50, 56.22, 56.20, 56.14, 45.03, 41.09, 40.54, 40.35, 40.07, 39.87, 39.75, 39.65, 38.94, 38.67, 38.58, 34.24, 33.39, 32.79, 31.93, 31.28, 31.16, 30.08, 29.89, 29.36, 29.24, 29.23, 29.21, 29.16, 28.44, 24.70, 24.65, 24.60, 12.87. MS (ESI): calculated for C15H23N [M + H]+, 218.19; found, 218.14.
2,3-Dimethyl-N-((S)-1-phenylethyl)cyclopentanamine (29d)
Diastereomeric mixture 29d was prepared (yield 63%) from 2,3-dimethylcyclopentanone and (S)-1-phenylethanamine following a similar reaction procedure described for 18a. 1H NMR (500 MHz, CDCl3) δ 7.49–7.14 (m, 5H), 3.82–3.70 (m, 1H), 3.00 (dt, J = 7.7, 6.6 Hz, 1H), 1.85–1.78 (m, 1H), 1.73 (m, 1H), 1.69–1.53 (m, 4H), 1.35 (d, J = 6.6 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 146.62, 128.73, 128.31, 126.72, 58.85, 56.57, 43.21, 40.09, 31.83, 31.14, 24.53, 20.90, 14.02. MS (ESI): calculated for C15H23N [M + H]+, 218.19; found, 218.28.
General Procedure for 19–22, 24–27, and 30–36. (Method B)
To a solution of an N-(1-phenylethyl)cyclopentylamine (18a,b, 23a,b, 29a–d; 1 mmol) in dry dichloromethane (10 mL) was added an isocyanate (1 mmol), and the mixture was stirred at room temperature for 3 h. The solvent was removed by vacuum distillation, and 95% TFA in water (5 mL) was added to the reaction mixture. After being stirred for 1 h at room temperature, the reaction mixture was concentrated under vacuum, redissolved in toluene, and then dried by vacuum distillation. After the sample was dissolved in dry dichloromethane (40 mL), malonyl chloride (1.1 mmol) was added dropwise under vigorous stirring at room temperature for 5 min. The resulting pale yellow solution was stirred for an additional 1 h and washed with brine (50 mL). The organic layer was dried over anhydrous MgSO4 and concentrated at reduced pressure to a small volume. The resultant reaction mixture was purified with silica gel (24 g) using an Agilent 971-FP purification system to give analytically pure compounds (50–80% yield).
Spectral Data of the Most Active Compounds. (S)-1-(3-Chlorophenethyl)-3-(1-cyclohexylethyl)pyrimidine-2,4,6-(1H,3H,5H)-trione (43) and (R)-1-(3-Chlorophenethyl)-3-(1-cyclohexylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (45)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.26–7.07 (m, 4H), 4.50 (m, 1H), 4.09 (m, 2H), 3.60 (s, 2H), 2.90 (m, 2H), 2.04 (m, 1H), 1.87 (m, 1H), 1.76 (m, 1H), 1.66 (m, 2H), 1.36 (d, J = 6.9 Hz, 3H), 1.32–1.06 (m, 4H), 0.86 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 164.85, 164.64, 150.91, 139.75, 134.27, 129.81, 129.13, 127.21, 126.95, 51.35, 42.50, 39.88, 39.20, 33.57, 30.83, 29.83, 26.07, 25.79, 25.71, 16.29. HRMS (ESI): calculated for C20H25ClN2O3 [M – H]−, 375.1481; found, 375.1494. 43, HPLC purity: 99.2%, [α]25D (c 2, CHCl3) = −6.8, chiral HPLC (Daicel AD-RH, 4.6–150 mm, 5 µm; 70% acetonitrile/30% 0.05 M pH 2 phosphate buffer; 0.4 mL/min; 268 nm) tR = 30.933 min; >99% ee. 45, HPLC purity: 99.9%, [α]25D (c 2, CHCl3) = +7.0; chiral HPLC (Daicel AD-RH, 4.6–150 mm, 5 µm; 70% acetonitrile/30% 0.05 M pH 2 phosphate buffer; 0.4 mL/min; 268 nm) tR = 28.017 min; >99% ee.
