

Abstract
Background
As a major economic trait in chickens, egg weight (EW) receives widespread interests in breeding, production and consumption. However, limited information is available for underlying genetic architecture of longitudinal trend in EW. Herein, we measured EWs at nine time points from onset of laying to 60 week of age, and conducted comprehensive genome-wide association studies (GWAS) in 1,534 F2 hens derived from reciprocal crosses between White Leghorn and Dongxiang chickens.
Results
Egg weights at all ages except the first egg weight (FEW) exhibited high SNP-based heritability estimates (0.47 ~ 0.60). Strong pair-wise genetic correlations (0.77 ~ 1.00) were found among all EWs. Nine separate univariate genome-wide screens suggested 73 signals showing significant associations with longitudinal EWs. After multivariate and conditional analyses, four variants on three chromosomes remained independent contributions. The minor alleles at two loci exerted consistent and positive substitution effects on EWs, and other two were negative. The four loci together accounted for 3.84 % of the phenotypic variance for FEW and 7.29 ~ 11.06 % for EWs from 32 to 60 week of age. We obtained five candidate genes, of which NCAPG harbors a non-synonymous SNP (rs14491030) causing a valine-to-alanine amino-acid substitution. Genome partitioning analysis indicated a strong linear correlation between the variance explained by each chromosome and its length, which provided evidence that EW follows a highly polygenic nature of inheritance.
Conclusions
Identification of significant genetic causes that together implicate EWs at different ages will greatly advance our understanding of the genetic basis behind longitudinal EWs, and would be helpful to illuminate the future breeding direction on how to select desired egg size.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1945-y) contains supplementary material, which is available to authorized users.
Keywords: Longitudinal egg weights, GWAS, SNP-based heritability, Polygene, Chicken
Background
In poultry industry, egg weight has always been an economically important trait, and is usually regarded as the major breeding objective and research goal [1–4]. As a sensory feature at first glance, egg weight demonstrates a critical impact on consumption due to diverse preferences towards size worldwide [5, 6]. For breeders, egg weight has been reported to affect chick quality including hatching weight, fitness and performance [7, 8]. Notably, owing to the significant effect of hen age, egg weight shows consecutive increase during the whole laying period [9]. Eggs with extreme size in the later stage may obstruct automatic packing and consumption. Therefore, investigation of the genetic architecture underlying egg weights at different ages would have both economic and biological importance.
Multiple lines of evidence have suggested that egg weights are mainly determined by genetic factors and they all show considerable heritability estimates [4, 10, 11]. Currently, 77 quantitative trait loci (QTL) involved in egg weight, located on 17 different chromosomes, have been deposited in the AnimalQTLdb (http://www.animalgenome.org/cgi-bin/QTLdb/index). However, a majority of reported QTLs are mapped with wide confidence intervals by low-density linkage analysis in the past [12]. In general, it is very difficult to identify potential causal variants in QTL mapping studies, mainly due to the relatively small number of recombinants generated from two original parents in a limited number of generations [13]. To improve the precision of gene-level mapping, genome-wide association study (GWAS) based on linkage disequilibrium (LD) between SNPs and causal QTLs/genes is proposed as a more powerful approach identifying genetic links between phenotypes and genotypes [14, 15]. So far, two leading studies have successfully refined associated intervals affecting egg weight via moderate-density SNP chips [3, 16]. Recent advances in next-generation sequencing technologies enable the discovery of a large number of SNP markers as well as the development of high-density SNP platforms [17]. In chickens, the availability of 600 K Affymetrix Chicken SNP array could contribute to narrowing down candidate genomic segments and pinpointing several dominating causal variants.
Despite the great advantages of GWAS, most studies to date utilized only phenotypes measured at a single time point [3, 16, 18–20]. In practical genetic studies, some traits can be measured repeatedly over a period of time, and how to analyze these longitudinal data has received increasing attention [21–23]. Recently, the longitudinal GWAS has been proposed to assess whether some significant SNPs are associated with the process that a trait develops over time [21, 24]. Numerous studies have provided growing evidence that the longitudinal design could offer the opportunity for the identification of time-dependent and consistent loci [25, 26]. Moreover, joint analyses for measurements at multiple time points could increase the statistical power over cross-sectional approach [27, 28], owing to their effectiveness in incorporating the correlation structure of multiple measurements and alleviating the multiple testing burden [21, 29, 30]. Genome-wide assessment of longitudinal egg weight data would be beneficial to distinguish between genetic contributions reflecting constant and specific effects.
In the present study, we implemented univariate, multivariate and conditional GWASs using 600 K Affymetrix Chicken SNP array in a total of 1,534 F2 chickens with observations for egg weights at different ages. In addition, we examined the genetic architecture of egg weight by partitioning genetic variation according to chromosome. The main goals of our current work were to assess the feasibility of longitudinal data as an intriguing resource in GWAS, pinpoint associated loci and genes that contribute to the phenotypic variability and longitudinal trend in egg weight, and provide valuable insights into the genetic basis of longitudinal egg weights.
Results
Phenotype statistics and genetic parameters
Descriptive statistics for nine egg weight variables are presented in Table 1. We observed that egg weight displayed a curvilinear increase with advancing hen age. Particularly, all phenotypic values conformed to the normal distribution after rank-based inverse normal transformation, and these transformed values were used for all primary analyses. We quantified the additive genetic variations in liability to egg weights at different ages captured by all eligible GWAS markers. Univariate GCTA analyses revealed that all egg weight traits, except first egg weight (FEW), had highly heritable patterns (Table 2), and the highest SNP-based heritability estimate was found in EW36 (h2snp = 0.60). Moreover, bivariate GCTA analyses indicated that egg weights at multiple ages were highly and positively interrelated, especially for egg weights at two neighboring time points. As the beginning of entire laying stage, FEW showed slightly lower genetic correlations with egg weights at the following ages (rg < 0.90), compared with those among egg weights from 32 to 60 week of age (rg > 0.90).
Table 1.
Descriptive statistics for nine egg weights in the F2 population
| Trait (g)a | N | Mean | SD | CV (%) | Min | Max |
|---|---|---|---|---|---|---|
| FEW | 1,477 | 35.80 | 3.86 | 10.80 | 25.00 | 49.72 |
| EW32 | 1,473 | 45.81 | 3.49 | 7.62 | 24.99 | 57.23 |
| EW36 | 1,464 | 47.44 | 3.83 | 8.06 | 37.07 | 62.20 |
| EW40 | 1,476 | 48.66 | 3.78 | 7.77 | 38.80 | 64.55 |
| EW44 | 1,420 | 49.44 | 4.06 | 8.22 | 37.68 | 63.82 |
| EW48 | 1,226 | 51.40 | 4.14 | 8.06 | 39.11 | 65.76 |
| EW52 | 1,225 | 51.79 | 4.44 | 8.56 | 36.94 | 69.85 |
| EW56 | 1,348 | 53.05 | 4.43 | 8.36 | 39.70 | 67.74 |
| EW60 | 1,336 | 53.13 | 4.53 | 8.53 | 38.27 | 71.71 |
N number of samples, Mean arithmetic mean, SD standard deviation, CV coefficient of variance, Min minimum, Max maximum
aFEW = first egg weight; EW32, EW36, EW40, EW44, EW48, EW52, EW56, EW60 = egg weight per four weeks from 32 to 60 week of age
Table 2.
