

Abstract
Individuals with Angelman syndrome (AS) suffer sleep disturbances that severely impair quality of life. Whether these disturbances arise from sleep or circadian clock dysfunction is currently unknown. Here, we explored the mechanistic basis for these sleep disorders in a mouse model of Angelman syndrome (Ube3am−/p+ mice). Genetic deletion of the maternal Ube3a allele practically eliminates UBE3A protein from the brain of Ube3am−/p+ mice, because the paternal allele is epigenetically silenced in most neurons. However, we found that UBE3A protein was present in many neurons of the suprachiasmatic nucleus—the site of the mammalian circadian clock—indicating that Ube3a can be expressed from both parental alleles in this brain region in adult mice. We found that while Ube3am−/p+ mice maintained relatively normal circadian rhythms of behavior and light-resetting, these mice exhibited consolidated locomotor activity and skipped the timed rest period (siesta) present in wild-type (Ube3am+/p+) mice. Electroencephalographic analysis revealed that alterations in sleep regulation were responsible for these overt changes in activity. Specifically, Ube3am−/p+ mice have a markedly reduced capacity to accumulate sleep pressure, both during their active period and in response to forced sleep deprivation. Thus, our data indicate that the siesta is governed by sleep pressure, and that Ube3a is an important regulator of sleep homeostasis. These preclinical findings suggest that therapeutic interventions that target mechanisms of sleep homeostasis may improve sleep quality in individuals with AS.
SIGNIFICANCE STATEMENT Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by loss of expression of the maternal copy of the UBE3A gene. Individuals with AS have severe sleep dysfunction that affects their cognition and presents challenges to their caregivers. Unfortunately, current treatment strategies have limited efficacy due to a poor understanding of the mechanisms underlying sleep disruptions in AS. Here we demonstrate that abnormal sleep patterns arise from a deficit in accumulation of sleep drive, uncovering the Ube3a gene as a novel genetic regulator of sleep homeostasis. Our findings encourage a re-evaluation of current treatment strategies for sleep dysfunction in AS, and suggest that interventions that promote increased sleep drive may alleviate sleep disturbances in individuals with AS.
Keywords: Angelman syndrome, EEG, imprinting, sleep, suprachiasmatic nucleus, Ube3a
Introduction
Angelman syndrome (AS) is a severe neurodevelopmental disorder characterized by developmental delay, seizures, lack of speech, ataxia, and profound sleep disturbances (Williams et al., 2006; Dan, 2009; Thibert et al., 2013). These sleep disturbances include reduced need for sleep, irregular sleep/wake cycles, and difficulty initiating and maintaining sleep. While sleep disturbances are more severe during childhood, they persist in roughly 70% of adults with AS (Walz et al., 2005; Pelc et al., 2008; Larson et al., 2015). The finding that AS individuals can exhibit dampened rhythms of circulating melatonin (Takaesu et al., 2012) has led to the suggestion that circadian clock dysfunction contributes to the sleep disturbance. However, melatonin treatments have had limited success in improving sleep quality in individuals with AS (Braam et al., 2008; Pelc et al., 2008). Thus, there is a need to better understand the neurobiology underlying disturbed sleep in individuals with AS to guide more effective behavioral therapies and pharmacological treatments.
The sleep deficits in AS presumably arise from a deficiency in UBE3A, as mutation or deletion of the maternally inherited allele of the UBE3A gene causes AS (Kishino et al., 1997; Matsuura et al., 1997; Sutcliffe et al., 1997; Williams et al., 2010; Mabb et al., 2011). The paternally inherited UBE3A allele is epigenetically silenced in almost all neurons (Albrecht et al., 1997; Rougeulle et al., 1997; Yamasaki et al., 2003); thus, the loss of a functional maternal UBE3A allele causes a nearly complete loss of UBE3A protein in most regions of the mammalian brain (Gustin et al., 2010; Judson et al., 2014). In mice, this epigenetic silencing of the paternal allele is found in neurons by the first postnatal week, while glial cells preserve biallelic expression of Ube3a into adulthood (Judson et al., 2014). The UBE3A protein (also known as E6-AP) is an E3 ubiquitin ligase that targets substrate proteins for proteasomal degradation (Scheffner et al., 1993). In Drosophila, loss of the UBE3A protein disrupts circadian patterns of locomotor activity (Wu et al., 2008; Gossan et al., 2014). In mammals, maternal deletion or RNAi-mediated knockdown of Ube3a can cause subtle alterations in circadian rhythmicity, presumably by stabilizing the core clock protein BMAL1 (Gossan et al., 2014; Shi et al., 2015). Together, these findings suggest a role for UBE3A in regulating circadian clock function in individuals with AS, potentially contributing to the sleep disturbances in individuals with AS.
The epigenetic mechanisms silencing expression of the paternal Ube3a allele are nearly identical in mouse and human brains (Nicholls and Knepper, 2001; Lalande and Calciano, 2007). Mouse models of AS (Ube3am−/p+ mice) display a number of traits that resemble core features of AS, including seizure susceptibility, ataxias, memory impairments, and sleep deficits (Jiang et al., 1998; Miura et al., 2002; Colas et al., 2005; van Woerden et al., 2007; Egawa et al., 2012; Huang et al., 2013). To provide insight into sleep dysregulation in individuals with AS, we therefore used the mouse model to explore mechanisms by which maternal Ube3a deletions might contribute to disrupted sleep regulation. We find that maternal Ube3a deletion does not significantly alter circadian rhythmicity, but markedly disrupts sleep homeostasis, especially the accumulation of sleep pressure. Thus, our findings support a role for Ube3a as a novel sleep-regulatory gene, and suggest that deficient sleep-pressure accumulation, rather than altered circadian clock function, is likely the major defect underlying sleep disturbances in AS.