(S)-1-(4-Chlorophenethyl)-3-(1-cyclohexylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (44) and (R)-1-(4-Chlorophenethyl)-3-(1-cyclohexylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (46)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 4.49 (m, 1H), 4.08 (m, 2H), 3.60 (s, 2H), 2.88 (m, 2H), 2.04 (m, 1H), 1.87 (m, 1H), 1.76 (m, 1H), 1.66 (m, 2H), 1.35 (d, J = 6.9 Hz, 3H), 1.31–1.06 (m, 4H), 0.85 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 164.85, 164.65, 151.06, 136.19, 132.56, 130.37, 128.66, 51.34, 42.56, 39.88, 39.19, 33.27, 30.81, 29.85, 26.06, 25.79, 25.70, 16.28. HRMS (ESI): calculated for C20H25ClN2O3 [M -H]−, 375.1148; found, 375.1494. 44, HPLC purity: 97.8%, [α]25D (c 2, CHCl3) = −6.3, chiral HPLC (Daicel AD-RH, 4.6–150 mm, 5 µm; 70% acetonitrile/30% 0.05 M pH 2 phosphate buffer; 0.4 mL/min; 268 nm) tR = 33.217 min; >99% ee. 46, HPLC purity: 99.3%, [α]25D (c 2, CHCl3) = +6.5, chiral HPLC (Daicel AD-RH, 4.6–150 mm, 5 µm; 70% acetonitrile/30% 0.05 M pH 2 phosphate buffer; 0.4 mL/min; 268 nm) tR = 37.199 min; >99% ee.
1-((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)-3-(3-chlorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (49) and 1-((1S,2R,4R)-Bicyclo[2.2.1]heptan-2-yl)-3-(3-chlorophenethyl)-pyrimidine-2,4,6(1H,3H,5H)-trione (51)
White powder; mp 155–157 °C; 1H NMR (500 MHz, CDCl3) δ 7.28–7.11 (m, 4H), 4.69–4.56 (m, 1H), 4.18–4.03 (m, 2H), 3.64 (dd, J = 9.2, 20.9 Hz, 2H), 2.90 (t, J = 7.8 Hz, 2H), 2.66–2.58 (m, 1H), 2.37 (t, J = 4.5 Hz, 1H), 2.22 (m, 1H), 1.81 – 1.65 (m, 2H), 1.59–1.50 (m, 1H), 1.47 (m, 1H), 1.45–1.37 (m, 2H), 1.36–1.31 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 165.90, 164.52, 152.15, 139.84, 134.29, 129.82, 129.09, 127.15, 126.93, 59.65, 42.82, 42.08, 40.90, 37.73, 37.53, 33.66, 29.49, 28.77, 23.86. HRMS (ESI): calculated for C19H21ClN2O3 [M – H]−, 359.1168; found, 359.1172. 49, HPLC purity: 99.0%; [α]25D (c 2, CHCl3) = +9.4 chiral HPLC (Daicel OD-RH, 4.6–150 mm, 5 µm; 60% acetonitrile/40% 0.1 M aqueous KPF6; 0.5 mL/min; 268 nm) tR = 26.400 min, >99% ee. 51, HPLC purity: 99.0%; [α]25D (c 2, CHCl3) = −9.1, chiral HPLC (Daicel OD-RH, 4.6–150 mm, 5 µm; 60% acetonitrile/40% 0.1 M aqueous KPF6; 0.5 mL/min; 268 nm) tR = 22.867 min, 97.2% ee.