Summary of genetic analysis for egg weights at different ages
| Traita | FEW | EW32 | EW36 | EW40 | EW44 | EW48 | EW52 | EW56 | EW60 |
|---|---|---|---|---|---|---|---|---|---|
| FEW | 0.36 (0.04) | 0.89 (0.04) | 0.91 (0.04) | 0.79 (0.05) | 0.87 (0.05) | 0.82 (0.05) | 0.86 (0.05) | 0.77 (0.06) | 0.80 (0.05) |
| EW32 | 0.53 | 0.59 (0.04) | 1.00 (0.01) | 0.98 (0.01) | 1.00 (0.01) | 0.99 (0.01) | 0.95 (0.02) | 0.94 (0.02) | 0.93 (0.02) |
| EW36 | 0.48 | 0.78 | 0.60 (0.04) | 0.97 (0.01) | 0.99 (0.01) | 0.99 (0.01) | 0.96 (0.02) | 0.94 (0.02) | 0.95 (0.02) |
| EW40 | 0.44 | 0.78 | 0.76 | 0.54 (0.04) | 1.00 (0.01) | 0.98 (0.01) | 0.96 (0.02) | 0.97 (0.02) | 0.96 (0.02) |
| EW44 | 0.44 | 0.71 | 0.7 | 0.74 | 0.47 (0.04) | 0.99 (0.01) | 0.97 (0.02) | 0.99 (0.02) | 0.99 (0.02) |
| EW48 | 0.43 | 0.74 | 0.72 | 0.76 | 0.74 | 0.56 (0.05) | 0.98 (0.01) | 1.00 (0.02) | 1.00 (0.01) |
| EW52 | 0.45 | 0.71 | 0.71 | 0.74 | 0.72 | 0.78 | 0.56 (0.05) | 1.00 (0.01) | 0.99 (0.01) |
| EW56 | 0.39 | 0.67 | 0.67 | 0.72 | 0.7 | 0.75 | 0.76 | 0.49 (0.05) | 1.00 (0.01) |
| EW60 | 0.41 | 0.68 | 0.67 | 0.71 | 0.69 | 0.74 | 0.76 | 0.76 | 0.53 (0.05) |
Diagonal: heritability estimates. Lower triangle: phenotypic correlations. Upper triangle: genetic correlations. Standard errors of the estimates are in parentheses
aFEW = first egg weight; EW32, EW36, EW40, EW44, EW48, EW52, EW56, EW60 = egg weight per four weeks from 32 to 60 week of age
Identification of candidate loci by GWAS
As a preliminary, we conducted nine separate association tests using univariate method for longitudinal egg weights. In total, 73 genome-wide significant SNPs located on chromosome 1, 3 and 4 (GGA1, GGA3 and GGA4) were successfully identified for these nine traits (Additional file 1: Table S1). These excess signals of association were entirely attributable to two chromosomal regions (GGA1: 167.4 ~ 174.4 Mb and GGA4: 73.1 ~ 77.2 Mb) and a single locus (rs316497033 on GGA3). Of all significant association signals, 45 SNPs were responsible for only one trait and other 28 hits affected multiple phenotypes. In particular, four loci around rs14491030 were found to be implicated in all egg weights except FEW. The global view of the putative P-values for all SNPs affecting EW36 is given in Fig. 1, and the Manhattan and quantile-quantile (QQ) plots for the remaining traits are provided in Additional file 2: Figure S1. The genome-wide discovery analyses yielded a small genomic inflation factor (λ) for each egg weight, ranging from 0.981 to 1.014. After adjustment by sample size, the standardized λ1000 values varied from 0.987 to 1.010. These results indicated negligible inflation of the observed genome-wide association signals caused by population stratification. Notably, moderate deviations within the upper tail of the distribution were found in all QQ plots, in spite of stringent quality control and inclusion of up to top five PCs. Thus, the departure due to a great number of weakly associated SNPs was indicative of polygenic inheritance.
Fig. 1.

Manhattan plot (left) and quantile-quantile plot (right) of the observed P-values for egg weight at 36 weeks of age (EW36). The Manhattan plot indicates -log10 (observed P-values) for genome-wide SNPs (y-axis) plotted against their respective positions on each chromosome (x-axis), and the horizontal green and black lines depict the genome-wide suggestive (1.69 × 10−5) and significant (8.43 × 10−7) threshold, respectively. For quantile-quantile plot, the x-axis shows the expected -log10-transformed P-values, and the y-axis represents the observed -log10-transformed P-values. The raw and adjusted genomic inflation factors (λ and λ1000) are shown on the top left in the QQ plot
In total, we obtained 270 genome-wide suggestive loci from nine independent univariate analyses. To enhance the statistical power, a joint GWAS analysis of nine traits was performed by fitting these SNPs into a multivariate model. Consequently, a total of seven significant hits on GGA1 and GGA4 provided compelling evidence for associations with longitudinal egg weights (Additional file 1: Table S2). We then performed stepwise conditional GWASs to prioritize separately associated SNPs owing to the potentially strong linkage disequilibrium (LD) between neighboring variants. After two-round adjustment in the multi-traits GWASs, two significant SNPs, rs314058619 on GGA1 and rs14491030 on GGA4, were uncovered to be independent signals. Meanwhile, another locus, rs14916609 (Padjust = 8.15 × 10−5) on GGA1, survived the genome-wide suggestive threshold of multivariate model. Considering that rs314058619 and rs14916609 are separated by 1.5 Mb and the pair-wise LD between them is relatively low at D’ = 0.37 and r2 = 0.09 (a LD block extends only for a short distance), we treated rs14916609 as another separate signal. Taken together, we obtained three significantly independent loci associated with all egg weights from multivariate analysis.
After that, we repeated conditional analyses in the single-trait model, through including the three aforementioned alleles as covariates in an orderly manner (rs314058619, rs14491030 and rs14916609). After two-step univariate conditional tests on rs314058619 and rs14491030, the third SNP rs14916609 was still significantly associated with all traits except FEW. This locus seemed to have independent effects on these traits, despite that it showed only suggestive significance inferred by two-round multivariate conditional GWASs. The finding further strengthened the validity regarding rs14916609 as an independent hit. It should be noted that the significance levels of loci nearby the peak signals were substantially attenuated after adjusting for the three independent hits. In order to provide visual support of independent associations, we interrogated the signals in the 1.0 Mb genomic region surrounding the independent loci (500 kb each side). Since rs14916609 and rs14491030 affected more phenotypes, we illustrated their regional association plots for EW36 and EW60 to compare the difference of putative significance levels before (upper panel in Fig. 2) and after (lower panel in Fig. 2) conditioning on the two hits. Moreover, conditional analyses on three aforementioned hits revealed that an additional SNP rs316497033 on GGA3 remained significantly involved in EW56. Therefore, we performed further analyses conditional on the genotype at rs316497033 site and found no more genome-wide significant signals. Finally, the four loci were considered as independent associations with longitudinal egg weights after a set of three GWASs.
Fig. 2.
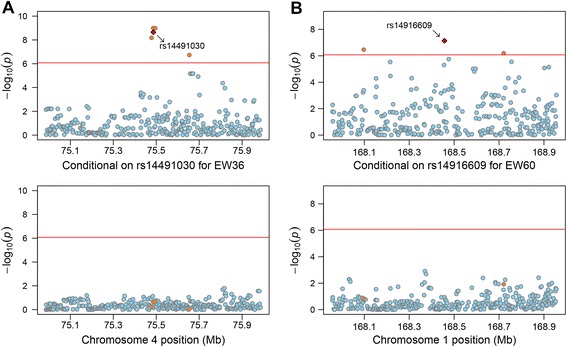
Regional association plots of two loci associated with EWs at 36 and 60 week of age (EW36 and EW60). For each plot, the -log10 (observed P-values) of SNPs (y-axis) are presented according to their chromosomal positions (x-axis). The horizontal red line depicts the genome-wide significance level (8.43 × 10−7). The significant SNP after univariate, multivariate and conditional GWASs is represented by a red diamond and is labeled by its rs number. a Regional association results for EW36 before (upper) and after (lower) conditioning on rs14491030. b Regional association results for EW60 before (upper) and after (lower) conditioning on rs14916609
Allelic contribution to phenotypic variation
For the four resulting loci, the allelic substitution effects and phenotypic variance explained by them were estimated for nine egg weight traits (Table 3). The minor allele at each locus is treated as the effect allele according to the GEMMA definition. The effect alleles at two SNPs, rs314058619 and rs14491030, were associated with the increase in egg weights at each time point, as opposite to other two variants. The genetic effects of the each allele at multiple ages had the same direction but slightly discordance, suggesting the presence of potential time-dependent impact. Through fitting the four variants into mixed model simultaneously, they together explained 3.84 % of the phenotypic variance for FEW and 7.29 ~ 11.06 % for EW from 32 to 60 week of age. Notably, rs14491030 exhibited the largest allelic substitution effect on egg weight at each age. At nine different ages, substituting one copy of allele A by allele G at rs14491030 site would cause 0.444 to 0.618 SD/allele increase in egg weight, in which the SD represents the standard deviation of EW at corresponding age. The corresponding phenotypic variance explained by this variant ranged from to 1.32 to 3.88 %. We compared the actual phenotypic difference between three genotypes at each locus and found that the three phenotypes showed significant segregation (Fig. 3). The comparative results revealed that the homozygote of effect allele possessed the highest or lowest egg weight at the same time point and the heterozygote are medium, suggesting that individual phenotype was more severely affected by homozygous effect allele. It should be noted that the homozygote of effect allele at rs14491030 site showed unstable EWs with advancing hen ages compared with the heterozygote and the homozygote of alternative allele, and other three SNPs yielded concordant change trends for egg weights at multiple ages.
