

Abstract
Background
The accurate delimitation of species is essential to numerous areas of biological research. An unbiased assessment of the diversity, including the cryptic diversity, is of particular importance for the below ground fauna, a major component of global biodiversity. On the British Isles, the epigeic earthworm Lumbricus rubellus, which is a sentinel species in soil ecotoxicology, consists of two cryptic taxa that are differentiated in both the nuclear and the mitochondrial (mtDNA) genomes. Recently, several deeply divergent mtDNA lineages were detected in mainland Europe, but whether these earthworms also constitute cryptic species remains unclear. This information is important from an evolutionary perspective, but it is also essential for the interpretation and the design of ecotoxicological projects. In this study, we used genome-wide RADseq data to assess the reproductive isolation of the divergent mitochondrial lineages of L. rubellus that occur in sympatry in multiple localities in Central Europe.
Results
We identified five divergent (up to 16 % net p-distance) mitochondrial lineages of L. rubellus in sympatry. Because the clustering of the RADseq data was according to the population of origin and not the mtDNA lineage, reproductive isolation among the mtDNA lineages was not likely. Although each population contained multiple mtDNA lineages, subdivisions within the populations were not observed for the nuclear genome. The lack of fixed differences and sharing of the overwhelming majority of nuclear polymorphisms between localities, indicated that the populations did not constitute allopatric species. The nucleotide diversity within the populations was high, 0.7–0.8 %.
Conclusions
The deeply divergent mtDNA sympatric lineages of L. rubellus in Central Europe were not reproductively isolated groups. The earthworm L. rubellus, which is represented by several mtDNA lineages in continental Europe, apparently is a single highly polymorphic species rather than a complex of several cryptic species. This study demonstrated the critical importance of the use of multilocus nuclear data for the unbiased assessment of cryptic diversity and for the delimitation of species in soil invertebrates.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-015-0488-9) contains supplementary material, which is available to authorized users.
Keywords: Species delimitation, Cryptic species, RADseq, mtDNA, Lumbricus rubellus, Soil diversity
Background
Species delimitation aims to identify species-level biological diversity while delineating interspecific boundaries and estimating the number of species [1, 2]. The accurate delimitation of species is of paramount importance in numerous fields, including evolutionary biology, systematics, biogeography, conservation biology and many areas of experimental biology [3]. Traditionally, species have been identified based on morphological traits. However, a large portion of the biological diversity may be impossible to detect by relying only on morphological characters [4]. These difficulties are most apparent in taxonomic groups that include closely related, recently diverged species, which form complexes of cryptic species. The morphology-based species delimitation may also severely underestimate the overall diversity in taxonomic groups with morphological uniformity or with a paucity of taxonomically useful morphological characters. Today, many biologists agree that species may be separately evolving metapopulation lineages [5, 6], which is a deliberately loose definition to proceed beyond the unresolvable debate about the species concepts. This lineage-based interpretation of species shifts the focus to genetic data and the other nonmorphological characters. The DNA data may be a source of valuable additional information to develop new and more accurate species delimitation methods that should be used by alpha taxonomists [4]. Distinguishing between the two groups of criteria that are used for species delimitation, the pattern-oriented and the process-oriented criteria, is useful [7]. The pattern-oriented criteria reflect the effect of a lineage existence, e.g., monophyly, diagnosability or formation of distinct genotypic clusters, whereas the process-oriented criteria identify the evolutionary cause of the lineage existence, e.g., the reproductive isolation or occupation of a distinct niche. Species treated as separate evolutionary lineages can be delimited based on these criteria even when the species definition or concept is debated [6]. The use of multiple criteria is recommended to increase the chance to detect recently separated lineages and to obtain clear evidence of the lineages as separate entities [7, 8].
In the past, the molecular identification of species involved primarily mitochondrial DNA (mtDNA) sequences. The most commonly used mitochondrial marker has been Cytochrome Oxidase I (COI), which is a standard in the DNA barcoding of animal species, under the sometimes questioned [9] assumptions of low variation within species and high differentiation between species [10]. Within species, high mtDNA differentiation is often observed between allopatric populations; it may or may not be accompanied by a differentiation in the nuclear markers. Sympatric mtDNA divergence is less common, and divergent sympatric lineages often show reproductive isolation and divergence in the nuclear genome (e.g., [11]). However, mtDNA differentiation is often reported to be discordant with the differentiation based on the nuclear genetic markers, and multiple explanations have been proposed for this pattern [2, 12, 13]. Thus, the joint analysis of mitochondrial and nuclear markers in sympatry is more likely to provide a robust test to identify cryptic species and assess species boundaries.
The delimitation of species is of primary importance in the study of belowground fauna. Global biodiversity is determined to a large extent by the belowground communities, and soil is one of the most species-rich terrestrial habitats [14]. A high percentage of species is estimated to remain undescribed for most soil taxa, and this lack of information is likely due to a lack of taxonomic knowledge and expertise, particularly in the case of small body-sized animals [15]. High cryptic diversity has been detected with DNA based methods in soil invertebrates, including springtails [16–18], earthworms [19, 20], mites [21] and centipedes [22]. Some of these soil invertebrates are considered sentinel species in ecotoxicology. The knowledge of their taxonomy, including the cryptic diversity, is critical for the proper design and for the interpretation of ecotoxicological experiments. The divergent evolution of cryptic species may lead to physiological differences, e.g., differential sensitivity to environmental stressors, including pollution. Individuals belonging to separate evolutionary lineages may display significant differences in the sensitivity to toxicants, and such a phenomenon was reported for the evolutionary lineages of the aquatic oligochaete Tubifex tubifex, which consisted of five cryptic species. The sensitivity to Cd, which was assessed based on the mortality and time to death, differed among these lineages [23]. The distribution of genetic lineages in nature can be expected to be shaped by pollution when the different levels of resistance of the mtDNA lineages to toxicants are considered. Thus, more sensitive lineages would be lost at more polluted sites, as predicted by the genetic erosion hypothesis, which posits the loss of genetic diversity because of pollution [24].
The species of lumbricid earthworms often consist of highly divergent mitochondrial lineages [19, 20, 25, 26]. The epigeic earthworm Lumbricus rubellus Hoffmeister, 1843, a sentinel species in ecotoxicology, is found in the UK as two distinct mtDNA lineages, A and B. The mtDNA sequence divergence between these lineages was high, over 8 % at COI (K2P distance) and 14 % at COII (uncorrected p-distance) [19, 27]. Recently, differentiation in the nuclear markers was also found between individuals from the two mtDNA lineages in sympatry, which implied reproductive isolation and supported the cryptic species hypothesis [27]. Several other deeply divergent mitochondrial lineages have been found within mainland Europe [28]; however, whether these continental lineages are reproductively isolated and represent cryptic species is not known. In Poland, we have identified highly divergent mtDNA lineages in earthworms in sympatry, which represent several of the lineages observed across Europe. Thus, the conditions were favorable to test for reproductive isolation between the mtDNA lineages of L. rubellus from continental Europe.
In this work, we tested whether the sympatric mitochondrial lineages of L. rubellus that were found in Poland represented cryptic species. We expected the nuclear clustering to be concordant with the sympatric mtDNA lineages if the mtDNA lineages corresponded to reproductively isolated groups. Additionally, this study aimed to estimate the genetic diversity of the L. rubellus populations and to compare the haplotypes found in Poland with the mtDNA lineages observed across Europe. The mitochondrial lineages were characterized based on COI and ATP6 sequences, whereas the multilocus genotype data that were generated by the Restriction site Associated DNA Sequencing approach (RADseq) [29] were used to estimate the differentiation in nuclear DNA. The distribution of the divergent mitochondrial lineages was related to environmental data, including data on pollution.