1-((1R,2S,4S)-Bicyclo[2.2.1]heptan-2-yl)-3-(4-chlorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (50) and 1-((1S,2R,4R)-Bicyclo[2.2.1]heptan-2-yl)-3-(4-chlorophenethyl)-pyrimidine-2,4,6(1H,3H,5H)-trione (52)
White powder; mp 121–123 °C; 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.3 Hz, 2H), 4.60 (m, 1H), 4.06 (m, 2H), 3.61 (dd, J = 21.1, 10.1 Hz 2H), 2.87 (t, J = 7.8 Hz, 2H), 2.58 (t, J = 3.8 Hz, 1H), 2.35 (t, J = 4.5 Hz, 1H), 2.19 (m, 1H), 1.78–1.61 (m, 2H), 1.58–1.47 (m, 1H), 1.47–1.29 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 165.90, 164.53, 152.17, 136.27, 132.56, 130.33, 128.68, 59.64, 42.91, 42.06, 40.92, 37.72, 37.54, 33.35, 29.44, 28.76, 23.82. HRMS (ESI): calculated for C19H21ClN2O3 [M – H]−, 359.1168; found, 359.1183. 50, HPLC purity: 99.0%; [α]25D (c 2, CHCl3) = +14.5, chiral HPLC (Daicel OD-RH, 4.6–150 mm, 5 µm; 60% acetonitrile/40% 0.1 M aqueous KPF6; 0.5 mL/min; 268 nm) tR = 25.367 min, >99% ee. 52, HPLC purity: 100%; [α]25D (c 2, CHCl3) = −14.7, chiral HPLC (Daicel OD-RH, 4.6–150 mm, 5 µm; 60% acetonitrile/40% 0.1 M aqueous KPF6; 0.5 mL/min; 268 nm) tR = 23.367 min, 97.0% ee.
1-Cyclopentyl-3-(3,5-dichlorophenethyl)pyrimidine-2,4,6-(1H,3H,5H)-trione (85)
White powder; mp 144–146 °C; 1H NMR (500 MHz, CDCl3) δ 7.25 (t, J = 1.9 Hz, 1H), 7.16 (d, J = 1.9 Hz, 2H), 5.15 (m, 1H), 4.09–4.02 (m, 2H), 3.66 (s, 2H), 2.92–2.80 (m, 2H), 1.95 (m, 4H), 1.85 (m, 2H), 1.66–1.52 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 164.69, 164.49, 150.85, 141.12, 135.02, 127.49, 127.05, 54.39, 42.36, 40.11, 33.51, 28.72, 25.57. HRMS (ESI): calculated for C17H18Cl2N2O3 [M – H]−, 367.0622; found, 367.0637. HPLC purity: 94.5%.
1-Cyclopentyl-3-(3,4-dichlorophenethyl)pyrimidine-2,4,6-(1H,3H,5H)-trione (86)
White powder; mp 131–133 °C; 1H NMR (500 MHz, CDCl3) δ 7.43–7.32 (m, 2H), 7.12 (d, J = 8.2 Hz, 1H), 5.24–5.08 (m, 1H), 4.13–4.03 (m, 2H), 3.66 (s, 2H), 2.93–2.78 (m, 2H), 2.04–1.90 (m, 4H), 1.89–1.79 (m, 2H), 1.67–1.54 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 164.71, 164.52, 150.87, 138.03, 132.50, 130.91, 130.85, 130.52, 128.40, 54.36, 42.48, 40.12, 33.18, 28.71, 25.56. HRMS (ESI): calculated for C17H18Cl2N2O3 [M – H]−, 367.0622; found, 367.0637. HPLC purity: 98.0%.
1-(3,5-Bis(trifluoromethyl)phenethyl)-3-cyclopentylpyrimi-dine-2,4,6(1H,3H,5H)-trione (87)
White powder; mp 163–166 °C; 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 1H), 7.73 (s, 2H), 5.27–4.99 (m, 1H), 4.21–4.04 (m, 2H), 3.66 (s, 2H), 3.17–2.93 (m, 2H), 2.00–1.75 (m, 6H), 1.66–1.51 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 164.57, 164.54, 150.83, 140.26, 131.84 (q, J = 33.3 Hz), 129.19 (m), 123.23 (q, J = 272.8 Hz), 120.88 (m), 54.38, 42.16, 40.03, 33.66, 28.68, 25.53. HRMS (ESI): calculated for C19H18F6N2O3 [M – H]−, 435.1149; found, 435.1169. HPLC purity: 98.5%.
1-((1-(4-Chlorophenyl)cyclopropyl)methyl)-3-cyclopentyl-pyrimidine-2,4,6(1H,3H,5H)-trione (96)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.21 (s, 4H), 5.00 (p, J = 8.7 Hz, 1H), 4.06 (s, 2H), 3.51 (s, 2H), 1.92–1.81 (m, 2H), 1.79–1.66 (m, 4H), 1.59–1.43 (m, 2H), 1.07–0.98 (m, 2H), 0.87–0.78 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 164.75, 150.82, 140.85, 132.80, 131.52, 128.32, 54.21, 48.83, 40.03, 28.56, 25.49, 25.04, 12.05. HRMS (ESI): calculated for C19H21ClN2O3 [M – H]−, 359.1168; found, 359.1186. HPLC purity: 96.1%.