Table 3.
Contributions of four associated SNPs to egg weights at different ages
| SNP | rs14916609 | rs314058619 | rs316497033 | rs14491030 | |
|---|---|---|---|---|---|
| Chromosome | GGA1 | GGA1 | GGA3 | GGA4 | |
| Position (bp)a | 168,456,799 | 169,930,974 | 23,254,004 | 75,486,534 | |
| EA/AA | G/A | T/G | A/G | G/A | |
| EAF | 0.41 | 0.47 | 0.21 | 0.06 | |
| FEW | beta (SE)b | −0.201 (0.051) | 0.095 (0.047) | −0.129 (0.055) | 0.366 (0.086) |
| CPV (%)c | 1.92 | 0.30 | 0.55 | 1.32 | |
| EW32 | beta (SE) | −0.284 (0.051) | 0.204 (0.046) | −0.132 (0.054) | 0.601 (0.082) |
| CPV (%) | 3.75 | 2.10 | 0.57 | 3.88 | |
| EW36 | beta (SE) | −0.259 (0.053) | 0.135 (0.047) | −0.200 (0.055) | 0.499 (0.083) |
| CPV (%) | 2.92 | 0.82 | 1.42 | 2.50 | |
| EW40 | beta (SE) | −0.269 (0.051) | 0.167 (0.046) | −0.204 (0.054) | 0.567 (0.081) |
| CPV (%) | 3.45 | 1.49 | 1.68 | 3.51 | |
| EW44 | beta (SE) | −0.283 (0.053) | 0.189 (0.047) | −0.195 (0.056) | 0.503 (0.085) |
| CPV (%) | 3.62 | 1.73 | 1.39 | 2.74 | |
| EW48 | beta (SE) | −0.266 (0.057) | 0.270 (0.051) | −0.190 (0.061) | 0.455 (0.092) |
| CPV (%) | 3.00 | 3.43 | 1.22 | 2.03 | |
| EW52 | beta (SE) | −0.303 (0.056) | 0.258 (0.050) | −0.277 (0.060) | 0.549 (0.092) |
| CPV (%) | 4.02 | 3.13 | 2.65 | 3.09 | |
| EW56 | beta (SE) | −0.283 (0.054) | 0.271 (0.048) | −0.317 (0.057) | 0.453 (0.087) |
| CPV (%) | 3.63 | 3.54 | 3.38 | 2.12 | |
| EW60 | beta (SE) | −0.286 (0.053) | 0.241 (0.048) | −0.272 (0.057) | 0.469 (0.085) |
| CPV (%) | 3.81 | 2.89 | 2.53 | 2.31 | |
EA effect allele (minor allele), AA alternative allele (major allele), EAF effect allele frequency, FEW first egg weight, EW32, EW36, EW40, EW44, EW48, EW52, EW56, EW60 egg weight per four weeks from 32 to 60 week of age
aAlleles are indexed to the forward strand of Galgal4 assembly
bEstimated allelic substitution effect per copy of the effect allele (EA) based on inverse-normal transformed scale under an additive model, expressed in SD unit/allele; SE = standard error of the beta
cCPV = contribution to phenotypic variance (%)
Fig. 3.
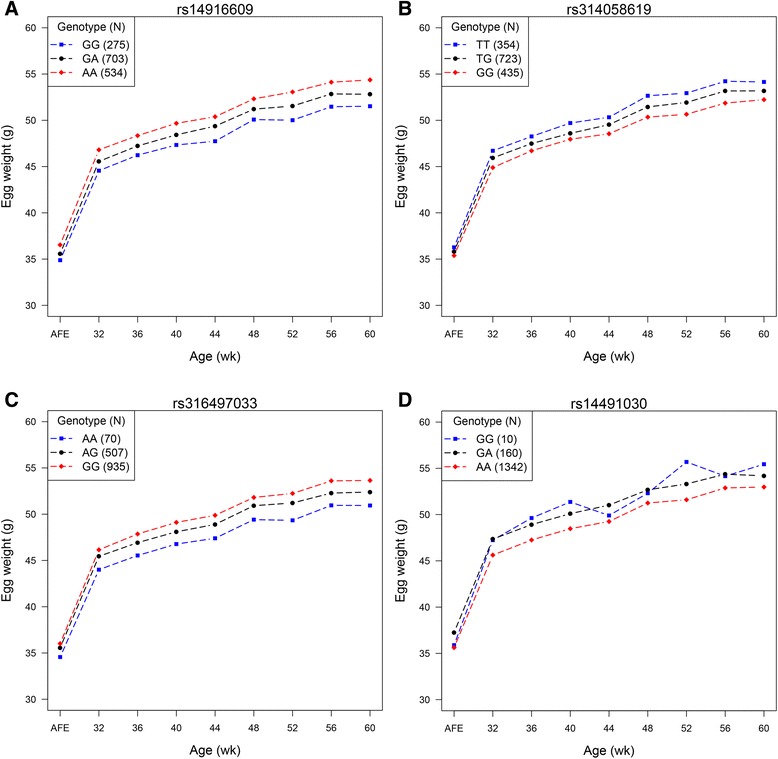
Consistent phenotypic differences contributed by four significant SNPs. Blue square, black circle and red diamond denote minor-allele homozygote, heterozygote and major-allele homozygote, respectively. Number of samples for each genotype is indicated in the top left corner. a Segregating phenotypes between three genotypes at rs14916609. b Segregating phenotypes between three genotypes at rs314058619. c Segregating phenotypes between three genotypes at rs316497033. d Segregating phenotypes between three genotypes at rs14491030
Annotation of significant SNPs
Gene annotation of the four significant SNPs allowed us to identify genes related to longitudinal egg weights. Considering that the putative variants may be in high LD with a causal locus genuinely associated with phenotype, we inferred the LD block around the sentinel SNP using Haploview. Four blocks containing the four hits were identified and the detailed information is summarized in Table 4. To investigate the functional characteristics of the four blocks, we scanned these regions with Biomart system. Five different genes overlapping with the blocks were integrated into final gene set. The VEP tool revealed that the four SNPs were encompassed by four aforementioned genes, and three of them lies within introns and one resides in exon (Table 4). Notably, a SNP, rs14491030 significantly present at the lowest minor allele frequency, is a non-synonymous mutation in exon 14 of NCAPG and mediates a valine-to-alanine amino-acid substitution (V674A). According to the putative SIFT score (0.74), the change of amino-acid was predicted as tolerated to protein structure and function.
Table 4.