Materials and Methods
Animals and circadian behavior.
Ube3am−/p+ mice (Jiang et al., 1998) and their wild-type littermates were used for circadian behavior and sleep studies and were obtained from Jackson Laboratory (stock #016590). Mice were 6–8 weeks old when studies began, and 10–12 months old when killed for suprachiasmatic nucleus (SCN) dissection and quantitative PCR (qPCR) analysis. Mice were individually housed and maintained under a 12 h light/12 h dark cycle (LD 12:12, light: ∼150 lux) unless indicated otherwise, with ad libitum access to food (Purina Rodent Chow #5001) and water, in rooms maintained at 22 ± 2°C ambient temperature. To monitor behavioral rhythms, mice were transferred to cages, equipped with running wheels, inside of secondary light-controlled enclosures (Evans et al., 2013). Wheel-running activity was monitored continuously and analyzed using Clocklab (Actimetrics). At least 2 weeks were allowed between manipulations of the lighting conditions to ensure mice were stably entrained. Circadian periods were determined using regressions through activity onsets across the 21 d period in constant darkness (χ2 periodogram analysis produced identical results), and phase shifts were measured as described previously (DeBruyne et al., 2006).
Rhythmic gene expression.
At the indicated times in the LD 12:12 cycle, mice were killed and the SCN/anterior hypothalami were dissected out and flash-frozen. RNA was extracted using TRI Reagent (Molecular Research Center), including 1 μl of GlycoBlue (Ambion) per SCN as a precipitation carrier, and reverse-transcribed using the high-capacity cDNA reverse transcription kit (Life Technologies). qPCR was performed using SsoAdvanced Sybr Green Supermix (Bio-Rad) on a CFX96 real-time detection system (Bio-Rad). All data are plotted relative to Gapdh as a loading control (DiTacchio et al., 2011). Dbp, RevErbα, and Bmal1 primers were described in (Wu et al., 2008), and the Per1 and Cry1 primers were described previously (Anand et al., 2013). Rhythms in Bmal1-Luc U2OS cells were monitored using a Lumicycle (Actimetrics) starting 48 h after transfecting cells with 10 pmol of AllStars negative control (Qiagen) or siRNAs directed against Ube3a (Qiagen, catalog #GS7337) as described previously (Yamazaki and Takahashi, 2005; Baggs et al., 2009). Knockdown was verified via Western blot with anti-UBE3A antibodies from Sigma-Aldrich (catalog #SAB1404508).
Immunohistochemistry.
Ten-to-sixteen-week-old male and female mice (Ube3am+/p+ or Ube3am−/p+) were housed under a LD 12:12 cycle and given ad libitum access to food and water before perfusion. At the indicated zeitgeber times (ZTs), mice were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.) before transcardial perfusion with room-temperature PBS immediately followed by room-temperature phosphate-buffered paraformaldehyde, pH 7.3. Perfused brains were removed from the skull and postfixed overnight at 4°C. Fixed brains were cryoprotected in 10, 20, and 30% sucrose in PBS at 4°C for 12 h each, before being frozen in dry ice and sectioned coronally at 40 μm on a freezing sliding microtome (Thermo Scientific). Sections were stored in a cryopreservative solution (v/v: 45% PBS, 30% ethylene glycol, 25% glycerol) at −20°C before free-floating immunohistochemistry.
Tissue sections were washed several times in PBS and blocked in PBS with 5% normal goat serum and 0.2% Triton X-100 (NGST) for 1 h at room temperature. Blocked tissue sections were incubated with primary antibodies diluted in NGST for 48 h at 4°C. Sections were then washed several times in PBS containing 0.2% Triton X-100 before incubation with secondary antibodies (diluted in NGST) for 1 h at room temperature. The following primary antibodies were used: mouse anti-UBE3A (1:1000, clone 3E5; Sigma-Aldrich, catalog #SAB1404508) and rabbit anti-PER2 (1:5000, clone R38; Millipore, catalog #AB2202). The following secondary antibodies were used at a concentration of 1:500: goat anti-mouse IgG1 Alexa 568 (Millipore, catalog #A21124), goat anti-mouse IgG2A Alexa 488 (Millipore, catalog #A21131), goat anti-rabbit Alexa 568 (Millipore, catalog #A11011), and goat anti-rabbit Alexa 633 (Millipore, catalog #A21071). In all experiments, 4′,6-diamidino-2-phenylindole (DAPI) was added during the secondary antibody incubation at a concentration of 700 ng/ml for nuclear counterstaining. Perfusions, tissue sectioning, and immunostaining were performed by experimenters blind to genotype. Brain sections compared within figures were stained within the same experiment under identical conditions.
Confocal microscopy and image analysis.