Results
mtDNA
The two analyzed mtDNA fragments totaled 1016 bp (COI: 453 bp and ATP6: 563 bp). Among the 123 sequenced L. rubellus individuals originating from four populations (OL2, OL4, OL5 and TR), 276 polymorphic sites defined 27 unique haplotypes, 10 of which were observed only for one individual (Table 1). ATP6 showed higher sequence diversity than COI (Additional file 1: Tables A1, A2). The haplotypes formed five deeply divergent lineages (Fig. 1); four of the lineages had been previously described (A1, A2, A3, and E), and one lineage was new and named C2 (Fig. 2, Additional file 1: Figure A1). The sequence divergence between the C2 haplotypes and a haplotype from Serbia assigned to the C lineage was 7.5–8.5 %, which led us to distinguish C2 as a separate lineage. An additional mtDNA lineage, which we named D2, was detected in five individuals from another geographic region, and this lineage had not been genotyped with nuclear markers (Fig. 2). The net sequence divergence between the observed lineages was substantial and ranged from 1.3 % between the A2 and A3 lineages to 16 % between the C2 and E lineages (Table 2).
Table 1.
The mtDNA variation in Lumbricus rubellus populations
| Site | N | S | H | Hd | π |
|---|---|---|---|---|---|
| OL2 | 31 | 174 | 8 | 0.815 ± 0.045 | 0.0277 ± 0.0072 |
| OL4 | 31 | 71 | 7 | 0.811 ± 0.044 | 0.0152 ± 0.0029 |
| OL5 | 31 | 62 | 4 | 0.578 ± 0.081 | 0.0167 ± 0.0033 |
| TR | 30 | 254 | 13 | 0.855 ± 0.056 | 0.0960 ± 0.0085 |
| ALL | 123 | 276 | 27 | 0.923 ± 0.012 | 0.0580 ± 0.0065 |
Shown results were based on concatenated mtDNA data (COI: 453 bp and ATP6:563 bp). Site - sampling site, N - number of analyzed individuals, S - number of polymorphic nucleotide positions, H - number of haplotypes, Hd - haplotype (gene) diversity (mean ± SD), π - nucleotide diversity (mean ± SD)
Fig. 1.
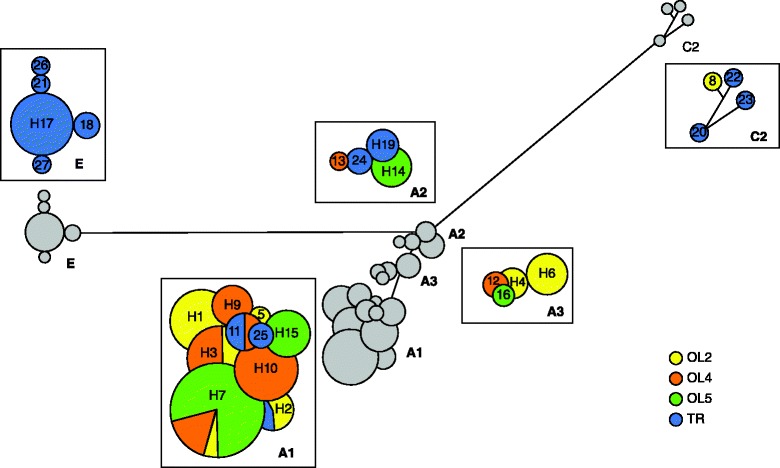
Haplotype network of mtDNA (COI + ATP6) sequences of Lumbricus rubellus. The network shows the divergent mtDNA lineages (A1, A2, A3, C2, and E). Circles represent distinct haplotypes, which are marked with the labels H1-H27 in the enlarged insertions. The size of each circle is proportional to the total number of individuals that showed that haplotype, and the haplotype distributions within the populations are indicated as pie charts. The smallest circle corresponds to n = 1
Fig. 2.
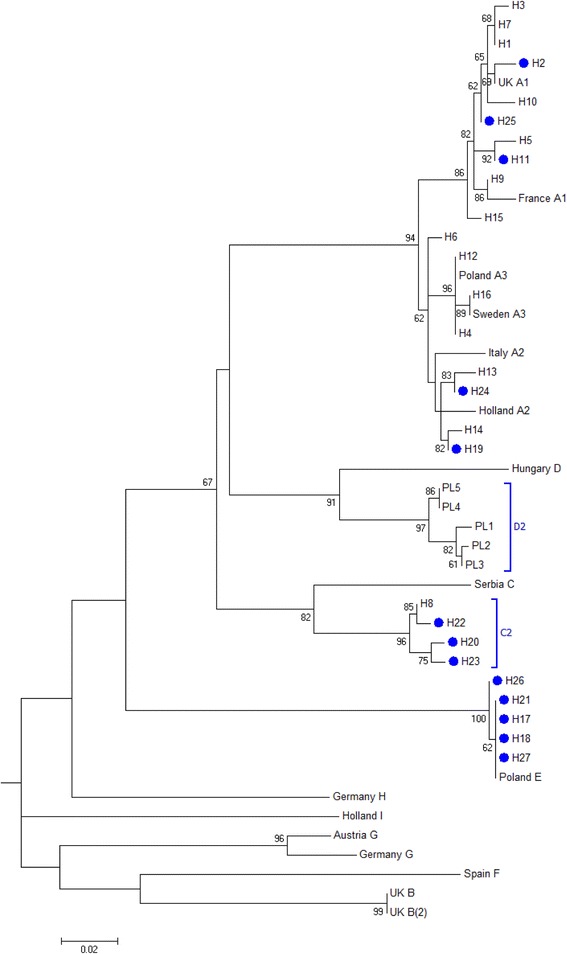
Maximum-likelihood tree based on the COI sequences of Lumbricus rubellus collected across Europe. Haplotypes observed in the studied Polish populations are marked with the labels H1-H27. Blue circle labels represent the haplotypes found in the TR population. Blue brackets represent the new mtDNA lineages (C2 and D2). The COI sequence of Hirudo medicinalis [GenBank: EF446709.1] was used as the outgroup to root the tree. Bootstrap percentages ≥ 50 are shown at the branch points
Table 2.
Evolutionary divergence between mitochondrial lineages of Lumbricus rubellus
| A1 | A2 | A3 | C2 | E | |
|---|---|---|---|---|---|
| A1 | 0.032 | 0.028 | 0.118 | 0.157 | |
| A2 | 0.041 | 0.013 | 0.114 | 0.148 | |
| A3 | 0.039 | 0.023 | 0.113 | 0.150 | |
| C2 | 0.133 | 0.128 | 0.130 | 0.160 | |
| E | 0.162 | 0.152 | 0.156 | 0.170 |
Mean pairwise sequence divergence - below diagonal; Net sequence divergence - above diagonal; p-distance
More than one lineage was detected in each studied population, with four lineages co-occurring in the TR population, which also included the most divergent haplotypes (17.5 % uncorrected sequence divergence; Additional file 1: Table A3). The A lineages predominated in the Olkusz area but were found in all studied populations. In contrast to the mtDNA lineages, most mtDNA haplotypes were unique, with only four haplotypes shared by at least two populations and no haplotypes shared by all populations (Fig. 1). Consequently, the mtDNA differentiation (FST) among all population pairs was substantial and highly significant (Table 3). The highest within-population variation, both for the haplotype (Hd = 0.855 ± 0.056; mean ± SD) and for the nucleotide (π = 0.096 ± 0.008; mean ± SD) diversity, was found in the TR population. Among the Olkusz populations, the mtDNA diversity increased with the level of pollution, and the OL2 population was the most diverse (Table 1).
Table 3.