Genomic regions surrounding four significant SNPs
| Tag SNP | Chromosome | Length of block (kb) | SNP number in the block | Nearest genea | Ensembl gene ID | Location (kb)b |
|---|---|---|---|---|---|---|
| rs14916609 | GGA1 | 57.9 | 19 | CAB39L | ENSGALG00000017005 | Intron |
| CDADC1 | ENSGALG00000017004 | Downstream_18.2 | ||||
| rs314058619 | GGA1 | 4.4 | 2 | FKHR (FOXO1) | ENSGALG00000017034 | Intron |
| rs316497033 | GGA3 | 15.2 | 8 | KCNG3 | ENSGALG00000009919 | Intron |
| rs14491030 | GGA4 | 17.2 | 9 | NCAPG | ENSGALG00000014425 | Exon 14 |
aIdentification of the gene according to Ensembl genes database 76
bDistance in kb of SNP for the gene
Genome partitioning of genetic variation
We implemented an exploratory analysis through partitioning the genetic variation onto chromosome segments to further illustrate the genetic architecture of egg weight. Owing to relatively small sample size in the F2 population, parameter estimates for some traits in the joint model could not converge. However, the strong genetic correlations between nine egg weight phenotypes provided evidence that they had great potential to share a consistent genetic architecture. Therefore, we only exemplified the partitioning spectrum of EW36 due to its highest heritability. The estimates of variance contributed by each chromosome exhibited a strong linear relationship with the length of the chromosome for EW36 (R2 = 0.695, Fig. 4a), and no chromosome was found to show exceptional contribution. For three chromosomes, GGA1, GGA3 and GGA4, each of them explained more than 5.00 % of phenotypic variance, demonstrating higher genetic contributions than other chromosome segments (Fig. 4b). To quantify the effects of the four resulting variants on EW36, we fitted these SNPs as covariates and repeated the genome partitioning analysis. When compared with the results before adjustment, we found that the variance explained by GGA1 dropped from 9.01 to 6.39 % (Fig. 4b). The same estimate for GGA4 showed the largest decrease from 7.81 to 4.17 %. Particularly, a slight reduction in the estimated proportion of variance captured by GGA3 was found, and the estimates for other 27 chromosomes almost remained the same.
Fig. 4.
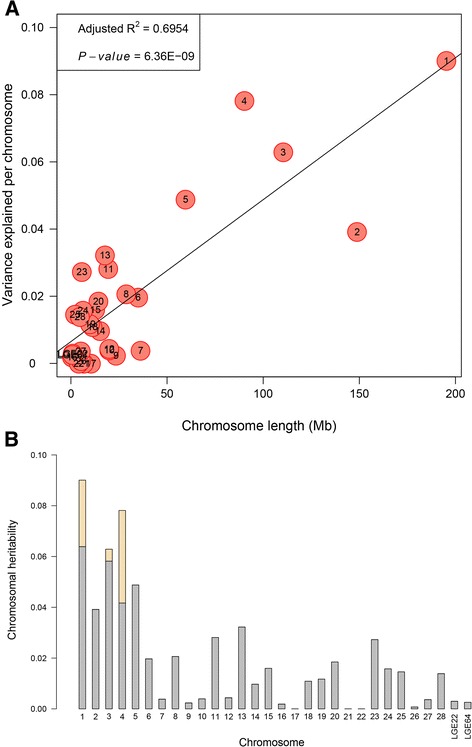
Genome partitioning for egg weight at 36 weeks of age (EW36) by joint analysis. a The estimated proportion of variance captured by each chromosome against its size. The characters in the circles are the chromosome numbers. b Contributions of GWAS SNPs partitioned by chromosome. The whole bars indicate the estimates of variance explained by each chromosome, in which the three wheat bars represent the same values by four resulting loci
Discussion
Although many genetic contributions to egg weight at a single time point have been unearthed by general GWAS approach [1, 3, 16], less is known about genetic architecture of longitudinal egg weights. To elucidate potential genetic loci that affect egg weight over time, we conducted comprehensive genome-wide association analyses for egg weights at multiple time points in an F2 segregating population, by utilizing the high-density 600 K SNP arrays. The current work not only provided a pioneering genome-wide association scan shedding light on longitudinal egg weight data, but also could deepen our understanding of genetic architecture underlying how egg weight develops over time. Through incorporating the associated loci into human-engineered breeding programs, we can select eggs with proper sizes to meet consumer needs, and help to predict the weight change in the following production.
Currently, conducting GWAS in an F2 population has been a routine approach to detect causal loci and genes [18, 20, 31]. GWAS approach is generally based on LD patterns in experimental populations, and the population structure is an important factor influencing the extent and range of LD [32]. Three previous studies [20, 33, 34] suggested that crossbred populations show a smaller extent and rapider decay of LD by distance as well as smaller haplotype block size than pure lines. The lower level of LD could enable that the associated SNPs are located in close proximity to the casual locus due to the tight linkage between them. Therefore, the F2 population and high-density array in our study would be beneficial for high-resolution mapping of genuinely causal loci.
All QQ plots displayed moderate leftward deflections of the observed distribution, suggesting the presence of numerous weakly associated variants. This phenomenon is often attributed to “spurious inflation”, and would be expected under polygenic architecture [35, 36]. Genetic analyses by GCTA tool revealed highly heritable patterns of egg weight. The high SNP-based heritability estimates showed that a substantial proportion of phenotypic variance could be captured by eligible GWAS SNPs, providing compelling support for polygenic model. Meanwhile, the strong genetic correlations between nine egg weight phenotypes indicated that these traits may share the similar genetic components or be influenced by some pleiotropic genomic regions [37, 38]. The results were also supported by the fact that the four independent SNPs exerted consistent effects on all egg weights. In poultry industry, maintaining good stability without significant increase after laying peak is another great concern [3, 4]. We previously conducted a genetic analysis for the increment traits of egg weight with advancing hen age [4], in which the increment of EW was defined as the difference between two EWs at neighboring ages. The low heritability estimates reflect that EWs at different ages may share similar genetic causes, and calculating the difference values may eliminate the same additive effects between them. Therefore, according to the two studies by us, how to improve the stability in egg weight seemed to be a considerable challenge by general genetic methods.
To further decipher the heritable architecture in egg weight, we partitioned the genetic variance onto different chromosomes based on estimated chromosomal GRMs [39, 40]. Notably, three macrochromosomes GGA1, GGA3 and GGA4 accounted for the largest genetic variance (>5.00 %), producing corroborative support for the GWAS results that four loci on them were identified as significant associations with egg weights. Moreover, a strong and positive linear correlation between variance explained by chromosomes and their length was found, suggesting the presence of a large number of genomic loci mediating weak effects [35]. The finding again consolidated the polygenic pattern in egg weight, and was consistent with other traits in animals [41, 42] and human [40, 43, 44]. The result reflected that longer chromosomes are likely to occupy more informative and effective markers, leading to a polygenic nature of egg weight [39]. Overall, despite that there were four SNPs with relatively large effects contributing to longitudinal egg weights, and our results demonstrated that many genetic variants each with a small effect widely spread across the whole genome. To pursue additional genetic loci with small or modest effect sizes, a larger population and high-throughput sequencing technology would be required.
Compared with some previous strategies leveraging only cross-sectional phenotype data [3, 16, 18–20], we took advantage of longitudinal egg weight data at nine time points. In total, we captured four promising loci with strong evidence for influencing egg weight over time. While we failed to detect some time-dependent variants, again suggesting that egg weight at different stages was likely to be affected by the same genetic loci. Regarding the four SNPs, only rs14491030 was discovered to be responsible for all phenotypes except FEW via nine univariate analyses. This result indicated that the separate use of cross-sectional measurements might miss several loci which had weak effect sizes at one time point but large effects at other stages [26]. Cross-sectional surveys only identified time-dependent loci, but not provided insights into how such factors influenced traits over time [27, 45]. The longitudinal method was capable of permitting the investigation of genetic variants with consistent effects [26, 46]. In addition, univariate analysis for EW56 discovered a novel effective locus missed by multivariate method. The reason could attribute to smaller effect sizes of this mutation at other ages that had low power to uncover it. Moreover, it should be noted that multivariate test only used individuals with intact data records at each time point [29]. Considering that the number of analyzed individuals was 783 in multi-trait analysis, only one half of total sample size, thus some missing phenotypes at different ages may have a negative impact on the investigation of candidate variants [29, 47]. In consequence, to enhance the power detecting variants with consistent effects, we must enable more complete phenotypic information for each individual.