Immunostained sections containing the SCN were imaged using a Zeiss LSM 710 confocal microscope equipped with ZEN Imaging Software (Zeiss). Single-plane optical slices 1.8 μm thick were acquired using a 20× objective (Plan-Apochromat 20×/0.8 M27) at a pixel dwell time of 3.15 μs with 4× averaging. Images containing the SCN, third ventricle, and surrounding hypothalamic regions were acquired by tiling 3 × 3 micrographs of 1024 × 1024 pixels that were stitched using ZEN software. Images compared within figures were acquired using identical acquisition parameters. All images to be compared underwent identical manipulations.
Quantification of immunofluorescent signals was performed in ImageJ (http://imagej.nih.gov/ij/). Regions bounding the left and right SCN were created manually for each image using the DAPI channel by an experimenter blind to genotype. The regions were applied to the UBE3A or PER2 channels, and the mean gray values within SCN regions were measured. An oval region of a standard size across all images (150 × 300 μm) was also placed in the adjacent lateral hypothalamus, and the average intensity within this region was subtracted from the average intensity in the SCN to normalize the SCN intensity value. Values in the lateral hypothalamus regions did not change across ZTs. Sections containing rostral, central, and caudal regions of the SCN were imaged for all conditions in a balanced fashion and included for quantification.
Surgery.
Electroencephalogram (EEG) and electromyogram (EMG) electrodes for polysomnographic recording of sleep–wake states were implanted in anesthetized mice. Electrodes were implanted by surgeons blind to the experiment. A prefabricated head mount (Pinnacle Technologies) was used to position four stainless-steel epidural screw electrodes. Frontal and ground electrodes were located 1.5 mm anterior to bregma and 1.5 mm on either side of the central suture, and parietal/occipital and common references were located 3.5 mm posterior to bregma and 1.5 mm on either side of the central suture. Electrical continuity between the screw electrodes and headmount was aided by the use of silver epoxy. EMG activity was monitored using stainless-steel Teflon-coated wires integrated into the headmount and inserted bilaterally into the nuchal muscle. The headmount (integrated 2 × 3 pin grid array) was secured to the skull with dental acrylic. Over the next 36 h, mice were given Buprenex (buprenorphine; 2 mg/kg subcutaneously) once every 12 h to control pain. All procedures were performed on a heating pad; mice were allowed to recover for ≥14 d before being transferred to sleep-recording chambers.
EEG/EMG recordings.
Following recovery, mice were placed in a sleep-recording chamber and connected to a lightweight tether connected to a low-resistance commutator mounted over the cage (Pinnacle Technologies), allowing complete freedom of movement throughout the cage. Except for the recording tether, conditions in the recording chamber were identical to those in the home cage. Mice were allowed ≥7 additional days to acclimate to the tether. Recording of EEG and EMG waveforms began at ZT 0 (light onset in the 12 h light/dark cycle). Following a 24 h baseline recording, mice were sleep deprived during the first 6 h of the light phase (ZT 0–6) by gentle handling (introduction of novel objects into the cage, tapping on the cage, and, when necessary, delicate touching) and allowed an 18 h recovery opportunity (ZT 6–24). Data acquisition was performed on a PC running Sirenia Acquisition software (Pinnacle Technologies), a software system designed specifically for polysomnographic recording in rodents. EEG signals were low-pass filtered with a 30 Hz cutoff and collected continuously at a sampling rate of 400 Hz. After collection, all waveforms were classified in 2 s epochs by a trained observer blind to the genotype and experimental condition (using both EEG leads and EMG) as wake (low-voltage, high-frequency EEG; high-amplitude EMG), nonrapid-eye-movement (NREM) sleep (high-voltage, mixed-frequency EEG; low-amplitude EMG), or rapid-eye-movement (REM) sleep [low-voltage EEG with a predominance of theta activity (6–10 Hz); very low amplitude EMG]. EEG epochs determined to have artifact (interference caused by scratching, movement, eating, or drinking) was excluded from analysis. Artifact comprised <5% of all recordings used for analysis. Analysis of NREM delta power and NREM spectral distribution was accomplished by applying a fast Fourier transformation to raw EEG waveforms. Only epochs classified as NREM sleep were included in this analysis. Delta power was measured as spectral power in the 0.5–4 Hz frequency range, and expressed as a percentage of total spectral power in the EEG signal (0.5–100 Hz) during that time period.
Wave incidence.
Analysis was performed using custom-written functions in Igor Pro 6.2 (WaveMetrics) as previously described (Ehlen et al., 2013). Briefly, raw EEG signals from the frontal-parietal derivation were bandpass filtered in the frequency range indicated using a Butterworth fourth-order bandpass filter (Igor Pro routine FilterIIR). Peaks in the filtered data were detected as negative deflections between two zero crossings. The amplitudes (maximum negative deflection from zero crossing) of all peaks detected in an entire 24 h period were then determined. The upper 30% of peak amplitudes that occurred in epochs scored as wake were then counted and expressed as peaks per minute (wave incidence). The wave incidence data were then combined into 2 h bins for graphing and statistical analysis. Short durations of wake (<10 epochs) were excluded from analysis.
Statistics.
Data are expressed as mean ± SEM unless otherwise specified. Quantified data were plotted in Microsoft Excel or GraphPad Prism 6 (GraphPad Software), and statistical analysis was performed using StatPlus (AnalystSoft) or GraphPad Prism 6. Two-tailed Student's t tests, one-way ANOVA, and two-way ANOVA were performed as indicated, using corrections for multiple comparisons and post hoc tests as specified in the text. For all statistical tests, p < 0.05 was considered statistically significant.