Pairwise genetic differentiation between Lumbricus rubellus populations
| OL2 | OL4 | OL5 | TR | |
|---|---|---|---|---|
| OL2 | - | 0.1518 | 0.2892 | 0.1595 |
| OL4 | 0.1146 | - | 0.2457 | 0.1635 |
| OL5 | 0.1436 | 0.1828 | - | 0.2839 |
| TR | 0.1278 | 0.1608 | 0.1808 | - |
mtDNA FST based on haplotype frequency - above diagonal; RADseq FST based on SNP allele frequency - below diagonal. All values were significant (10,100 permutations; p < 0.05, after strict Bonferroni correction)
RADseq
The RADseq data were obtained for 100 individuals, 25 individuals per population. After stringent quality control in Stacks, our data set consisted of 1101 RADseq loci (~96,800 bp) that contained 5712 biallelic SNPs. The genetic diversity was highest in the TR population (H = 4.28 ± 0.07; Hd = 0.436 ± 0.008; π = 0.0081 ± 0.0002; mean ± SE), which also showed the highest number of private polymorphisms (SxTR = 1379). Similar to the mtDNA results, the genetic diversity among the Olkusz populations increased with the level of pollution, and the genetic diversity was highest in population OL2 (Table 4). The RADseq-based differentiation between the earthworm populations was significant, and the pairwise FST ranged from 0.1146 to 0.1828 (Table 3). Although the populations were significantly differentiated in allele frequencies, the overwhelming majority of polymorphic positions were shared among localities, and fixed differences were not observed between the localities (Table 4). The Bayesian clustering identified a clear population structure, the individuals were grouped into four clusters according to their populations of origin with a low level of admixture observed mainly between the neighboring OL2 and OL4 sites (Fig. 3, Additional file 1: Figure A2). The four clusters revealed by Structure were recovered also in the Neighbor-Joining tree that was based on the genetic distances between individuals calculated from all 5712 RADseq SNPs (Fig. 4). We did not identify a clear pattern of isolation by distance or a correlation between the level of pollution and the genetic differentiation between the populations (Additional file 1: Figure A3, Additional file 1: Table A4).
Table 4.
Polymorphism of Lumbricus rubellus populations estimated from the RADseq data (1101 RAD tags that contained 5712 SNPs)
| Site | H | Hd | π | S | Sf | Sx | Ss |
|---|---|---|---|---|---|---|---|
| OL2 | 3.68 ± 0.06a | 0.427 ± 0.008a | 0.0081 ± 0.0002a | 3239 | 0 | 541 | 2696 |
| OL4 | 3.23 ± 0.05b | 0.395 ± 0.008b | 0.0074 ± 0.0002ab | 2775 | 0 | 372 | 2403 |
| OL5 | 2.69 ± 0.04c | 0.360 ± 0.008c | 0.0068 ± 0.0002b | 2284 | 0 | 310 | 1974 |
| TR | 4.28 ± 0.07d | 0.436 ± 0.008a | 0.0081 ± 0.0002a | 3816 | 0 | 1379 | 2437 |
Site – sampling site, H - number of haplotypes (mean ± SE), Hd - haplotype (gene) diversity (mean ± SE), π - nucleotide diversity (mean ± SE); S – number of polymorphic sites, Sf – number of fixed differences, Sx – number of polymorphic sites unique for a population, Ss – number of polymorphic sites shared with other populations. Means with different letters are significantly different (t-test; p < 0.05, after strict Bonferroni correction)
Fig. 3.
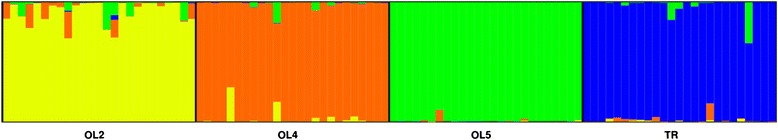
Population genetic structure of Lumbricus rubellus. The graph shows the results of the Structure analysis of the RAD tags (single SNP selected from each of 1101 RAD tags). Each vertical bar represents a different individual from one of the four populations
Fig. 4.
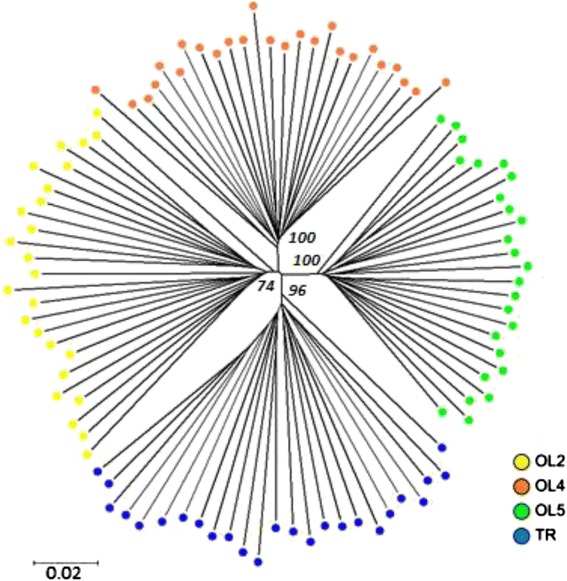
Neighbor-Joining tree generated from the between-individual distance matrix (uncorrected p-distance) based on all 5712 SNPs from the RADseq data. Each dot represents an individual Lumbricus rubellus earthworm, and the color represents the population of origin. Bootstrap support values indicate grouping of individuals according to the population of origin
The clustering of the RADseq data according to the population of origin and not the mtDNA lineage indicated the lack of reproductive isolation between the mtDNA lineages. Although each population contained multiple mtDNA lineages, subdivisions within the populations were not observed in the nuclear genome, not even in the separate Structure analysis of the most diverse TR population.
Discussion
Our analyses of the L. rubellus individuals sampled from multiple sites in Poland revealed deeply divergent mtDNA lineages that occurred in sympatry. However, these divergent lineages were not reproductively isolated as evidenced by patterns of clustering in the nuclear data and therefore did not represent cryptic species. The situation we observed in Poland contrasts with that in the UK because the two main mtDNA lineages of L. rubellus in the UK, A and B, whose divergence was comparable to that observed in our study, were also differentiated at nuclear microsatellite markers [27]. The morphological data further supported the hypothesis that the two British lineages represent cryptic species [30]. Thus, the level of mtDNA divergence in L. rubellus within and between reproductively isolated lineages may be similar. This similarity is not surprising because the divergence at which reproductive isolation evolves varies extensively between and within taxonomic groups [31, 32]. Additionally, Torres-Leguizamon et al. [33] found different patterns of mitochondrial and nuclear structuring in another earthworm species that consist of two highly divergent (8.7 %) mtDNA lineages, Apporectodea icterica. This finding suggested the random interbreeding of the mtDNA lineages. Because the nuclear markers of earthworms examined in the present study clustered according to the sample location, we theoretically could have sampled several microallopatric species that share mtDNA lineages. However, the genetic data did not support such a hypothesis. First, fixed differences were not detected among the localities in the nuclear genome; second, the overwhelming majority of polymorphisms were shared among the localities; and third, the signatures of genetic admixture between populations, particularly those separated by small distances in the Olkusz area, were detected. Because our data indicate no reproductive isolation between the lineages, a question arises which processes and mechanisms might explain the presence of highly divergent mtDNA lineages of L. rubellus in sympatry? In the following sections, we discuss several plausible, although not mutually exclusive, hypotheses.
The highly divergent mtDNA lineages in Poland might be the result of admixture that followed the postglacial recolonization among previously geographically but not reproductively isolated lineages derived from separate glacial refugia. Such scenario was suggested for the British L. rubellus [27] as well as other earthworm species [19, 33]. Recent studies [28, 34] note that cryptic refugium may have occurred on the northwestern coasts of Europe, where one or more of the L. rubellus mtDNA lineages could have survived the periods of unfavorable climate during the Pleistocene. The area of present-day Poland may have been colonized from refugia located in central, southern and eastern Europe [35, 36], and such a pattern appears common because zones of contact between divergent evolutionary lineages have been described for multiple taxa in Poland [37–40]. However, the large genetic distances among the mtDNA lineages of L. rubellus indicated that their origin predated the Last Glacial Maximum. Nevertheless, multiple cycles of changes in the ranges of the earthworms during the Pleistocene may have caused major changes in the distribution of the ancient mtDNA lineages, which resulted in their sorting into separate refugia. The changes in range may also have prevented the accumulation of reproductive isolation mechanisms between geographically separated populations because of the opportunities for multiple contacts and genetic exchange [41, 42]. The sampling of earthworms in potential refugial areas would provide a direct test of the multiple refugia hypothesis, and a single lineage in a particular area would suggest that the situation observed in Poland resulted from a postglacial admixture. Moreover, admixture might also have contributed to the high nuclear polymorphism detected in our study.