In total, four LD blocks tagged by the resulting SNPs were inferred, and contained five candidate genes of potentially functional relevance. Two SNPs (rs14916609 and rs314058619) on GGA1 are segregated by 1.5 Mb and located in the intronic regions of CAB39L (calcium binding protein 39-like) and FKHR (forkhead box O1, also known as FOXO1), respectively. The CAB39L gene has been proposed to participate in the mTOR signaling pathway. It is increasingly apparent that mTOR signaling can affect most cellular functions as a central controller of cell growth (mass accumulation) and proliferation [48, 49]. Numerous studies have reported that FKHR is a member of FOXO subfamily and an important mediator of the insulin signaling pathway [50, 51]. This pathway could stimulate protein synthesis and cell growth by activation of mTOR [52]. For birds, the unfertilized yolk is a single cell (ovum), and the deutoplasm part will be gradually accumulated during oogenesis. The polymorphisms in the two genes may result in the change of egg weight through influencing the deposition of yolk and egg white.
A non-synonymous mutation (rs14491030) located within the NCAPG (non-SMC condensin I complex, subunit G) was unveiled in the current study. In chickens, many independent studies reported that multiple genomic segments containing this gene were identified to be implicated in both egg weight and body weight [1, 3, 16, 19, 20, 53]. However, a recent study revealed that another candidate gene CCKAR (cholecystokinin A receptor) located around 2.7 Mb away from NCAPG is mainly responsible for body weight [54], by establishing a 16-generation advanced intercross line (AIL) between a fast and a relatively slow growing strain. Because accumulated recombination events in the AIL are expected to provide better resolution for QTL mapping [55], CCKAR might be a more promising causal gene. Notably, the gene NCAPG has been suggested to be associated with many growth and weight traits in several livestock species [56–60]. Therefore, we could not ignore the potentially conserved function of NCAPG for body weight in chicken, owing to a particularly interactive relationship between egg weight and body weight [7, 61], i.e., the larger egg would cause higher hatch weight and the heavier chicken would produce larger eggs. We speculated that NCAPG gene may exert a pleiotropic effect involved in the two phenotypes simultaneously. Consequently, the impact on body weight of the gene for egg weight identified by us requires further investigation.
Another gene CDADC1 (cytidine and dCMP deaminase domain containing 1) is located about 18.2 kb upstream of SNP rs14916609, but its function and mechanism is not yet clear. One previous study indicated that this gene may be an important factor regulating testicular development and spermatogenesis in human [62]. Due to its function on reproductive performance, we suspected that the gene may highlight a potential role on oogenesis. On GGA3, a gene KCNG3 (potassium voltage-gated channel, subfamily G, member 3) indexed by rs316497033 encodes a subunit of the potassium voltage-gated channel. It is known to be mainly involved in cell volume and smooth muscle contraction [63], and these biological functions may reflect potential connection with changed egg weight due to different alleles.
Conclusions
In summary, we performed univariate, multivariate and conditional GWASs for longitudinal egg weight data using high-density 600 K SNP arrays, and suggested that longitudinal analysis had higher power to dig out variants that influence phenotype variability over time. Our study provided evidence that egg weight appears to be highly heritable and polygenic, and shares the similar genetic determinants and mechanisms at different ages. Four significant loci and five candidate genes were detected with significant effect on egg weights. In particular, NCAPG may influence both egg weight and body weight simultaneously in a pleiotropic manner. These promising loci and genes could be helpful to engineer practical breeding programs and produce the desired egg size according to diverse consumption markets.
Methods
Resource population
An F2 resource population was developed by reciprocal crosses between White Leghorn (WL) and Dongxiang chickens (DX), representing a popular commercial breed and a Chinese indigenous strain, respectively. The two pure lines were maintained for about ten years at the experimental farm, and WL eggs weighed about 14 g more than DX eggs at the same age. Six WL males were mated with 133 DX females and six DX males were mated with 80 WL females, to generate 1,029 and 552 birds of the F1 generation, respectively. Then, 25 males and 407 females from WL/DX cross and 24 cocks and 235 hens from DX/WL cross were selected to produce the F2 population. A total of 3,749 F2 individuals (1,893 pullets and 1,856 cockerels) with full pedigrees originating from 590 full-sib families were created in the same hatch. The population was maintained in three-tier single-hen cages and reared in the same environment with feed and water ad libitum at the experimental farm of Jiangsu Institute of Poultry Science. Finally, to ensure sufficient phenotypic information and accurate pedigree, we chose 1,534 hens from 49 half-sib families and 550 full-sib families for SNP genotyping.
Phenotypic measurements
To characterize the genetic architecture of egg weight developmentally, we collected egg weight data at nine different time points. Firstly, the first egg weight (FEW) was recorded when hens started laying, mainly considering that FEW is conceptually defined as weight of first egg for each hen, and it is very difficult to collect eggs on several consecutive days due to the physiologically unstable state of hens at onset of laying. Subsequently, egg weights (EW) were measured on two consecutive days per four weeks from 32 to 60 week of age, and the average for two days was regarded as the phenotypic value for each hen. Cracked, soft-shell and double-yolk eggs were not used in our study. Descriptive statistics were calculated with the MEANS procedure of the SAS software package (SAS Institute Inc., 2006) using all available records. For traits deviating from normality, we conducted the rank-based inverse normal transformations (INTs) using SAS ahead of association tests [64]. Briefly, the procedure involves first transforming all observations to ranks and then converting these ranks to follow a standard normal distribution with a mean of 0 and an SD of 1. These transformed values were then used for the downstream analyses including GWAS discovery and heritability estimation.
Genotyping, quality assurance and imputation
The whole blood samples were collected from brachial veins of chickens by standard venipuncture. Genomic DNA was isolated using standard phenol/chloroform extraction method and genotyped with the 600 K Affymetrix Axiom Chicken Genotyping Array (Affymetrix, Inc. Santa Clara, CA, USA). From an initial set of 580,954 SNPs [17], 7,883 SNPs with unknown genomic positions and 112 SNPs with redundant genomic coordinates were removed. Subsequently, first-pass quality control and genotype calling from the raw data in the form of CEL files were implemented with Affymetrix Power Tools v1.16.0 (APT) software using the Axiom GT1 algorithm. Only samples with dish quality control (DQC) of 0.82 or better and call rate > 97 % were included into the downstream analyses. An R script supplied by Affymetrix was run to compute the SNP QC metrics and filter out individual SNPs falling below given thresholds. All parameters were set to the default values recommended by Affymetrix. After these QC steps, 1,512 samples and 532,299 SNPs remained. In addition, we excluded 6,402 SNPs on sex chromosomes considering that current statistical methods are more powerful to detect the associations between phenotypes and autosomal genotypes. To improve the power of association analyses, we dropped 67,330 SNPs with minor allele frequency (MAF) < 5 % and 22,700 SNPs deviating from Hardy-Weinberg equilibrium (HWE) test P < 1 × 10−6 using the PLINK v1.90 package [65]. Some sporadic missing genotypes were imputed using the BEAGLE v4.0 procedure [66], then only SNPs with imputation quality score R2 > 0.5 underwent the next analyses step. Finally, a total of 1,512 samples and 435,867 SNPs were eligible for inclusion in the following GWASs.
Association analysis
Prior to GWAS, we conducted a principal component analysis (PCA) to eliminate spurious associations due to the presence of potential cryptic relatedness or hidden population stratification. Considering that clusters of highly correlated SNPs may distort the resulting PCs, we first pruned the full SNP set to 41,130 independent SNPs via the --indep-pairwise 25 5 0.2 command (PLINK), and then calculated top five PCs as covariates into the mixed model. To establish proper thresholds for genome-wide suggestive and significant associations, we corrected for multiple testing using the simpleM method [67] accounting for linkage disequilibrium (LD) relationships among SNPs. Using simpleM we estimated the effective number of independent tests as Meff = 59,308, thus the genome-wide suggestive and significant P-values were 1.69 × 10−5 (1.00/59,308) and 8.43 × 10−7 (0.05/59,308), respectively.