Study approval.
All animal procedures were approved by the institutional animal care and use committees of either the Morehouse School of Medicine or the University of North Carolina School of Medicine, and performed in accordance with the guidelines of the U.S. National Institutes of Health.
Results
Circadian clock function is largely intact in AS model mice
To assess whether perturbed circadian rhythms might contribute to sleep disturbances associated with AS, we placed Ube3am−/p+ mice (congenic on C57BL/6J background) and wild-type (Ube3am+/p+) littermate controls in cages equipped with running wheels, and continuously recorded wheel-running behavior. Activity levels were similar between the two genotypes (Fig. 1A,B), suggesting that the impaired motor coordination and reduced activity in open-field tests reported in Ube3am−/p+ mice (Jiang et al., 1998; Huang et al., 2013) does not impair wheel-running activity. We asked whether maternal loss of Ube3a alters the period of circadian behavioral rhythms in constant darkness, or the clock's ability to adjust its timing to aberrant light-schedules such as simulated jet lag, as a defect in either could signify impaired clock function. We found that neither circadian period nor the ability of the clock to shift in response to altered lighting paradigms was significantly changed in Ube3am−/p+ mice (Fig. 1C–E). Thus, at a behavioral level, the circadian system appears to function normally in Ube3am−/p+ mice.
Figure 1.

Circadian behavior is largely intact in AS model mice. A, Double-plotted wheel-running records obtained from wild-type (Ube3am+/p+) and AS model (Ube3am−/p+) mice on 12 h light/dark cycle (LD) and in constant darkness (DD). Running activity is indicated by tick marks, and subsequent days are plotted to the right and below each previous day. The shading illustrates the light cycle schedules. B, Average daily wheel-running counts over the last 14 d in LD. Means ± SEM, n = 11 Ube3am+/p+ or 9 Ube3am−/p+ mice (two-tailed t test, p > 0.05). C, Average circadian free-running periods (±SEM) measured over 21 d in DD from the same animals in B (two-tailed t test, p > 0.05). D, Circadian resetting in response to 4 h light pulses applied early (ZT 13–17) or late (ZT 20–24) during the night preceding DD. The number of hours the circadian clock was shifted is plotted as mean ± SEM from 11 Ube3am+/p+ and 8 Ube3am−/p+ mice (two-tailed t test, p > 0.05). E, Example records (left) and plot of activity onset timing (right) during re-entrainment to a simulated 6 h advance jet lag. The activity onset plot represents mean ± SEM from 21 Ube3am+/p+ and 19 Ube3am−/p+ mice, combined from two independent experiments. Activity onset time was not significantly different between genotypes on any day (two-tailed t test on each day, p > 0.05).
Because behavioral circadian rhythms are linked to the rhythmic expression of several “clock genes” in the SCN, we also examined the expression patterns of several clockwork genes in the SCN of Ube3am−/p+ and wild-type mice (Zylka et al., 1998; Partch et al., 2014; Fig. 2A). Maternal loss of Ube3a had no marked effect on diurnal expression of the clock genes Per1, RevErbα, Cry1, and Bmal1, or the circadian output gene Dbp (Fig. 2A). Likewise, rhythms in immunoreactivity of the clock protein PER2 were also indistinguishable in SCN of Ube3am−/p+ and wild-type mice (Fig. 2B,C). Thus, the molecular clockwork in the SCN of Ube3am−/p+ mice displays no obvious differences from that of littermate controls, consistent with their unaltered circadian behavioral rhythmicity.
Figure 2.
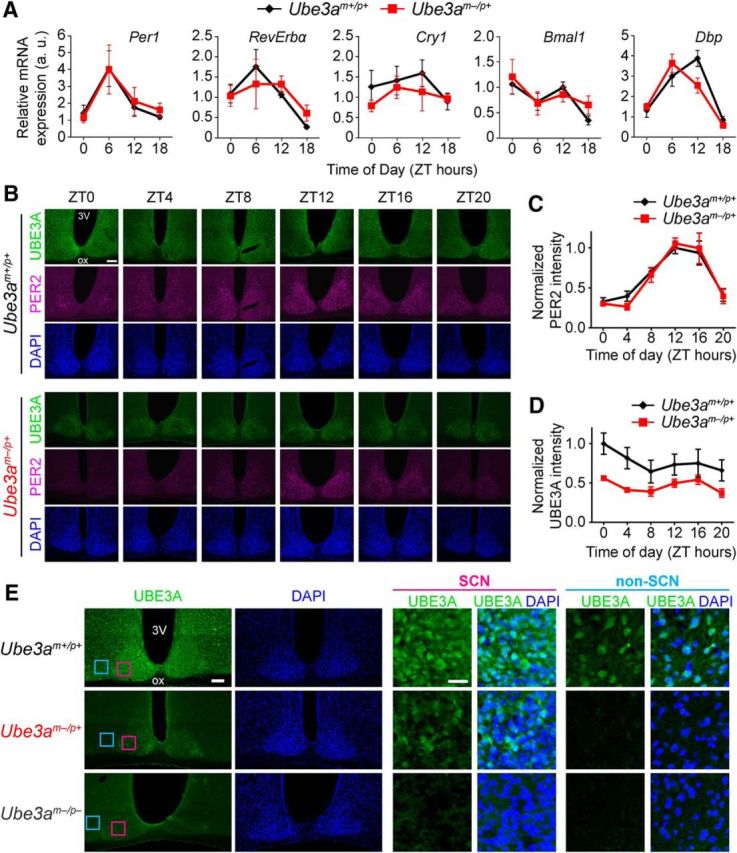
AS model mice display normal molecular rhythmicity and persistent UBE3A expression in the SCN. A, mRNA levels of the indicated genes measured by qPCR across the day in SCN. Each data point represents the mean ± SEM from four to six mice per genotype and time. B, Representative images of UBE3A and PER2 immunostaining in coronal sections through the SCN from adult Ube3am+/p+ or Ube3am−/p+ mice perfused at the indicated ZTs. DAPI was used as a nuclear counterstain. 