The highly divergent sympatric mtDNA lineages might also be a simple consequence of a large effective population size (Ne); the estimates of the Ne may be inflated when coupled with population subdivision and low migration rates [43]. Earthworms are generally considered highly polymorphic organisms that are characterized by a large Ne and very low migration rates [44]. High intraspecific mtDNA divergence, which often exceeds 5 % in COI, is not limited to L. rubellus and has been reported in numerous earthworm species, including Allolobophora chlorotica, Aporrectodea rosea, Octolasion lacteum, Dendrobaena octaedra, L. castaneus, and L. terrestris (e.g., [19, 20, 45]). Earthworms appear to also be highly polymorphic in the nuclear genome, although most available estimates are based on microsatellites [25, 27, 46, 47] and are therefore difficult to compare among taxa. However, the genome-wide synonymous nucleotide diversity in Allolobophora chlorotica exceeds 1 % [48], which is comparable with the values obtained in our study (0.7–0.8 %). These values might underestimate the true diversity because many polymorphic RADseq loci were filtered out due to the high incidence of missing data. This could result from mutations in the restriction recognition sites but could be also simply due to the random loss of RAD tags during the preparation of RADseq library and random variation in coverage depth. The multiple divergent mtDNA lineages caused by long genealogies in a large population and a high mtDNA mutation rate might be particularly plausible in L. rubellus. This species is an obligate cross-fertilizing hermaphrodite: each individual passes its mtDNA to its progeny, which increases the ratio of nuclear to mitochondrial Ne. For a large Ne, even colonization from a single refugium could explain our results. Additionally, this hypothesis can be tested by directly sampling populations in multiple putative refugial areas. The comparison of the nuclear diversity between the recently recolonized areas and the refugial areas should indicate whether the colonization was accompanied by a reduction in genetic diversity, as postulated by many models of range expansion [49].
The evaluation of the two hypotheses presented above would require additional sampling and data on genome wide variation and differentiation among populations. However, the RADseq markers are less than ideal for such purposes because a large fraction of the loci are not usable due to the high frequency of missing data. This problem has been previously recognized as a serious issue in highly polymorphic species [50, 51]. Therefore, alternative approaches to estimate nucleotide variation could focus either on the protein-coding genes that harbor extensive synonymous variation and can be analyzed using various targeted resequencing methods [52] or on the ultraconserved [53] or conserved elements [54], which also capture more polymorphic flanking regions.
The high intraspecific mtDNA divergence may be due to an introgression from a related species, as commonly observed in animals [13]. However, the mtDNA sequences of L. rubellus found in our study were highly divergent from the sequences of the related species, L. castaneus and L. terrestris, that were available in GenBank, which eliminated the possibility of introgression from currently known Lumbricus lineages. Nevertheless, introgression from an undescribed or extinct lineage remains a viable option.
The multiple divergent mtDNA lineages might also be maintained by natural selection, particularly selection acting in a highly heterogeneous environment like soil. A recent study of Kozancioğlu and Arnqvist [55] suggested that negative frequency-dependent selection (NFDS), a form of balancing selection that favors rare variants, could maintain mtDNA polymorphism [56]. Kozancioğlu and Arnqvist [55] showed an increase in the rare mtDNA haplotype frequency and a decrease in the common haplotype frequency in experimental populations of the seed beetle (Callosobruchus) over the course of 10 generations. NFDS is expected under conditions with environmental heterogeneity, genotype-by-environment interactions and competition for resources, which are conditions likely to be common in earthworm populations.
Soil contamination may also affect the distribution of genetic lineages in nature. If the degrees of sensitivity to soil pollution differ among mtDNA lineages, some lineages will be lost in polluted areas, which reduces variation and is consistent with the genetic erosion hypothesis [24]. For example, Andre et al. [57] investigated the highly differentiated populations of L. rubellus from a Pb-polluted habitat near Cwmystwyth in Wales, UK. The predominant linage differed by study site depending on the level of contamination, and this pattern supported the loss of distinct mtDNA lineages due to pollution. In our research, four mtDNA lineages occurred in the least polluted TR site. In contrast, the E lineage was not found at any of the polluted Olkusz sites. However, either experimental manipulations or field data from multiple pollution gradients are necessary to demonstrate that individuals carrying the mtDNA of the E lineage are more sensitive to metal pollution. On the other hand, among the three contaminated OL sites, the most polluted site OL2 was characterized by relatively high haplotype and nucleotide diversity and the largest number of the polymorphic sites and private SNPs; therefore, this result did not support the genetic erosion hypothesis and was consistent with a pattern we identified also for the rove beetle Staphylinus erythropterus, inhabiting the same gradient [58].
In ecotoxicology, earthworms are used for standard toxicity tests. The recommended and most commonly used species are Eisenia fetida and Eisenia andrei (e.g., [59, 60]). However, the taxonomy of these species is not clear because of cryptic diversity: The earthworm E. fetida has been suggested to be a species complex. Rӧmbke et al. [26] reported two distinct mtDNA COI clusters of E. fetida that were separated by a p-distance of 11.2 %. Based on the assumption that an uncorrected p-distance > 10 % indicates species level differentiation, these authors hypothesized that E. fetida consisted of cryptic species; this result calls the quality and the comparability of ecotoxicological tests into question because cultures of Eisenia earthworms are rarely barcoded [26]. Nuclear markers were not applied to confirm the mtDNA clustering of the E. fetida reported by Rӧmbke et al. [26], although previous analysis of nuclear 28S gene indicated possibility that E. fetida from Ireland might be a cryptic species [61]. Therefore, the findings of our study are particularly relevant because we showed that high mtDNA divergence, even values exceeding 15 %, did not necessarily indicate the presence of cryptic earthworm species. Thus, in addition to crossbreeding experiments, we recommend the use of multilocus nuclear data to test for cryptic species in E. fetida.
The results of our study have consequences for the estimation of the diversity of soil fauna and the delimitation of species. As emphasized by Emerson et al. [62], the soil mesofauna is more diverse than previously thought, and describing the cryptic diversity of soil remains a challenge for ecologists. Thus, the choice of the proper molecular techniques is of crucial importance. The identification of cryptic diversity in the previously mentioned soil invertebrates was often based on a small number of loci [17, 18, 21, 22], although the accuracy of species delimitation is known to depend on the number of loci sampled [63]. Although the cryptic diversity of earthworms has long been a matter of debate, the conclusions about cryptic species remain primarily based on mitochondrial data, which is sometimes complemented with a single nuclear gene or morphological traits (e.g., [45, 61, 64]). In this study, the NGS methods were used to our advantage and showed that genome-wide data supplied valuable knowledge for the study of cryptic diversity.
Conclusions
The highly divergent mtDNA lineages of the earthworm Lumbricus rubellus that sympatrically co-occurred in multiple localities in Poland did not constitute reproductively isolated groups. We concluded that L. rubellus, which is represented by several mtDNA lineages in continental Europe, is a single highly polymorphic species rather than a complex of several cryptic species. This study demonstrated the critical importance of multilocus nuclear data for the unbiased assessment of cryptic diversity and species delimitation in soil invertebrates.
Methods
Sampling
The earthworms were sampled from the smelting and mining area in southern Poland in the vicinity of the zinc and lead smelter ‘Bolesław’ that is close to Olkusz along a well-studied metal pollution gradient [65, 66]. No specific permissions were required because L. rubellus is not a protected species and sampling was not done in a protected area. Sufficient numbers of L. rubellus were found at three sites with different levels of pollution: OL2 (50°17′44′′ N, 19°29′27′′ E), OL4 (50°19′05′′ N, 19°30′32′′ E), and OL5 (50°19′46′′ N, 19°32′44′′ E). The soil at these sites was primarily contaminated with Cd, Pb and Zn (Table 5). As a reference, we used the TR site (49°49′14′′ N, 20°01′22′′ E) in Trzemeśnia, which is also in southern Poland and approximately 65 km from the Olkusz area. We sampled only adult earthworms with a fully developed clitellum. The earthworms were collected alive with the use of horse dung traps installed on 15 × 15 m plots. The specimens were washed with distilled water, starved for 48 h and then preserved in 96 % ethanol. Additional earthworms were collected to analyze the metal concentration in the tissues (Table 5). The genomic DNA was extracted from the anterior body segment tissues using the Wizard® Genomic DNA Purification Kit (Promega, Madison, USA).