We firstly performed univariate tests of association for SNPs having MAF ≥ 0.05 using an exact mixed model approach implemented in the GEMMA v0.94 software [68]. The centered relatedness matrix was calculated by those independent SNPs for all cases. And then each SNP was tested for additive association with each trait by modeling the effects of genotypes and the additional covariates including top five PCs as fixed effects, and random polygenic effects as random effects, mainly considering that our samples were from highly structured populations with strong family relatedness. The manhattan plot and quantile-quantile (QQ) plot depicting -log10-transformed observed P-values were generated using the “gap” and “qqman” packages in R [69, 70]. We calculated the genomic inflation factor λ to judge the extent of false positive signals [71]. Furthermore, given that the λ becomes larger with increasing of sample size, we also estimated an adjusted inflation factor by standardizing to a sample size of 1,000 [72, 73]. The univariate linear mixed model for each SNP marker and two compute methods for λ are as follows:
| 1 |
| 2 |
| 3 |
for the equation (1), y is an n × 1 vector of phenotypic values for n individuals, W is an n × c matrix of covariates (fixed effects, i.e., top five PCs) including a column vector of 1, α is a c × 1 vector of corresponding coefficients including the intercept, x is an n × 1 vector of marker genotypes at the locus tested, β is the corresponding effect size of the marker and all effects are reported for the minor allele in each marker, u is an n × 1 vector of random polygenic effects with a covariance structure as u ∼ N(0, KVg), where K represents a known n × n genetic relatedness matrix derived from SNP markers and Vg is the polygenic additive variance, and ε is an n × 1 vector of random residuals with ε ∼ N(0, IVe), where I is an n × n identity matrix, and Ve is the residual variance component. We used the Wald test statistic for each SNP to test the null hypothesis β = 0, where the best linear unbiased estimate (BLUE) of β and the corresponding sampling variance are obtained by solving the mixed model equations (MME) based on estimated Vg and Ve.
In the formula (2), T2i is the estimated Wald statistic and asymptotically distributed as a chi-square with 1 degree of freedom under the null hypothesis, and usually called observed χ2 statistics, and 0.455 is the expected median of the standard χ2 distribution under the null hypothesis of no association. For the formula (3), n is the sample size and λ is the estimate from the total samples.
Similarly, we applied a multivariate association analysis that directly models nine measurements on an individual [29], to capture genetic variants affecting the longitudinal egg weights consistently over time. Considering that insignificant SNPs from univariate analyses are not likely to exceed the significance level of multivariate test and handling all SNPs simultaneously would increase computational burden, thus we only analyzed those suggestive SNPs from the univariate results in a multivariate model. For each SNP marker, a multivariate linear mixed model could be fitted in the following form:
| 4 |
where Y is an n by d matrix of d phenotypes for n individuals, W = (w1,⋯,wc) is an n × c matrix of covariates (fixed effects, i.e., top five PCs) including a column of 1 s, A is a c by d matrix of corresponding coefficients including the intercept, x is an n-vector of marker genotypes, β is a d vector of marker effect sizes for the d phenotypes. It should be noted that G is an n by d matrix of random effects with G ~ MVNn×d(0, K, Vg) where Vg is a d by d symmetric positive definite matrix of genetic variance component, and E is an n by d matrix of residual errors with E ~ MVNn×d(0, I, Ve) where Ve is a d by d symmetric positive definite matrix of residual variance component (K and I are the same in the two models). MVNn×d(0, V1, V2) denotes the n × d matrix normal distribution with mean 0, row covariance matrix V1 (n by n) and column covariance matrix V2 (d by d).
Owing to the different number of statistic tests, we further adjusted the significance level in multivariate testing accounting for the nine independent inquiries in usual single-phenotype strategy. For the multivariate model, we corrected the genome-wide suggestive and significant cut-offs as 1.52 × 10−4 and 7.59 × 10−6 through multiplying the raw thresholds by the number of phenotypes [29].
Notably, a genomic region containing a cluster of neighboring SNPs in strong LD is usually associated with a phenotype for high-density array. To demarcate independent association signals across the putative regions, we run stepwise conditional analyses both in multiple and single models, through fitting the genotypes (coded as 0, 1 or 2 alleles) at the strongest signal identified by the multivariate analysis as covariates [74]. The process was repeated until no SNP reached genome-wide significance threshold.
Estimation of variance explained
For egg weight at each time point, univariate restricted maximum likelihood (REML) implemented in GCTA v1.24 program [75] was performed to estimate the heritability explained by the eligible SNPs (h2snp) for GWAS using the same inverse-normal transformed EW values. We also quantified the pair-wise genetic and phenotypic correlations among egg weights at multiple time points with the bivariate mixed model [76]. A genetic relationship matrix (GRM) was derived from all genotyped SNPs on autosomes and two linkage groups, and top five PCs calculated by the GCTA tool were included as covariates to account for the potential population structure. For those genome-wide significant SNPs, we estimated the phenotypic variance contributed by these associated loci in the following mixed linear model:
| 5 |
where y is an n × 1 vector of phenotypic values for n individuals, b is a vector of fixed effects with its incidence matrix X, gG is a vector of aggregate effects of all SNPs, and Var(gG) = AGσ2G with AG being the SNP-derived GRM and σ2G being the additive genetic variance, e is a random residual term with e ∼ N(0, Iσ2e) where σ2e represents the residual variance and I represents an identity matrix. In this model, we partitioned σ2G into the contributions of selected SNP loci () and other variants () of the whole genome, and the contribution to phenotypic variance (CPV) of selected SNPs is defined as CPV with σ2P being the phenotypic variance.
In addition, we partitioned the chicken genome into 28 autosomes and two linkage groups, and jointly estimated their contributions to phenotypic variance for traits of interest using the similar model to formula (5). Before that, we built the GRM for each chromosome and next fitted 30 GRMs simultaneously in a joint analysis [39, 77]. A regression analysis was done by R to evaluate the relationship between the variance explained by each chromosome and its length.
Linkage disequilibrium analysis and genes identification
In general, GWAS does not distinguish a genuine causal locus from those statistically significant loci within a strong LD region. Therefore, in order to characterize potential candidate genes responsible for a trait, we conducted LD analyses and inferred the haplotype blocks containing peak SNPs by Haploview v4.2 [78]. A block is derived using the solid spine algorithm, and defined as that the first and last SNPs in a region are in strong LD (D’ ≥0.8) with all intermediate SNPs, but the intermediate SNPs are not necessarily in LD with each other. Subsequently, we performed functional annotation on the significant SNPs and searched for candidate genes in the blocks based on the Galgal4 assembly, using Variant Effect Predictor (VEP) and Biomart tools supported by Ensembl [79, 80].
Acknowledgements
This work was funded in part by grants of the National High Technology Research and Development Program of China (2013AA102501), National Natural Science Foundation of China (31320103905), Program for Changjiang Scholars and Innovative Research Team in University (IRT1191), and China Agriculture Research Systems (CARS-41).