3V, Third ventricle; ox, optic chiasm. Scale bar, 100 μm. C, Quantification of PER2 immunofluorescence intensity in SCN of adult Ube3am+/p+ or Ube3am−/p+ mice killed at 4 h intervals across the day. There was no difference between the two genotypes in rhythmic expression patterns of PER2 in the SCN (two-way ANOVA, main effect of genotype: F(1,48) = 0.113, p = 0.738; main effect of ZT: F(5,48) = 24.0, p < 0.0001). Each point represents the mean ± SEM of five mice per genotype and time, normalized to peak intensity (ZT 12, Ube3am+/p+). D, Quantification of UBE3A immunofluorescence intensity in the same sections as C. There was no significant main effect of ZT on UBE3A abundance within each genotype (two-way ANOVA: F(5,48) = 1.65, p = 0.166). However, there was an effect of genotype on UBE3A expression (two-way ANOVA: F(1,48) = 24.2, p < 0.0001). Each point represents the mean ± SEM of five mice per genotype and time, normalized to peak intensity (ZT 0, Ube3am+/p+). E, Low-magnification and high-magnification images of immunofluorescent staining for UBE3A and DAPI in coronal sections through the SCN from adult Ube3am+/p+, Ube3am−/p+, and Ube3am−/p− mice. High-magnification images show expression of UBE3A in a subset of SCN neurons (pink boxes in left images), but not in a neighboring region of non-SCN hypothalamus (blue boxes) in Ube3am−/p+ mice. DAPI counterstain demarcates the dense population of cells that make up the SCN. Scale bars: left, 100 μm; right, 25 μm.
Previous work had established that, due to silencing of the paternal allele, Ube3a expression in mature neurons arises from the maternal allele (Rougeulle et al., 1997; Yamasaki et al., 2003; Judson et al., 2014). We therefore expected UBE3A protein to also be absent from the SCN of Ube3am−/p+ mice. To confirm this, we examined UBE3A protein expression patterns in wild-type and Ube3am−/p+ mice using an established immunofluorescence protocol that shows high specificity and sensitivity for UBE3A immunofluorescent signal (Judson et al., 2014). In wild-type mice, UBE3A protein was expressed in nearly all cells in the SCN and surrounding hypothalamus, and did not cycle in abundance (Fig. 2B,D). Much to our surprise, we found UBE3A protein was also robustly expressed throughout the SCN of Ube3am−/p+ mice (Fig. 2B,D,E). As expected in Ube3am−/p+ mice, regions surrounding the SCN exhibited no detectable UBE3A, but the SCN maintained constitutive UBE3A levels at ∼50% compared with wild-type SCN across the circadian day (Fig. 2D). Visual inspection suggested that both the number of cells expressing UBE3A and the intensity of labeling were reduced in the SCN of Ube3am−/p+ mice. We found UBE3A staining to be completely absent from the SCN of homozygous Ube3a-null mice (Fig. 2E), confirming the specificity of the antibody. The persistent expression of UBE3A in the SCN of Ube3am−/p+ mice must originate from the paternal allele, implying that, unlike in most other brain regions, Ube3a expression is biallelic in the SCN of adult mice.
Since UBE3A is still present in the SCN of maternally deleted Ube3a mice, it is possible that the remaining UBE3A is sufficient to maintain normal clock function in the SCN. Homozygous Ube3a knock-out mice on C57BL/6J background, however, are born at sub-Mendelian rates, and survivors exhibit morphological complications, including reduced body and brain weights (Jiang et al., 1998; M.C. Judson and B.D. Philpot, unpublished observations). These systemic influences of the mutation limit effective examination of behavioral circadian rhythms in adult homozygous Ube3a-null mice. We therefore opted to determine whether RNAi-mediated knockdown of Ube3a had substantial effects on circadian rhythmicity in the well established Bmal1-luciferase U2OS cell model (Baggs et al., 2009; Zhang et al., 2009). Consistent with similar experiments in NIH3T3 cells (Gossan et al., 2014), we found that UBE3A knockdown to <20% of control levels had a small but significant effect on circadian period, lengthening it by ∼36 min (scrambled siRNA negative control, 27.2 ± 0.1 h; Ube3a-siRNA treated, 27.8 ± 0.1 h; p < 0.05, Student's t test; n = 7–8 combined from two independent experiments). Thus, depletion of UBE3A beyond levels seen in the SCN of Ube3am−/p+ mice slightly alters circadian clock function, consistent with its proposed role in the mammalian circadian clockwork (Gossan et al., 2014; Shi et al., 2015). However, these data suggest that substantial dysfunction of the circadian clockwork is unlikely in homozygous Ube3a knock-out mice. Together, our data suggest (1) that maternal deletion of Ube3a does not markedly disrupt circadian clock function or its regulation of locomotor activity, and (2) that deficits in circadian rhythmicity may not be the major contributing mechanism for disturbed sleep patterns in AS.