1-(2-(3-Chlorophenyl)-2-methylpropyl)-3-cyclopentylpyri-midine-2,4,6(1H,3H,5H)-trione (98)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.46–7.00 (m, 4H), 5.20–4.97 (m, 1H), 4.08 (s, 2H), 3.61 (s, 2H), 1.97–1.73 (m, 6H), 1.61–1.49 (m, 2H), 1.36 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 165.00, 164.77, 151.22, 148.51, 134.09, 129.42, 126.69, 126.59, 124.55, 54.29, 51.06, 40.22, 40.15, 28.67, 26.94, 25.54. MS (ESI): calculated for C19H23ClN2O3 [M – H]−, 361.13; found, 361.23. HPLC purity: 99.1%.
1-Cyclopentyl-3-(2-methyl-2-(3-(trifluoromethyl)phenyl)-propyl)pyrimidine-2,4,6(1H,3H,5H)-trione (99)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.75–7.59 (m, 2H), 7.59–7.39 (m, 2H), 5.08 (p, J = 8.7 Hz, 1H), 4.12 (s, 2H), 3.61 (s, 2H), 1.89 (m, 2H), 1.85–1.72 (m, 4H), 1.58–1.48 (m, 1H), 1.42 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 164.98, 164.66, 151.15, 147.28, 130.37, 129.88, 128.61, 123.32, 123.06, 122.05, 54.27, 51.05, 40.25, 40.09, 28.65, 27.01, 25.51. MS (ESI): calculated for C20H23F3N2O3 [M – H]−, 395.16; found, 395.28. HPLC purity: 95.3%.
1-(2-(3-Chlorophenyl)propyl)-3-((±)-endo-norbornyl)-pyrimidine-2,4,6-(1H,3H,5H)-trione (102)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.27–7.05 (m, 4H), 4.64–4.49 (m, 1H), 4.11 (m, 1H), 3.97 (m, 1H), 3.67–3.44 (m, 2H), 3.36–3.17 (m, 1H), 2.68–2.45 (m, 1H), 2.37–2.28 (m, 1H), 2.14 (m, 1H), 1.85–1.57 (m, 3H), 1.57–1.49 (m, 1H), 1.46–1.35 (m, 2H), 1.34–1.17 (m, 5H); 13C NMR (126 MHz, CDCl3) δ 165.89, 165.85, 164.69, 152.33, 152.27, 145.13, 145.08, 134.24, 134.21, 129.74, 129.71, 127.79, 127.09, 125.71, 125.67, 59.62, 59.60, 47.87, 47.77, 47.76, 42.04, 41.96, 40.91, 40.81, 37.87, 37.81, 37.74, 37.70, 37.55, 37.49, 29.69, 29.35, 28.76, 23.83, 23.71, 18.53. MS (ESI): calculated for C20H23ClN2O3 [M – H]−, 373.13; found, 373.10. HPLC purity: 99.3%.
1-(1-(3-Chlorophenyl)propan-2-yl)-3-((±)-endo-norbornyl)-pyrimidine-2,4,6-(1H,3H,5H)-trione (103)
Colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.48–7.10 (m, 4H), 4.70–4.49 (m, 1H), 4.20–4.02 (m, 2H), 3.73–3.51 (m, 2H), 2.54 (d, J = 3.8 Hz, 1H), 2.34 (d, J = 4.6 Hz, 1H), 2.13–2.02 (m, 1H), 1.77–1.60 (m, 2H), 1.58–1.48 (m, 1H), 1.48–1.38 (m, 2H), 1.37 (s, 3H), 1.35 (s, 3H), 1.35–1.22 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 165.87, 165.01, 152.60, 148.54, 134.08, 129.41, 126.68, 126.60, 124.58, 59.72, 51.28, 41.94, 41.00, 40.24, 37.74, 37.52, 29.35, 28.75, 27.03, 26.92, 23.82. MS (ESI): calculated for C21H25ClN2O3 [M – H]−, 387.15; found, 387.28. HPLC purity: 96.9%.