Abbreviations
- GWAS
Genome-Wide Association Study
- SNP
Single nucleotide polymorphism
- QTL
Quantitative trait loci
- AIL
Advanced intercross line
- INT
Rank-based inverse normal transformation
- LD
Linkage disequilibrium
- REML
Restricted maximum likelihood
- BLUE
Best linear unbiased estimate
- MME
Mixed model equation
- GRM
Genetic relationship matrix
- MAF
Minor allele frequency
- HWE
Hardy-Weinberg equilibrium
- PCA
Principal component analysis
- VEP
Variant Effect Predictor
Additional files
Summary of genome-wide significant SNPs with respect to nine egg weights by univariate GWAS. Table S2. GWAS SNPs identified by nine-trait multivariate analysis. (XLSX 19 kb)
Manhattan plots (left) and quantile-quantile plots (right) for first egg weight (FEW) and egg weights at 32, 40, 44, 48, 52, 56 and 60 wk of age (EW32 and EW40~EW60). The Manhattan plots indicate -log10 (observed P-values) for genome-wide SNPs (y-axis) plotted against their respective positions on each chromosome (x-axis), and the horizontal green and black lines depict the genome-wide suggestive (1.69 × 10-5) and significant (8.43 × 10-7) threshold, respectively. For quantile-quantile plots, the x-axis shows the expected -log10-transformed P-values, and the y-axis represents the observed -log10-transformed P-values. The raw and adjusted genomic inflation factors (λ and λ_1000) are shown on the top left in the QQ plot. (A) ~ (H) indicate egg weights at eight different wk of age, respectively. (PDF 842 kb)
Footnotes
Guoqiang Yi and Manman Shen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NY and KW conceived the study and designed the project. GY, MS, JY, CS and ZD performed genetic and bioinformatics analyses. LQ, TD, MM, JL, JG, SC and LQ contributed to collecting samples and measuring phenotypic data. GY and MS wrote and NY revised the manuscript. All authors read and approved the final manuscript.
Authors' information
Not applicable.
Availability of data and materials
Not applicable.
Contributor Information
Guoqiang Yi, Email: yiguoqiang20@hotmail.com.
Manman Shen, Email: shenman2005@163.com.
Jingwei Yuan, Email: amstrongyuan@163.com.
Congjiao Sun, Email: suncongjiao@163.com.
Zhongyi Duan, Email: dzy806@163.com.
Liang Qu, Email: quliang19820603@163.com.
Taocun Dou, Email: 569843180@qq.com.
Meng Ma, Email: 779701121@qq.com.
Jian Lu, Email: lujian1617@163.com.
Jun Guo, Email: smileleaf@yeah.net.
Sirui Chen, Email: csr@cau.edu.cn.
Lujiang Qu, Email: quluj@cau.edu.cn.
Kehua Wang, Email: sqbreeding@126.com.
Ning Yang, Email: nyang@cau.edu.cn.
References
- 1.Sasaki O, Odawara S, Takahashi H, Nirasawa K, Oyamada Y, Yamamoto R, et al. Genetic mapping of quantitative trait loci affecting body weight, egg character and egg production in F2 intercross chickens. Anim Genet. 2004;35(3):188–94. doi: 10.1111/j.1365-2052.2004.01133.x. [DOI] [PubMed] [Google Scholar]
- 2.Savegnago RP, Caetano SL, Ramos SB, Nascimento GB, Schmidt GS, Ledur MC, et al. Estimates of genetic parameters, and cluster and principal components analyses of breeding values related to egg production traits in a White Leghorn population. Poult Sci. 2011;90(10):2174–88. doi: 10.3382/ps.2011-01474. [DOI] [PubMed] [Google Scholar]
- 3.Wolc A, Arango J, Settar P, Fulton JE, O'Sullivan NP, Preisinger R, et al. Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim Genet. 2012;43(Suppl 1):87–96. doi: 10.1111/j.1365-2052.2012.02381.x. [DOI] [PubMed] [Google Scholar]
- 4.Yi GQ, Liu WB, Li JY, Zheng JX, Qu LJ, Xu GY, et al. Genetic analysis for dynamic changes of egg weight in 2 chicken lines. Poult Sci. 2014;93(12):2963–9. doi: 10.3382/ps.2014-04178. [DOI] [PubMed] [Google Scholar]
- 5.Jones DR, Anderson KE, Davis GS. The effects of genetic selection on production parameters of single comb White Leghorn hens. Poult Sci. 2001;80(8):1139–43. doi: 10.1093/ps/80.8.1139. [DOI] [PubMed] [Google Scholar]
- 6.Koelkebeck KW, Bell DD, Carey JB, Anderson KE, Darre MJ. Egg marketing in national supermarkets: products, packaging, and prices--part 3. Poult Sci. 2001;80(4):396–400. doi: 10.1093/ps/80.4.396. [DOI] [PubMed] [Google Scholar]
- 7.Nangsuay A, Ruangpanit Y, Meijerhof R, Attamangkune S. Yolk absorption and embryo development of small and large eggs originating from young and old breeder hens. Poult Sci. 2011;90(11):2648–55. doi: 10.3382/ps.2011-01415. [DOI] [PubMed] [Google Scholar]
- 8.Enting H, Boersma WJ, Cornelissen JB, van Winden SC, Verstegen MW, van der Aar PJ. The effect of low-density broiler breeder diets on performance and immune status of their offspring. Poult Sci. 2007;86(2):282–90. doi: 10.1093/ps/86.2.282. [DOI] [PubMed] [Google Scholar]
- 9.Tumova E, Gous RM. Interaction of hen production type, age, and temperature on laying pattern and egg quality. Poult Sci. 2012;91(5):1269–75. doi: 10.3382/ps.2011-01951. [DOI] [PubMed] [Google Scholar]
- 10.Begli HE, Zerehdaran S, Hassani S, Abbasi MA, Ahmadi AR. Heritability, genetic and phenotypic correlations of egg quality traits in Iranian native fowl. Br Poult Sci. 2010;51(6):740–4. doi: 10.1080/00071668.2010.528750. [DOI] [PubMed] [Google Scholar]
- 11.Zhang LC, Ning ZH, Xu GY, Hou ZC, Yang N. Heritabilities and genetic and phenotypic correlations of egg quality traits in brown-egg dwarf layers. Poult Sci. 2005;84(8):1209–13. doi: 10.1093/ps/84.8.1209. [DOI] [PubMed] [Google Scholar]
- 12.Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41(Database issue):D871–9. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15:442. doi: 10.1186/1471-2164-15-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 15.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6(2):95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Li D, Liu J, Chen S, Qu L, Zheng J, et al. A genome-wide SNP scan reveals novel Loci for egg production and quality traits in white leghorn and brown-egg dwarf layers. PLoS ONE. 2011;6(12):e28600. doi: 10.1371/journal.pone.0028600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kranis A, Gheyas AA, Boschiero C, Turner F, Yu L, Smith S, et al. Development of a high density 600 K SNP genotyping array for chicken. BMC Genomics. 2013;14:59. doi: 10.1186/1471-2164-14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Zhao G, Liu R, Zheng M, Hu Y, Wu D, et al. The identification of 14 new genes for meat quality traits in chicken using a genome-wide association study. BMC Genomics. 2013;14:458. doi: 10.1186/1471-2164-14-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolc A, Arango J, Jankowski T, Dunn I, Settar P, Fulton JE, et al. Genome-wide association study for egg production and quality in layer chickens. J Anim Breed Genet. 2014;131(3):173–82. doi: 10.1111/jbg.12086. [DOI] [PubMed] [Google Scholar]
- 20.Gu X, Feng C, Ma L, Song C, Wang Y, Da Y, et al. Genome-wide association study of body weight in chicken F2 resource population. PLoS One. 2011;6(7):e21872. doi: 10.1371/journal.pone.0021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das K, Li J, Wang Z, Tong C, Fu G, Li Y, et al. A dynamic model for genome-wide association studies. Hum Genet. 2011;129(6):629–39. doi: 10.1007/s00439-011-0960-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Q, Chazaro I, Cui J, Guo CY, Demissie S, Larson M, et al. Genetic analyses of longitudinal phenotype data: a comparison of univariate methods and a multivariate approach. BMC Genet. 2003;4(Suppl 1):S29. doi: 10.1186/1471-2156-4-S1-S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan R, Zhang Y, Albert PS, Liu A, Wang Y, Xiong M. Longitudinal Association Analysis of Quantitative Traits. Genet Epidemiol. 2012. [DOI] [PMC free article] [PubMed]