Maternal Ube3a deletion eliminates the active-phase rest period
In contrast to the relatively normal circadian locomotor activity seen in Ube3am−/p+ mice, maternal loss of Ube3a dramatically altered the organization of their daily activity patterns (Fig. 3A). Wild-type mice typically became active at lights-off and remained active for the first half of the night. This primary bout of activity was followed by a rest period with little locomotor activity that lasted 2–3 h followed by a second bout of activity. This rest period (also known as “nocturnal break” or “siesta”) is typically observed in studies of sleep and locomotor activity in this strain of mice (C57BL/6J; Valentinuzzi et al., 1997; Challet et al., 2001; Dudley et al., 2003). The remarkably precise timing of the rest period is readily apparent in 14 d average-activity profiles of all wild-type mice in our study (Fig. 3B,C). Like wild-type mice, Ube3am−/p+ mice had an activity onset immediately after lights-off. However, locomotor activity in these mice was restricted to a single consolidated bout that persisted throughout most of the night. Unlike wild-type mice, Ube3am−/p+ mice did not display any consistent rest periods during the night (Fig. 3A,B). The absence of this rest period was highly penetrant; the genotype of each individual could be accurately predicted by its activity profile (Fig. 3C).
Figure 3.
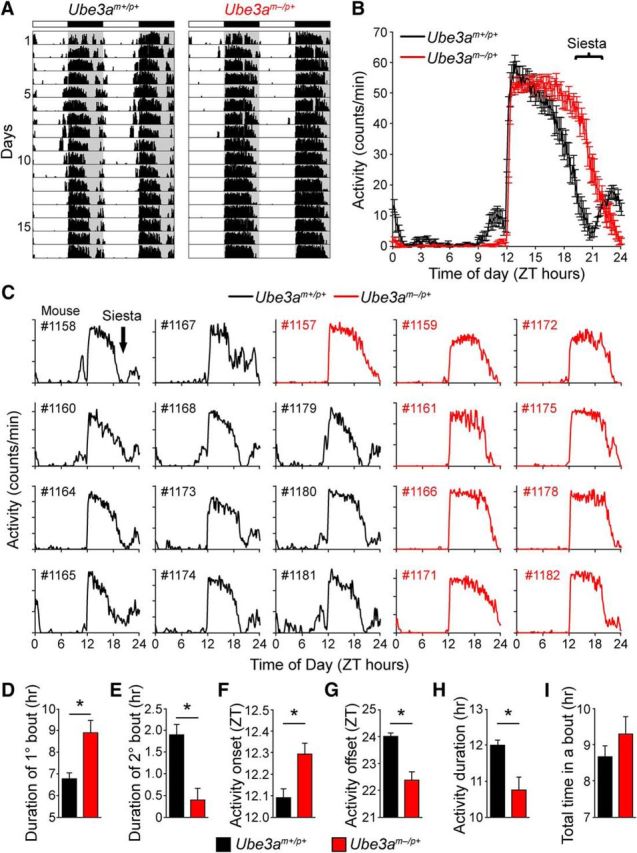
Reorganization of activity patterns in AS model mice eliminates the active-phase rest period. A, Typical wheel-running records depicting the pattern of activity over 17 d in 12 h light/dark cycle (LD12:12). Note the striking difference between genotypes in the nighttime activity pattern (gray shading). B, Average daily activity profiles over 14 d in LD12:12 of Ube3am+/p+ (black) and Ube3am−/p+ (red) mice (mean ± SEM, n = 9–11 mice per genotype). Bracket indicates the primary difference in activity profiles between genotypes. C, Individual 14 d average-activity profiles for each of the mice included in B. Note the siesta-like break in activity (black arrow) present in all the Ube3am+/p+ records (black), but absent from the Ube3am−/p+ records (red). D–I, Comparison of activity pattern characteristics between Ube3am+/p+ and Ube3am−/p+ mice on LD12:12. Means ± SEM are shown (n = 11 Ube3am+/p+; n = 8 Ube3am−/p+). Asterisk indicates significant differences (two-tailed t test on each day, p < 0.05). Bout calculations were derived from each individual's average-activity profile shown in C. A bout was defined as >30 min of continuous activity preceded and followed by a period of inactivity for >30 min, measured from 11 d average-activity profiles during LD12:12.
Overall daily activity levels were comparable to those of wild-type mice (Fig. 1B), suggesting that the Ube3am−/p+ mice were not simply more active. Instead, Ube3am−/p+ mice ended their nightly activity nearly 2 h earlier than wild-type mice, despite a slightly delayed activity onset (Fig. 3D-G). Thus, Ube3am−/p+ mice completed their main bout of activity at approximately the same time wild-type mice began their second bout of activity (Fig. 3B). Consequently, Ube3am−/p+ mice were active over a ∼2-h-shorter portion of the 12 h night (Fig. 3H), despite having comparable levels of total activity (Fig. 3I). Extending the dark period by 6 h (LD 6:18) lengthened the total duration of activity in both genotypes as expected, but did not induce a “second bout” of activity in Ube3am−/p+ mice (data not shown). This indicates that the missing rest period in Ube3am−/p+ mice is not due to a change in its timing relative to the normal 12 h light/dark cycle. We conclude that the loss of maternal Ube3a alters the nighttime distribution of locomotor behavior, eliminating a timed rest period that normally separates locomotor activity into discrete bouts.