Table 5.
Characteristics of the sites at which Lumbricus rubellus was sampled
| Site | Distance [km] | pHCaCl2 | OM [%] | Cd [mg kg−1] | Pb [mg kg−1] | Zn [mg kg−1] |
|---|---|---|---|---|---|---|
| OL2 | 2.5 | 4.12 ± 0.03 | 53.5 ± 0.4 | 49.1 ± 1.1 | 2 060 ± 37 | 3 960 ± 54 |
| 244 ± 120 | 743 ± 234 | 3568 ± 1158 | ||||
| OL4 | 5.3 | 3.46 ± 0.02 | 54.2 ± 2.0 | 14.8 ± 0.2 | 847 ± 38 | 966 ± 22 |
| 80.7 ± 33.0 | 209 ± 211 | 1672 ± 1602 | ||||
| OL5 | 7.7 | 4.29 ± 0.01 | 36.3 ± 0.7 | 12.1 ± 0.7 | 708 ± 12 | 756 ± 11 |
| 70.2 ± 35.7 | 71.9 ± 75.4 | 1125 ± 653 | ||||
| TR | ~65 | 5.33 ± 0.04 | 13.0 ± 0.1 | 1.77 ± 0.295 | 65.4 ± 1.10 | 170 ± 17 |
| 35.0 ± 17.6 | 1.35 ± 1.50 | 300 ± 72.3 |
The distance from the smelter, soil pH, organic matter content at ~10 cm depth (OM %), and metal concentrations [mg kg−1 dwt.]: total concentrations in soil (normal font) and concentrations in earthworm tissue (italics), are shown; mean ± SD (soil: n = 3; earthworms: n = 3–6). Some of the data in the table were obtained from [66]
Mitochondrial DNA
The fragments of the COI and the ATP6 mitochondrial genes were sequenced. The primers were designed with the Primer3 software [67, 68] based on the conservative fragments of the L. rubellus and L. terrestris mitochondrial genomes (Additional file 1: Table A5). The PCR reactions contained ~50–150 ng of DNA template, 0.5 μM of each primer, 1X Taq buffer with (NH4)2SO4, 1.5 mM of MgCl2, 0.2 mM of each dNTP, and 0.75 U of Taq polymerase (Thermo Fisher Scientific, Waltham, USA) in a total volume of 15 μl; the reactions were performed under the conditions shown in Additional file 1: Table A5. After the agarose gel visualization and the Exo-AP cleaning (Exonuclease I and Thermosensitive Alkaline Phosphatase; Thermo Fisher Scientific), the PCR products were sequenced using the BigDye® Terminator v3.1 Cycle Sequencing Kit, cleaned with Ethanol/EDTA precipitation and then analyzed on an ABI 3130xl Genetic Analyzer (Applied Biosystems). The raw sequences were aligned with the SeqScape® software (Applied Biosystems).
The COI sequences of L. rubellus that were sampled from across Europe by members of the Organisms and the Environment research group from the Cardiff School of Biosciences at the University of Cardiff were downloaded from GenBank [GenBank: KP642090-KP612109]. We selected unique sequences that represented the primary haplotype groups, and we used 16 sequences that originated from individuals sampled in 11 European countries (Austria, France, Germany, Holland, Hungary, Italy, Poland, Serbia, Spain, Sweden, and the UK). We also obtained the COI sequences from a few Polish individuals for which no RADseq data were generated, and these individuals originated from the OL3 site (located between OL2 and OL4; individuals PL1-PL4; 50°18’29” N, 19°29’45” E) and from central Poland (individual PL5; 51°32′44′′ N, 21°11′22′′ E).
RADseq
The genomic DNA was extracted from 25 individuals that were randomly selected from each population and was normalized to a concentration of 50 ng/μl using a Qubit® fluorometer. The RAD sequencing libraries were prepared according to the double-digest RADseq method described by Peterson et al. [69]. For each individual, 500 ng of genomic DNA were digested with SphI-HF and MseI restriction enzymes (NEB). After adapter ligation, the individual samples were pooled into four libraries, purified and size selected with the LabChip XT (LabChip XT DNA 300 Assay Kit; PerkinElmer, Waltham, USA). We selected the 346–406 bp fraction to not exceed ~120,000 RAD tags per earthworm. The libraries were amplified in PCR reactions (20 μl) that contained 1X Phusion buffer, 200 μM of each dNTP, 1.0 μM each of PCR1 and PCR2 primers, 0.5 U of Phusion HF polymerase (Thermo Fisher Scientific) and 2.5 μl of the size-selected library under the following conditions: 98 °C for 30 s, followed by10 cycles at 98 °C for 10 s, at 62 °C for 30 s, and at 72 °C for 30 s, and with a final extension at 72 °C for 5 min. The size distribution of the amplified libraries was checked on a Bioanalyzer (HS DNA chips; Agilent Technologies). The libraries were sequenced on an Illumina HiSeq 2000 sequencer (single end, 100 bp) at the Center for Genome Research and Biocomputing of Oregon State University, USA (see Additional file 1 for details).
The raw Illumina reads were analyzed with the Stacks software [70, 71]. To avoid incorrect barcode-individual matches, we first removed all reads that had at least one barcode base with quality < 10 Phred. Subsequently, the reads were demultiplexed and cleaned with process_radtags.pl (Additional file 1: Table A6), and the SphI recognition site sequence (CATGC) was removed (Additional file 1: Note A1). The loci were reconstructed with denovo_map.pl with the following parameters: −m 4, −M 4, −n 4 (see Additional file 1: Table A7 for the Stacks commands and Additional file 1: Table A8 for comparison of results for different Stacks parameters). The MySQL database was used for graphical visualization and data filtering. For further analyses, we used the loci found in all four populations that were genotyped in at least 75 % of the individuals of each population, had at least 5× coverage in each individual, and contained no more than 10 SNPs. The sequencing resulted in ~100,000 RAD tags per individual, with mean coverage 28 reads per RAD tag (Additional file 1: Figure A4). Numerous RAD tags were discarded because they were not present in the required % of individuals (Additional file 1: Figure A5).
Statistical analyses
The earthworms that were sampled from the different sites were assumed to represent local populations. To assess the population genetic diversity in the mtDNA, we estimated the haplotype diversity, nucleotide diversity and the number of polymorphic sites using DnaSP [72]. The measures of population differentiation (mtDNA FST) were calculated based on haplotype frequencies with Arlequin 3.5 (statistical significance was assessed with 10,100 permutations; [73]). The relationships among the haplotypes were illustrated with a Median-Joining haplotype network that was constructed with Network 4.6 [74]. To show genetic differences between the identified haplotypes, we calculated the pairwise distances (p-distance and K2P distance) in MEGA 6 [75]. To relate the haplotypes found in Poland (including haplotypes PL1-PL4 from the additional sites) to the mtDNA lineages observed across Europe, we reconstructed a phylogenetic tree of the Polish and the primary European haplotypes with the Maximum Likelihood (ML) method in MEGA 6 (HKY + G model; 1000 bootstraps). The model was selected using the Bayesian Information criterion. Additionally, a Bayesian tree was constructed in MrBayes (5 mln generations; GTR + G model).