- 24.Furlotte NA, Eskin E, Eyheramendy S. Genome-wide association mapping with longitudinal data. Genet Epidemiol. 2012;36(5):463–71. doi: 10.1002/gepi.21640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith EN, Chen W, Kahonen M, Kettunen J, Lehtimaki T, Peltonen L, et al. Longitudinal genome-wide association of cardiovascular disease risk factors in the Bogalusa heart study. PLoS Genet. 2010;6(9):e1001094. doi: 10.1371/journal.pgen.1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Hong Y, Gao J, Xiao S, Ma J, Zhang W, et al. Genome-wide association study reveals constant and specific loci for hematological traits at three time stages in a White Duroc x Erhualian F2 resource population. PLoS ONE. 2013;8(5):e63665. doi: 10.1371/journal.pone.0063665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Z, Shen X, Pan W. Longitudinal analysis is more powerful than cross-sectional analysis in detecting genetic association with neuroimaging phenotypes. PLoS ONE. 2014;9(8):e102312. doi: 10.1371/journal.pone.0102312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Mei L, Yang K, Rotter JI, Guo X. Identifying association under a previous linkage peak on chromosome 16 for body mass index using cross-sectional and longitudinal data of the Framingham Heart Study. BMC Proc. 2009;3(Suppl 7):S101. doi: 10.1186/1753-6561-3-s7-s101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11(4):407–9. doi: 10.1038/nmeth.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S, Xing EP. Statistical estimation of correlated genome associations to a quantitative trait network. PLoS Genet. 2009;5(8):e1000587. doi: 10.1371/journal.pgen.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo C, Qu H, Ma J, Wang J, Li C, Yang C, et al. Genome-wide association study of antibody response to Newcastle disease virus in chicken. BMC Genet. 2013;14:42. doi: 10.1186/1471-2156-14-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3(4):299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- 33.Lu D, Sargolzaei M, Kelly M, Li C, Vander Voort G, Wang Z, et al. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front Genet. 2012;3:152. doi: 10.3389/fgene.2012.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu W, Dekkers JC, Lee WR, Abasht B. Linkage disequilibrium in crossbred and pure line chickens. Genet Sel Evol. 2015;47:11. doi: 10.1186/s12711-015-0098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19(7):807–12. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9(4):e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14(7):483–95. doi: 10.1038/nrg3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014;10(3):e1004198. doi: 10.1371/journal.pgen.1004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43(6):519–25. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Lee T, Kim J, Cho MC, Han BG, Lee JY, et al. Ubiquitous polygenicity of human complex traits: genome-wide analysis of 49 traits in Koreans. PLoS Genet. 2013;9(3):e1003355. doi: 10.1371/journal.pgen.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdollahi-Arpanahi R, Pakdel A, Nejati-Javaremi A, Moradi Shahrbabak M, Morota G, Valente BD, et al. Dissection of additive genetic variability for quantitative traits in chickens using SNP markers. J Anim Breed Genet. 2014;131(3):183–93. doi: 10.1111/jbg.12079. [DOI] [PubMed] [Google Scholar]
- 42.Santure AW, De Cauwer I, Robinson MR, Poissant J, Sheldon BC, Slate J. Genomic dissection of variation in clutch size and egg mass in a wild great tit (Parus major) population. Mol Ecol. 2013;22(15):3949–62. doi: 10.1111/mec.12376. [DOI] [PubMed] [Google Scholar]
- 43.Lee SH, DeCandia TR, Ripke S, Yang J, Sullivan PF, Goddard ME, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet. 2012;44(3):247–50. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467(7317):832–8. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costanza MC, Beer-Borst S, James RW, Gaspoz JM, Morabia A. Consistency between cross-sectional and longitudinal SNP: blood lipid associations. Eur J Epidemiol. 2012;27(2):131–8. doi: 10.1007/s10654-012-9670-1. [DOI] [PubMed] [Google Scholar]
- 46.Mei H, Chen W, Jiang F, He J, Srinivasan S, Smith EN, et al. Longitudinal replication studies of GWAS risk SNPs influencing body mass index over the course of childhood and adulthood. PLoS ONE. 2012;7(2):e31470. doi: 10.1371/journal.pone.0031470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stephens M. A unified framework for association analysis with multiple related phenotypes. PLoS ONE. 2013;8(7):e65245. doi: 10.1371/journal.pone.0065245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–75. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 49.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 50.Nakae J, Oki M, Cao Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008;582(1):54–67. doi: 10.1016/j.febslet.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 51.Wondisford AR, Xiong L, Chang E, Meng S, Meyers DJ, Li M, et al. Control of Foxo1 gene expression by co-activator P300. J Biol Chem. 2014;289(7):4326–33. doi: 10.1074/jbc.M113.540500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 53.Schreiweis MA, Hester PY, Settar P, Moody DE. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Anim Genet. 2006;37(2):106–12. doi: 10.1111/j.1365-2052.2005.01394.x. [DOI] [PubMed] [Google Scholar]
- 54.Dunn IC, Meddle SL, Wilson PW, Wardle CA, Law AS, Bishop VR, et al. Decreased expression of the satiety signal receptor CCKAR is responsible for increased growth and body weight during the domestication of chickens. Am J Physiol Endocrinol Metab. 2013;304(9):E909–21. doi: 10.1152/ajpendo.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Darvasi A, Soller M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics. 1995;141(3):1199–207. doi: 10.1093/genetics/141.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, et al. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics. 2009;183(3):951–64. doi: 10.1534/genetics.109.106476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, et al. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009;10:43. doi: 10.1186/1471-2156-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, et al. Association, effects and validation of polymorphisms within the NCAPG - LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011;12:103. doi: 10.1186/1471-2156-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, et al. Four loci explain 83 % of size variation in the horse. PLoS ONE. 2012;7(7):e39929. doi: 10.1371/journal.pone.0039929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Signer-Hasler H, Flury C, Haase B, Burger D, Simianer H, Leeb T, et al. A genome-wide association study reveals loci influencing height and other conformation traits in horses. PLoS ONE. 2012;7(5):e37282. doi: 10.1371/journal.pone.0037282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niknafs S, Nejati-Javaremi A, Mehrabani-Yeganeh H, Fatemi SA. Estimation of genetic parameters for body weight and egg production traits in Mazandaran native chicken. Trop Anim Health Prod. 2012;44(7):1437–43. doi: 10.1007/s11250-012-0084-6. [DOI] [PubMed] [Google Scholar]
- 62.Liu Q, Liu J, Cao Q, Sha J, Zhou Z, Wang H, et al. NYD-SP15: a novel gene potentially involved in regulating testicular development and spermatogenesis. Biochem Genet. 2006;44(7–8):409–23. doi: 10.1007/s10528-006-9038-x. [DOI] [PubMed] [Google Scholar]
- 63.Moreno-Dominguez A, Cidad P, Miguel-Velado E, Lopez-Lopez JR, Perez-Garcia MT. De novo expression of Kv6.3 contributes to changes in vascular smooth muscle cell excitability in a hypertensive mice strain. J Physiol. 2009;587(Pt 3):625–40. doi: 10.1113/jphysiol.2008.165217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beasley TM, Erickson S, Allison DB. Rank-based inverse normal transformations are increasingly used, but are they merited? Behav Genet. 2009;39(5):580–95. doi: 10.1007/s10519-009-9281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–23. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao X, Becker LC, Becker DM, Starmer JD, Province MA. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol. 2010;34(1):100–5. doi: 10.1002/gepi.20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao JH. gap: Genetic Analysis Package. R package version 11–12 2014, http://cran.r-project.org/web/packages/gap/index.html.
- 70.Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. R package version 11–12 2014, http://cran.r-project.org/web/packages/qqman/index.html.
- 71.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 72.Freedman ML, Reich D, Penney KL, McDonald GJ, Mignault AA, Patterson N, et al. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36(4):388–93. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 73.Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177(1):577–85. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28(19):2540–2. doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44(4):369–75. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 79.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011;2011:bar030. doi: 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]