1-(3,5-Bis(trifluoromethyl)phenethyl-3-((±)-endo-norbornyl)pyrimidine-2,4,6(1H,3H,5H)-trione (104)
White powder; mp 74–77 °C; 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 1H), 7.73 (s, 2H), 4.72–4.57 (m, 1H), 4.22–4.07 (m, 2H), 3.77–3.56 (m, 2H), 3.06 (t, J = 7.8 Hz, 2H), 2.61 (d, J = 4.1 Hz, 1H), 2.36 (t, J = 4.6 Hz, 1H), 2.18 (m, 1H), 1.83–1.63 (m, 2H), 1.60–1.49 (m, 1H), 1.49–1.28 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 165.76, 164.60, 152.11, 140.31, 131.83 (q, J = 33.4 Hz), 129.20 (d, J = 3.7 Hz), 123.25 (q, J = 272.8 Hz), 120.88 (p, J = 3.7 Hz), 59.70, 42.36, 42.09, 40.80, 37.72, 37.52, 33.67, 29.52, 28.74, 23.81. HRMS (ESI): calculated for C21H20F6N2O3 [M – H]−, 461.1305; found, 461.1321. HPLC purity: 96.4%.
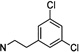
1-(2(3,5-Dichlorophenyl)ethyl)-3-((±)-endo-norbornyl)-pyrimidine-2,4,6(1H,3H,5H)-trione (105)
White powder; mp 150–151 °C; 1H NMR (500 MHz, CDCl3) δ 7.26 (s, 1H), 7.17 (s, 2H), 4.71–4.58 (m, 1H), 4.13–4.01 (m, 2H), 3.65 (dd, J = 21.2, 8.7 Hz, 2H), 2.87 (t, J = 7.8 Hz, 2H), 2.66–2.58 (m, 1H), 2.42–2.34 (m, 1H), 2.25–2.15 (m, 1H), 1.81–1.66 (m, 2H), 1.61–1.51 (m, 1H), 1.48–1.32 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 165.85, 164.55, 152.13, 141.15, 135.01, 127.51, 127.05, 59.70, 42.53, 42.11, 40.90, 37.76, 37.55, 33.50, 29.55, 28.80, 23.88. MS (ESI): calculated for C19H20Cl2N2O3 [M – H]−, 393.08; found, 393.26. HPLC purity: 98.1%.
1-((±)-exo-Norbornyl)-3-(3-(trifluoromethyl)phenethyl)-pyrimidine-2,4,6(1H,3H,5H)-trione (106), 1-((1R,2S,4S)-Bicyclo-[2.2.1]heptan-2-yl)-3-(3-(trifluoromethyl)phenethyl)-pyrimidine-2,4,6(1H,3H,5H)-trione (107), and 1-((1S,2R,4R)-Bicyclo[2.2.1]heptan-2-yl)-3-(3-(trifluoromethyl)phenethyl)-pyrimidine-2,4,6(1H,3H,5H)-trione (108)
White powder; mp 65–69 °C; 1H NMR (500 MHz, CDCl3) δ 7.51–7.26 (m, 4H), 4.59–4.45 (m, 1H), 4.10–3.93 (m, 2H), 3.55 (dd, J = 21.0, 12.9 Hz, 2H), 2.89 (t, J = 7.8 Hz, 2H), 2.52 (d, J = 3.7 Hz, 1H), 2.27 (d, J = 4.4 Hz, 1H), 2.11 (m, 1H), 1.71 −1.54 (m, 2H), 1.49–1.41 (m, 1H), 1.40–1.21 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 165.90, 164.58, 152.17, 138.80, 132.45, 130.84 (q, J = 32.0 Hz), 129.05, 125.69 (q, J = 3.7 Hz), 124.08 (q, J = 272.4 Hz), 123.63 (d, J = 4.1 Hz), 59.64, 42.74, 42.09, 40.86, 37.74, 37.54, 33.77, 29.50, 28.77, 23.84. HRMS (ESI): calculated for C20H21F3N2O3 [M – H]−, 393.1432; found, 393.1448. 106, HPLC purity: 97.9%. 107, HPLC purity: 96.8%, [α]25D (c 2, CHCl3) = −7.5, chiral HPLC (Daicel OD-RH, 4.6–150 mm, 5 µm; 60% acetonitrile/ 40% 0.1 M aqueous KPF6; 0.5 mL/min; 268 nm) tR = 18.800 min, 96.9% ee. 108, HPLC purity: 99.0%; [α]25D (c 2, CHCl3) = +7.7; chiral HPLC (Daicel OD-RH, 4.6–150 mm, 5 µm; 60% acetonitrile/ 40% 0.1 M aqueous KPF6; 0.5 mL/min; 268 nm) tR = 21.583 min; >99% ee.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Michael J. Fox Foundation (Therapeutics Development Initiative) and the RJG Foundation for financial support of this research.