The RADseq data were analyzed with the populations program of Stacks. The population genetic statistics, such as the number of haplotypes, haplotype diversity and nucleotide diversity, were estimated. The pairwise differentiation between populations (RADseq FST) was estimated with Arlequin based on the SNP allele frequency (statistical significance was assessed with 10,100 permutations of individuals between the populations). The number of polymorphic sites that were shared between the populations (Ss) and unique to the individual populations (Sx) as well as the number of fixed differences between the populations (Sf) were calculated in mstatspop [76]. A Bayesian clustering method, implemented in the Structure software [77–80], was used to examine the population structure with the estimation of the most likely number of genetically differentiated clusters and the fractions of the individual genotypes that were attributable to each cluster. For the Structure analysis, we used one randomly selected SNP per RAD tag (−−write_single_snp), which resulted in 1101 loci (Additional file 2: Structure input file). We tested K-values in the range of 1 to 8, with 20 replicates per value. The Structure analysis was run with 100,000 burn-in steps and 1,000,000 post-burn-in iterations per run. The admixture model and the correlated allele frequencies model were used. The lambda parameter was set to one (analysis with the estimated value of lambda, λ = 0.4, resulted in the same number of clusters). The optimal K was selected based on the inspection of the change in the probability value of the data for a given K (L(K)), analyzed with Structure Harvester [81], assuming the largest value for a correct K. For an additional examination of the population structuring, all SNPs were used to calculate a pairwise distance matrix between individuals (uncorrected p-distance) to construct a Neighbor-Joining tree in MEGA 6.
The isolation by distance was tested with a simple Mantel test. The effect of pollution on the degree of genetic differentiation between the populations was tested with a partial Mantel test while accounting for geographic distance [82]. The tests were performed with the use of the IBDWS ([83]; http://ibdws.sdsu.edu/; 10,000 randomizations). The following distance matrices were used in the study: pairwise FST values, log-transformed geographic distance (straight-line distance), and difference in Cd total soil concentration. To test the differences in the genome-wide genetic diversity between populations, we used t-tests with a strict Bonferroni correction for multiple comparisons.
Availability of supporting data
The mtDNA sequences of L. rubellus generated in this study are available on GenBank under accession numbers: KT731474 – KT731500 (COI), and KT731501 – KT731525 (ATP6). The RADseq data (denovo_map.pl output tsv files) are available in the Dryad Digital Repository at the http://dx.doi.org/10.5061/dryad.8070m. Raw Illumina reads are available at NCBI BioProject number PRJNA296755 (http://www.ncbi.nlm.nih.gov/bioproject/296755) or upon request to the corresponding author.
Additional files
Supplementary materials. This file includes supplementary materials to the main text: Note A1. Detailed description of the Illumina sequencing and Stacks analysis of RAD tags. Table A1. Variation at COI (453 bp) in Lumbricus rubellus populations. Table A2. Variation at ATP6 (563 bp) in Lumbricus rubellus populations. Table A3. Pairwise genetic distances between mtDNA haplotypes H1-H27 found in Poland. Table A4. Mantel test and partial Mantel test statistics for Lumbricus rubellus populations sampled at sites with different levels of metal pollution. Table A5. Information on mtDNA sequence markers of Lumbricus rubellus and the PCR conditions. Table A6. Quality control of Illumina reads from the HiSeq 2000. Table A7. The Stacks commands used to process the RADseq data. Table A8. Comparison of Stacks results for different analysis parameters. Figure A1. Bayesian tree based on the COI sequences of Lumbricus rubellus. Figure A2. The Ln P(D) in Structure analysis of Lumbricus rubellus. Figure A3. Relation between the genetic distance (RADseq FST) and the log(geographic distance) in Lumbricus rubellus populations; a reduced major axis regression based on the Mantel test. Figure A4. Final coverage per RAD tag (mean ± SD) for individual earthworms of Lumbricus rubellus. Figure A5. Effect of the Stacks parameters on the final number of usable RAD tags in Lumbricus rubellus. (PDF 650 kb)
Structure file. This is the main Structure input file generated by Stacks with the use of one SNP per RAD tag. (TSV 438 kb)
Acknowledgements
We thank Edyta Podmokła and Sebastian Żmudzki for their help with sampling earthworms and Barbara Kowalczyk for her contribution to COI sequencing. This study was supported by the Polish National Science Centre Grant No. 2011/03/N/NZ8/00013 and the Foundation for Polish Science International PhD Projects Programme co-financed by the EU European Regional Development Fund in the frame of the “Environmental Stress, Population Viability and Adaptation” project; MPD/2009-3/5. Support from Jagiellonian University in Kraków, DS/MND/WBiNoZ/INoŚ/11/2014, is also acknowledged.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IG designed the project, sampled the earthworms, prepared the RADseq libraries, collected and analyzed the data, and led the preparation of the manuscript. PS contributed to the project by providing the COI sequences of earthworms collected across Europe and assisting with the interpretation of the data and the preparation of the manuscript. WB participated in the design of the project, the analysis and the interpretation of the data, and the preparation of the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Contributor Information
Iwona Giska, Email: iwona.giska@uj.edu.pl.
Pierfrancesco Sechi, Email: pierfrancesco.sechi@ise.cnr.it.
Wiesław Babik, Email: wieslaw.babik@uj.edu.pl.
References
- 1.Fujita MK, Leache AD, Burbrink FT, McGuire JA, Moritz C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol Evol. 2012;27:480–488. doi: 10.1016/j.tree.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Carstens BC, Pelletier TA, Reid NM, Satler JD. How to fail at species delimitation. Mol Ecol. 2013;22:4369–4383. doi: 10.1111/mec.12413. [DOI] [PubMed] [Google Scholar]
- 3.Barley AJ, White J, Diesmos AC, Brown RM. The challenge of species delimitation at the extremes: diversification without morphological change in philippine sun skinks. Evolution. 2013;67:3556–3572. doi: 10.1111/evo.12219. [DOI] [PubMed] [Google Scholar]
- 4.Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, et al. Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2007;22:148–155. doi: 10.1016/j.tree.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 5.de Queiroz K. The general lineage concept of species, species criteria, and the process of speciation: a conceptual unification and terminological recommendations. In: Howard DJ, Berlocher SH, editors. Endless forms: species and speciation. New York: Oxford University Press; 1998. pp. 57–75. [Google Scholar]
- 6.de Queiroz K. Species concepts and species delimitation. Syst Biol. 2007;56:879–886. doi: 10.1080/10635150701701083. [DOI] [PubMed] [Google Scholar]
- 7.Reeves PA, Richards CM. Species delimitation under the general lineage concept: an empirical example using wild North American hops (Cannabaceae: Humulus lupulus) Syst Biol. 2011;60:45–59. doi: 10.1093/sysbio/syq056. [DOI] [PubMed] [Google Scholar]
- 8.Bacon CD, McKenna MJ, Simmons MP, Wagner WL. Evaluating multiple criteria for species delimitation: an empirical example using Hawaiian palms (Arecaceae: Pritchardia) BMC Evol Biol. 2012;12:23. doi: 10.1186/1471-2148-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meier R, Shiyang K, Vaidya G, Ng PKL. DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol. 2006;55:715–728. doi: 10.1080/10635150600969864. [DOI] [PubMed] [Google Scholar]