AS model mice have altered sleep regulation
Having determined that circadian rhythmicity was largely unaffected in Ube3am−/p+ mice, we explored whether changes in sleep contribute to their altered activity profiles. Overall, polysomnographic findings were consistent with the wheel-running activity of both genotypes (Fig. 4A,B). Both strains displayed high amounts of sleep during the day and low amounts during the 12 h night (Fig. 4B), typical of nocturnal rodents. Both wild-type and Ube3am−/p+ mice slept for most of the day, with no obvious differences in amounts of total sleep, wake, or overall sleep fragmentation between genotypes (data not shown). Thus, sleep amount was not dramatically altered in Ube3am−/p+ mice during the primary rest period for mice.
Figure 4.

AS model mice have altered sleep–wake distribution. A, Representative polysomnographic recordings for Ube3am+/p+ and Ube3am−/p+ mice in each vigilance state. B, Total sleep or wake in Ube3am+/p+ and Ube3am−/p+ mice, recorded over an uninterrupted 24 h period in 12 h light/dark cycle. Data are shown as percentage of each 2 h block spent asleep or awake within each mouse, averaged within genotypes (mean ± SEM; n = 6 Ube3am+/p+ or n = 8 Ube3am−/p+ mice). Note the difference in sleep/wake amounts between genotypes across the night (indicated by the gray shading). *p < 0.008 between genotypes at the indicated times (post hoc t test; Bonferroni corrected).
In contrast, sleep patterns during the dark were dramatically altered by the loss of maternal Ube3a. The distribution of sleep during the dark period in wild-type C57BL/6 mice was characteristic of this strain (Huber et al., 2000; Wisor et al., 2008; Paul et al., 2009). Wild-type mice slept little during the night, except for a rest period that peaked at ZT 21, in which sleep was predominant (Fig. 4B). The timing of this nighttime siesta corresponded well to the rest period in locomotor activity (compare Figs. 3B, 4B). In contrast to the wild-type mice, Ube3am−/p+ mice slept at a nearly constant level throughout the dark period (Fig. 4B; two-way ANOVA for genotype × time-of-day interactions in total sleep; p < 0.001). Ube3am−/p+ mice spent more time sleeping and less time awake over the first part of the night compared with wild-type controls, and conversely slept less than wild-type mice later in the night due to the lack of the siesta (Fig. 4B). The consistent level of sleep across the dark period suggests that the lack of siesta, and associated rest period in locomotor activity, may reflect a marked dysregulation in sleep homeostasis in Ube3am−/p+ mice.
To further explore sleep regulation in Ube3am−/p+ mice, we assessed the architecture of REM and NREM (or slow-wave) sleep across the 24 h day (Fig. 5A–F). The overall pattern of REM sleep displayed by wild-type mice mimicked the pattern of total sleep, with high levels during the day and low levels during the early night that increased to a peak during the siesta (Fig. 5A). The REM sleep pattern in Ube3am−/p+ mice was similar to the wild-type pattern, with the important exception that these mice displayed no increase in REM sleep during late night, when wild-type mice exhibit a siesta (Fig. 5A). The loss of siesta REM sleep resulted in a substantial (∼20%) reduction in the total amount of daily REM sleep in Ube3am−/p+ mice (Ube3am−/p+, 64 ± 2 min; wild type, 80 ± 5 min; t(12) = 2.72, p = 0.019). This decrease in REM sleep was due to a reduction in the number of REM bouts without a significant change in REM bout duration (Fig. 5B,C), perhaps secondary to the effects on NREM sleep regulation.
Figure 5.

REM and NREM sleep states are reduced and fragmented in AS model mice during the active phase. A, Time spent in REM sleep across the day in Ube3am+/p+ and Ube3am−/p+ mice. Two-way ANOVA revealed significant main effects of time and genotype (main effect of time-of-day, F(11,1) = 51.7, p < 0.001; main effect of genotype, F(11,1) = 8.2, p = 0.015), as well as a significant interaction of time-of-day and genotype (interaction: F(11,1) = 4.0, p < 0.001). B, Number of REM bouts across the day in Ube3am+/p+ and Ube3am−/p+ mice. Two-way ANOVA revealed a significant main effect of time-of-day (F(11,1) = 24.4, p < 0.001) but not genotype (F(11,1) = 1.8, p > 0.20), and a significant interaction of time and genotype (interaction: F(11,1) = 2.5, p = 0.006). C, Duration of REM bouts across the day in Ube3am+/p+ and Ube3am−/p+ mice. Two-way ANOVA revealed a statistically significant main effect of genotype (F(11,1) = 9.6, p = 0.009) but not time (F(11,1) = 2.3, p = 0.15), and no interaction of time-of-day and genotype (F(11,1) = 1.8, p = 0.057). D, Time spent in NREM sleep during this period. Two-way ANOVA revealed a significant main effect of time-of-day (F(11,1) = 31.3, p < 0.001) but not genotype (F(11,1) = 0.24, p = 0.63), and a significant interaction of time-of-day and genotype (F(11,1) = 4.3, p < 0.001). E, Number of NREM bouts across the day in Ube3am+/p+ and Ube3am−/p+ mice. Two-way ANOVA revealed a significant main effect of time (F(11,1) = 9.0, p < 0.001) but not genotype (F(11,1) = 2.4, p = 0.15), and a significant interaction of time-of-day and genotype (F(11,1) = 2.9, p = 0.002). F, Duration of NREM bouts across the day in Ube3am+/p+ and Ube3am−/p+ mice. Two-way ANOVA revealed a significant main effect of time-of-day (F(11,1) = 4.0, p < 0.001) but not genotype (F(11,1) = 1.3, p = 0.29), and a significant interaction of time and genotype (F(11,1) = 1.9, p = 0.044). Data in all panels are shown as percentages of time spent in each arousal state, bout number, or mean bout duration of each 2 h block within each mouse, averaged within genotypes (mean ± SEM; n = 6 Ube3am+/p+ or n = 8 Ube3am−/p+ mice). Asterisk indicates significant differences between genotypes at the indicated times (p < 0.05, post hoc t test; Bonferroni corrected).