ABBREVIATIONS USED
- LTCC
L-type calcium channel
- PD
Parkinson’s disease
- PYT
pyrimidinetrione
- SNc
substantia nigra pars compacta
- FLIPR
fluorometric imaging plate reader
- DMEM
Dulbecco’s modified Eagle medium
Footnotes
ASSOCIATED CONTENT
Supporting Information
Spectroscopic and analytical data of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Casamassima F, Hay AC, Benedetti A, Lattanzi L, Cassano GB, Perlis RH. L-type calcium channels and psychiatric disorders, a brief review. Am. J. Med. Genet, Part B. 2010;153B:1373–1390. doi: 10.1002/ajmg.b.31122. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 3.Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J. Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- 4.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda J-C, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 5.Zuccotti A, Clementi S, Reinbothe T, Torrente A, Vandael DH, Pirone A. Structural and functional differences between L-type calcium channels: crucial issues for future selective targeting. Trends Pharmacol. Sci. 2011;32:366–375. doi: 10.1016/j.tips.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Striessnig J, Bolz HJ, Koschak A. Channelopathies in CaV1.1, CaV1.3, and CaV1.4 voltage-gated L-type Ca2+ channels. Pfluegers Arch. 2010;460:361–374. doi: 10.1007/s00424-010-0800-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J. Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan CS, Guzman JN, Ilijic E, Mercer J, Rick C, Tkatch T, Meredith G, Surmeier DJ. Rejuvenation protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 9.Riederer P, Wuketich S. Time course of nigrostriatal degeneration in Parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J. Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- 10.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 11.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009;29:11011–11009. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jankovic J. Parkinson’s disease: clinical features and diagnosis. J. Neurol., Neurosurg. Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- 14.Epstein M. Calcium antagonists: still appropriate as first line antihypertensive agents. Am. J. Hypertens. 1996;9:110–121. doi: 10.1016/0895-7061(96)00013-1. [DOI] [PubMed] [Google Scholar]
- 15.Sullivan E, Tucker EM, Dale L. Measurement of [Ca2+] using the fluorometric imaging plate reader (FLIPR). Methods Mol. Biol. 1999;114:125–133. doi: 10.1385/1-59259-250-3:125. [DOI] [PubMed] [Google Scholar]
- 16.Minta A, Kao JPY, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- 17.Triggle DJ. 1,4-Dihydropyridines as calcium channel ligands and privileged structures. Cell. Mol. Neurobiol. 2003;23:293–303. doi: 10.1023/A:1023632419813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Triggle DJ, Hawthorn M, Gopalakrishnan M, Minarini A, Avery S, Rutledge A, Bangalore R, Zheng W. Synthetic organic ligands active at voltage-gated calcium channels. Ann. N.Y. Acad. Sci. 1991;635:123–138. doi: 10.1111/j.1749-6632.1991.tb36487.x. [DOI] [PubMed] [Google Scholar]
- 19.Chang C-C, Cao S, Kang S, Kai L, Tian X, Pandey P, Fernandez DS, Luan C-H, Surmeier DJ, Silverman RB. Antagonism of 4-substituted 1,4-dihydropyridine-3,5-dicarboxylates toward voltage-dependent L-type Ca2+ channels CaV1.3 and CaV1.2. Bioorg. Med .Chem. 2010;18:3147–3158. doi: 10.1016/j.bmc.2010.03.038. [DOI] [PubMed] [Google Scholar]