- 10.Hebert PDN, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270(Suppl 1):96–99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haine ER, Martin J, Cook JM. Deep mtDNA divergences indicate cryptic species in a fig-pollinating wasp. BMC Evol Biol. 2006;6:83. doi: 10.1186/1471-2148-6-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogner S, Laskemoen T, Lifjeld JT, Porkert J, Kleven O, Albayrak T, et al. Deep sympatric mitochondrial divergence without reproductive isolation in the common redstart Phoenicurus phoenicurus. Ecol Evol. 2012;12:2974–2988. doi: 10.1002/ece3.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21:3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x. [DOI] [PubMed] [Google Scholar]
- 14.Wolters V. Biodiversity of soil animals and its function. Eur J Soil Biol. 2001;37:221–227. doi: 10.1016/S1164-5563(01)01088-3. [DOI] [Google Scholar]
- 15.Decaëns T, Jiménez JJ, Gioia C, Measey GJ, Lavelle P. The values of soil animals for conservation biology. Eur J Soil Biol. 2006;42(Suppl 1):23–38. doi: 10.1016/j.ejsobi.2006.07.001. [DOI] [Google Scholar]
- 16.Timmermans MJTN, Ellers J, Marien J, Verhoeff SC, Ferwerda EB, Van Straalen NM. Genetic structure in Orchesella cincta (Collembola): strong subdivision of European populations inferred from mtDNA and AFLP markers. Mol Ecol. 2005;14:2017–2024. doi: 10.1111/j.1365-294X.2005.02548.x. [DOI] [PubMed] [Google Scholar]
- 17.Porco D, Bedos A, Greenslade P, Janion C, Skarżyński D, Stevens MI, et al. Challenging species delimitation in Collembola: cryptic diversity among common springtails unveiled by DNA barcoding. Invert Syst. 2012;26:470–477. doi: 10.1071/IS12026. [DOI] [Google Scholar]
- 18.Cicconardi F, Fanciulli PP, Emerson BC. Collembola, the biological species concept and the underestimation of global species richness. Mol Ecol. 2013;22:5382–5396. doi: 10.1111/mec.12472. [DOI] [PubMed] [Google Scholar]
- 19.King RA, Tibble AL, Symondson WOC. Opening a can of worms: unprecedented sympatric cryptic diversity within British lumbricid earthworms. Mol Ecol. 2008;17:4684–4698. doi: 10.1111/j.1365-294X.2008.03931.x. [DOI] [PubMed] [Google Scholar]
- 20.Klarica J, Kloss-Brandstätter A, Traugott M, Juen A. Comparing four mitochondrial genes in earthworms – implications for identification, phylogenetics, and discovery of cryptic species. Soil Biol Biochem. 2012;45:23–30. doi: 10.1016/j.soilbio.2011.09.018. [DOI] [Google Scholar]
- 21.Schäffer S, Pfingstl T, Koblmüller S, Winkler KA, Sturmbauer C, Krisper G. Phylogenetic analysis of European Scutovertex mites (Acari, Oribatida, Scutoverticidae) reveals paraphyly and cryptic diversity – a molecular genetic and morphological approach. Mol Phylogenet Evol. 2010;55:677–688. doi: 10.1016/j.ympev.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spelda J, Reip HS, Oliveira-Biener U, Melzer RR. Barcoding Fauna Bavarica: Myriapoda - a contribution to DNA sequence-based identifications of centipedes and millipedes (Chilopoda, Diplopoda) Zookeys. 2011;156:123–139. doi: 10.3897/zookeys.156.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturmbauer C, Opadiya GB, Niederstätter H, Riedmann A, Dallinger R. Mitochondrial DNA reveals cryptic oligochaete species differing in cadmium resistance. Mol Biol Evol. 1999;16:967–974. doi: 10.1093/oxfordjournals.molbev.a026186. [DOI] [PubMed] [Google Scholar]
- 24.Van Straalen N, Timmermans MJTN. Genetic variation in toxicant-stressed populations: an evaluation of the “genetic erosion” hypothesis. Hum Ecol Risk Assess. 2002;8:983–1002. doi: 10.1080/1080-700291905783. [DOI] [Google Scholar]
- 25.Dupont L, Lazrek F, Porco D, King RA, Rougerie R, Symondson WOC, et al. New insight into the genetic structure of the Allolobophora chlorotica aggregate in Europe using microsatellite and mitochondrial data. Pedobiologia. 2011;54:217–224. doi: 10.1016/j.pedobi.2011.03.004. [DOI] [Google Scholar]
- 26.Rӧmbke J, Aira M, Backeljau T, Breugelmans K, Domínguez J, Funke E, et al.: DNA barcoding of earthworms (Eisenia fetida/andrei complex) from 28 ecotoxicological test laboratories. Appl Soil Ecol 2015; doi:10.1016/j.apsoil.2015.02.010
- 27.Donnelly RK, Harper GL, Morgan AJ, Orozco-Terwengel P, Pinto-Juma GA, Bruford MW. Nuclear DNA recapitulates the cryptic mitochondrial lineages of Lumbricus rubellus and suggests the existence of cryptic species in an ecotoxicological soil sentinel. Biol J Linnean Soc. 2013;110:780–795. doi: 10.1111/bij.12171. [DOI] [Google Scholar]
- 28.Sechi P. An evolutionary history of the peregrine epigeic earthworm Lumbricus rubellus. Cardiff, Wales, United Kingdom: Doctoral dissertation, Cardiff University; 2013. [Google Scholar]
- 29.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;7:e40701. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donnelly RK, Harper GL, Morgan AJ, Pinto-Juma GA, Bruford MW. Mitochondrial DNA and morphological variation in the sentinel earthworm species Lumbricus rubellus. Eur J Soil Biol. 2014;64:23–29. doi: 10.1016/j.ejsobi.2014.07.002. [DOI] [Google Scholar]
- 31.Johns GC, Avise JC. A comparative summary of genetic distances in the vertebrates from the mitochondrial cytochrome b gene. Mol Biol Evol. 1998;15:1481–1490. doi: 10.1093/oxfordjournals.molbev.a025875. [DOI] [PubMed] [Google Scholar]
- 32.Meier R, Zhang G, Ali F. The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst Biol. 2008;57:809–813. doi: 10.1080/10635150802406343. [DOI] [PubMed] [Google Scholar]
- 33.Torres-Leguizamon M, Mathieu J, Decaëns T, Dupont L. Genetic structure of earthworm populations at a regional scale: inferences from mitochondrial and microsatellite molecular markers in Aporrectodea icterica (Savigny 1826) PLoS One. 2014;9:e101597. doi: 10.1371/journal.pone.0101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vega R, Fløjgaard C, Lira-Noriega A, Nakazawa Y, Svenning J-C, Searle JB. Northern glacial refugia for the pygmy shrew Sorex minutus in Europe revealed by phylogeographic analyses and species distribution modelling. Ecography. 2010;33:260–271. [Google Scholar]
- 35.Schmitt T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front Zool. 2007;4:1–13. doi: 10.1186/1742-9994-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmitt T, Varga Z. Extra-Mediterranean refugia: the rule and not the exception. Front Zool. 2012;9:22. doi: 10.1186/1742-9994-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Babik W, Branicki W, Crnobrnja‐Isailović J, Cogălniceanu D, Sas I, Olgun K, et al. Phylogeography of two European newt species - discordance between mtDNA and morphology. Mol Ecol. 2005;14:2475–2491. doi: 10.1111/j.1365-294X.2005.02605.x. [DOI] [PubMed] [Google Scholar]
- 38.Durka W, Babik W, Ducroz JF, Heidecke D, Rosell F, Samjaa R, et al. Mitochondrial phylogeography of the Eurasian beaver Castor fiber L. Mol Ecol. 2005;14:3843–3856. doi: 10.1111/j.1365-294X.2005.02704.x. [DOI] [PubMed] [Google Scholar]
- 39.Gratton P, Konopiński MK, Sbordoni V. Pleistocene evolutionary history of the Clouded Apollo (Parnassius mnemosyne): genetic signatures of climate cycles and a ‘time‐dependent’mitochondrial substitution rate. Mol Ecol. 2008;17:4248–4262. doi: 10.1111/j.1365-294X.2008.03901.x. [DOI] [PubMed] [Google Scholar]
- 40.Wójcik JM, Kawałko A, Marková S, Searle JB, Kotlík P. Phylogeographic signatures of northward post‐glacial colonization from high‐latitude refugia: a case study of bank voles using museum specimens. J Zool. 2010;281:249–262. [Google Scholar]
- 41.Hofreiter M, Serre D, Rohland N, Rabeder G, Nagel D, Conard N, et al. Lack of phylogeography in European mammals before the last glaciation. Proc Natl Acad Sci U S A. 2004;101:12963–12968. doi: 10.1073/pnas.0403618101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hewitt GM. Quaternary phylogeography: the roots of hybrid zones. Genetica. 2011;139:617–638. doi: 10.1007/s10709-011-9547-3. [DOI] [PubMed] [Google Scholar]
- 43.Charlesworth B, Charlesworth D, Barton NH. The effects of genetic and geographic structure on neutral variation. Annu Rev Ecol Evol Syst. 2003;34:99–125. doi: 10.1146/annurev.ecolsys.34.011802.132359. [DOI] [Google Scholar]