- 20.Kang S, Cooper G, Dunne SF, Luan C-H, Surmeier DJ, Silverman RB. Antagonism of L-type Ca2+ channels CaV1.3 and CaV1.2 by 1,4-dihydropyrimidines and 4H–pyrans as dihydropyridine mimics, Bioorg. Med. Chem. [Online early access] doi: 10.1016/j.bmc.2013.04.054. Published Online: Apr 28, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Kang S, Cooper G, Dunne SF, Dusel B, Luan C-H, Surmeier DJ, Silverman RB. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 2012;3:1146. doi: 10.1038/ncomms2149. [DOI] [PubMed] [Google Scholar]
- 22.Wohler F. Ueber kunstliche bildung des harnstoffs. Ann. Phys. Chem. 1828;88:253. [Google Scholar]
- 23.Biltz H, Wittek H. Alkylierte und acylierte barbitursauren. Ber. 1921;54B:1035. [Google Scholar]
- 24.(a) Poll T, Sobczak A, Hartmann H, Helmchen G. Diastereoface-discriminative metal coordination in asymmetric synthesis: d-pantolactone as practical chiral auxiliary for Lewis acid catalyzed Diels-Alder reactions. Tetrahedron Lett. 1985;26:3095–3098. [Google Scholar]; (b) Poll T, Abdel Hady AF, Karge R, Linz G, Weetman J, Helmchen G. N-Substituted hydroxysuccinhides from (S)-malic acid as new reagents for asymmetric Diels-Alder additions to enoates. Tetrahedron Lett. 1989;30:5595–5598. [Google Scholar]
- 25.Eda M, Takemoto T, Ono S-I, Okada T, Kosaka K, Gohda M, Matzno S, Nakamura N, Fukaya C. Novel potassium-channel openers: preparation and pharmacological evaluation of racemic and optically active N-(6-amino-3-pyridyl)-N′-bicycloalkyl-N″-cyanoguanidine derivatives. J. Med. Chem. 1994;37:1983–1990. doi: 10.1021/jm00039a011. [DOI] [PubMed] [Google Scholar]
- 26.(a) Bollu V, Boren BC, Dalgar JE, Flatt BT, Haq N, Hudson S, Mohan R, Morrissey M, Pratt B, Wang T-L. Triazole and Imidazole Derivatives for Use as TGR5 Agonists in the Treatment of Diabetes and Obesity. PCT Int. Appl. 2011 Jun 16;2011071565 [Google Scholar]; (b) Meier H, Bender E, Brueggemeier U, Flamme I, Karthaus D, Kolkhof P, Meibom D, Schneider D, Voehringer V, Fuerstner C, Keldenich J, Lang D, Pook E, Schmerk C. Preparation of 4-Arylimidazol-2-ones and 5-Aryl-1,2,4-triazolones as Vasopressin Receptor Inhibitors. PCT Int. Appl. 2007 Nov 29;2007134862 [Google Scholar]
- 27.Nugent TC, Ghosh AK, Wakchaure VN, Mohanty RB. Asymmetric reductive amination: convenient access to enantioen-riched alkyl-alkyl or aryl-alkyl substituted α-chiral primary amines. Adv. Synth. Catal. 2006;348:1289–1299. [Google Scholar]
- 28.Moss N, Gauthier J, Ferland J-M. The enantioselective synthesis of neopentylamine derivatives. Synlett. 1995;2:142–144. [Google Scholar]
- 29.(a) Frahm AW, Knupp G. Asymmetric Synthesis of cis-2-substituted cyclohexanamines with high optical purity. Tetrahedron Lett. 1981;22:2633–2636. [Google Scholar]; (b) Knupp G, Frahm AW. Synthesis and absolute configuration of 2-substituted cyclohexylamine. Chem. Ber. 1984;117:2076–2098. [Google Scholar]
- 30.Handrock R, RaoSchymanski R, Klugbauer N, Hofmann F, Herzig S. Dihydropyridine enantiomers block recombinant L-type Ca2+ channels by two different mechanisms. J. Physiol. 1999;521.1:31–42. doi: 10.1111/j.1469-7793.1999.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.