- 44.Marinissen JCY, Van den Bosch F. Colonization of new habitats by earthworms. Oecologia. 1992;91:371–376. doi: 10.1007/BF00317626. [DOI] [PubMed] [Google Scholar]
- 45.James SW, Porco D, Decaëns T, Richard B, Rougerie R, Erséus C. DNA barcoding reveals cryptic diversity in Lumbricus terrestris L., 1758 (Clitellata): resurrection of L. herculeus (Savigny, 1826) PLoS One. 2010;5:e15629. doi: 10.1371/journal.pone.0015629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velavan TP, Weller S, Schulenburg H, Michiels NK. High genetic diversity and heterogeneous parasite load in the earthworm Lumbricus terrestris on a German meadow. Soil Biol Biochem. 2009;41:1591–1595. doi: 10.1016/j.soilbio.2009.03.026. [DOI] [Google Scholar]
- 47.Novo M, Almodόvar A, Fernández R, Trigo D, Díaz Cosín DJ. Cryptic speciation of hormogastrid earthworms revealed by mitochondrial and nuclear data. Mol Phylogenet Evol. 2010;56:507–512. doi: 10.1016/j.ympev.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, et al. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature. 2014;515:261–263. doi: 10.1038/nature13685. [DOI] [PubMed] [Google Scholar]
- 49.Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annu Rev Ecol Evol Syst. 2009;40:481–501. doi: 10.1146/annurev.ecolsys.39.110707.173414. [DOI] [Google Scholar]
- 50.Gautier M, Gharbi K, Cezard T, Foucaud J, Kerdelhué C, Pudlo P, et al. The effect of RAD allele dropout on the estimation of genetic variation within and between populations. Mol Ecol. 2013;22:3165–3178. doi: 10.1111/mec.12089. [DOI] [PubMed] [Google Scholar]
- 51.Arnold B, Corbett-Detig RB, Hartl D, Bomblies K. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Mol Ecol. 2013;22:3179–3190. doi: 10.1111/mec.12276. [DOI] [PubMed] [Google Scholar]
- 52.Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 53.Faircloth BC, McCormack JE, Crawford NG, Harvey MG, Brumfield RT, Glenn TC. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst Biol. 2012;61:717–726. doi: 10.1093/sysbio/sys004. [DOI] [PubMed] [Google Scholar]
- 54.Lemmon AR, Emme SA, Lemmon EM. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst Biol. 2012;61:727–744. doi: 10.1093/sysbio/sys049. [DOI] [PubMed] [Google Scholar]
- 55.Kozancioğlu E, Arnqvist G. The maintenance of mitochondrial genetic variation by negative frequency-dependent selection. Ecol Lett. 2014;17:22–27. doi: 10.1111/ele.12195. [DOI] [PubMed] [Google Scholar]
- 56.Fijarczyk A, Babik W. Detecting balancing selection in genomes: limits and prospects. Mol Ecol. 2015;24:3529–3545. doi: 10.1111/mec.13226. [DOI] [PubMed] [Google Scholar]
- 57.Andre J, King RA, Stürzenbaum SR, Kille P, Hodson ME, Morgan AJ. Molecular genetic differentiation in earthworms inhabiting a heterogeneous Pb-polluted landscape. Environ Pollut. 2010;158:883–890. doi: 10.1016/j.envpol.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 58.Giska I, Babik W, Van Gestel CAM, Van Straalen NM, Laskowski R. Genome-wide genetic diversity of rove beetle populations along a metal pollution gradient. Ecotoxicol Environ Saf. 2015;119:98–105. doi: 10.1016/j.ecoenv.2015.04.048. [DOI] [PubMed] [Google Scholar]
- 59.OECD (Organisation for Economic Co-operation and Development): Guidelines for the testing of chemicals No. 222. Earthworm reproduction test (Eisenia fetida/andrei). Paris, France: OECD; 2004.
- 60.ISO (International Organization For Standardization) Soil quality – avoidance test for evaluating the quality of soils and the toxicity of chemicals. Test with Earthworms (Eisenia fetida/andrei) Geneva, Switzerland: ISO 17512–1; 2008. [Google Scholar]
- 61.Pérez-Losada M, Eiroa J, Mato S, Domínguez J. Phylogenetic species delimitation of the earthworms Eisenia fetida (Savigny, 1826) and Eisenia andrei Bouche, 1972 (Oligochaeta, Lumbricidae) based on mitochondrial and nuclear DNA sequences. Pedobiologia. 2005;49:317–324. doi: 10.1016/j.pedobi.2005.02.004. [DOI] [Google Scholar]
- 62.Emerson BC, Cicconardi F, Fanciulli PP, Shaw PJ. Phylogeny, phylogeography, phylobetadiversity and the molecular analysis of biological communities. Philos Trans R Soc Lond B Biol Sci. 2011;366:2391–2402. doi: 10.1098/rstb.2011.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knowles LL, Carstens BC. Delimiting species without monophyletic gene trees. Syst Biol. 2007;56:887–895. doi: 10.1080/10635150701701091. [DOI] [PubMed] [Google Scholar]
- 64.Pérez-Losada M, Ricoy M, Marshall JC, Domínguez J. Phylogenetic assessment of the earthworm Aporrectodea caliginosa species complex (Oligochaeta: Lumbricidae) based on mitochondrial and nuclear DNA sequences. Mol Phylogenet Evol. 2009;52:293–302. doi: 10.1016/j.ympev.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Azarbad H, Niklińska M, Van Gestel CAM, Van Straalen NM, Rӧling WFM, Laskowski R. Microbial community structure and functioning along metal pollution gradients. Environ Toxicol Chem. 2013;32:1992–2002. doi: 10.1002/etc.2269. [DOI] [PubMed] [Google Scholar]
- 66.Giska I, Van Gestel CAM, Skip B, Laskowski R. Toxicokinetics of metals in the earthworm Lumbricus rubellus exposed to natural polluted soils – relevance of laboratory tests to the field situation. Environ Pollut. 2014;190:123–132. doi: 10.1016/j.envpol.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 67.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 68.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. G3. 2011;1:171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Catchen JM, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rozas J. DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol. 2009;537:337–350. doi: 10.1007/978-1-59745-251-9_17. [DOI] [PubMed] [Google Scholar]
- 73.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 74.Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 75.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramos-Onsins SE, Ferretti L, Raineri E, Marmorini G, Burgos W, Vera G. mstatspop: statistical analysis using multiple populations to pipeline with ms. 2015. [Google Scholar]
- 77.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7:574–578. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hubisz M, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9:1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Earl DA, Von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 82.Smouse PE, Long JC, Sokal RR. Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst Zool. 1986;35:627–632. doi: 10.2307/2413122. [DOI] [Google Scholar]
- 83.Jensen JL, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genet. 2005;6:13. doi: 10.1186/1471-2156-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials. This file includes supplementary materials to the main text: Note A1. Detailed description of the Illumina sequencing and Stacks analysis of RAD tags. Table A1. Variation at COI (453 bp) in Lumbricus rubellus populations. Table A2. Variation at ATP6 (563 bp) in Lumbricus rubellus populations. Table A3. Pairwise genetic distances between mtDNA haplotypes H1-H27 found in Poland. Table A4. Mantel test and partial Mantel test statistics for Lumbricus rubellus populations sampled at sites with different levels of metal pollution. Table A5. Information on mtDNA sequence markers of Lumbricus rubellus and the PCR conditions. Table A6. Quality control of Illumina reads from the HiSeq 2000. Table A7. The Stacks commands used to process the RADseq data. Table A8. Comparison of Stacks results for different analysis parameters. Figure A1. Bayesian tree based on the COI sequences of Lumbricus rubellus. Figure A2. The Ln P(D) in Structure analysis of Lumbricus rubellus. Figure A3. Relation between the genetic distance (RADseq FST) and the log(geographic distance) in Lumbricus rubellus populations; a reduced major axis regression based on the Mantel test. Figure A4. Final coverage per RAD tag (mean ± SD) for individual earthworms of Lumbricus rubellus. Figure A5. Effect of the Stacks parameters on the final number of usable RAD tags in Lumbricus rubellus. (PDF 650 kb)
Structure file. This is the main Structure input file generated by Stacks with the use of one SNP per RAD tag. (TSV 438 kb)