The overall architecture of NREM sleep displayed by both genotypes of mice was reminiscent of their respective patterns of total sleep (Fig. 5D). Wild-type mice exhibited very little NREM sleep just after lights-off that progressively increased and peaked during the siesta. Ube3am−/p+ mice displayed a quite different pattern of NREM sleep throughout the night (Fig. 5D). Ube3am−/p+ mice spent more time in NREM sleep during the early night (ZT 14), and less NREM sleep during the late night (ZT 21). These differences reflect a change in sleep–wake architecture without alterations in total amounts of daily NREM sleep (total daily NREM sleep, Ube3am−/p+, 589 ± 15 min; wild type, 599 ± 19 min; t(12) = 0.4, p = 0.69).
Maternal Ube3a loss had a striking effect on the architecture of NREM sleep, as reflected by changes in both NREM bout number and duration. In wild-type mice, shorter and fewer bouts of NREM sleep occurred during the first few hours of the dark phase and progressively increased to a peak during the siesta (Fig. 5E,F). Ube3am−/p+ mice had NREM bout durations that were at their shortest near dark onset, but unlike wild-type mice, NREM bouts remained short for a majority of the dark period (Fig. 5F). The number of NREM bouts also remained high during the early night in Ube3am−/p+ mice. These results show that Ube3am−/p+ mice have more fragmented nighttime NREM sleep than wild-type mice, suggesting that maternal Ube3a loss disrupts mechanisms involved in initiating and maintaining NREM sleep during the night.
AS mice have reduced accumulation of sleep pressure
The “two-process” model of sleep regulation posits that sleep is primarily regulated by two biological processes: output signals from the circadian clock, independent of sleep–wake history; and homeostatic sleep “pressure,” which is driven by prior sleep–wake history (Borbély, 1982). In this context, the changes in daily sleep patterns in Ube3am−/p+ mice might be driven by alterations in either the circadian clock or homeostatic processes. Since circadian processes are largely preserved in Ube3am−/p+ mice, we decided to investigate the homeostatic drive to sleep.
NREM delta (0.5–4 Hz) power is a measure of sleep intensity and a well established marker of sleep pressure; it is positively correlated with prior time spent awake, and sleep deprivation is followed by increases in NREM delta power (Huber et al., 2000). We found that NREM delta power declined during the day at an equivalent rate for mice of both genotypes, suggesting that Ube3am−/p+ mice maintain a neurotypical ability to dissipate sleep pressure during the day (Fig. 6A). In contrast, marked differences in NREM delta power were observed during the night. In wild-type mice, NREM delta power increased throughout the night to a peak coincident with the siesta (Fig. 6A). In contrast, Ube3am−/p+ mice exhibited a considerably slower increase in delta power, and thus sleep-pressure accumulation, throughout the night (Fig. 6A). Normalizing 24 h average NREM delta power to the 24 h average waking delta power for each mouse revealed that NREM delta power was significantly lower in Ube3am−/p+ mice than in that of littermate controls (wild type, 179 ± 3%; Ube3am−/p+, 128 ± 3%; t(13) = 2.8, p = 0.008, Student's t test), indicating the effect on delta power was specific to NREM sleep.
Figure 6.

AS model mice exhibit reduced nighttime sleep-pressure accumulation. A, NREM sleep intensity, as measured by delta power, is significantly reduced in Ube3am−/p+ mice during the latter portion of the night (interaction of time-of-day and genotype, F(5,1) = 3.0, p < 0.02; main effect of time-of-day, F(5,1) = 7.3, p < 0.001; main effect of genotype, F(5,1) = 0.72, p = 0.42). Data represent NREM relative delta power (power 0.5–4 Hz/total EEG power) and are presented as 2 h averages (n = 6 Ube3am+/p+ or n = 8 Ube3am−/p+ mice) ± SEM. *p < 0.05 between genotypes at the indicated times (post hoc t test; Bonferroni corrected). B, The increase in I2–6 during the 12 h dark (wakeful) period is abolished in Ube3am−/p+ mice. Representative EEG recordings (black and red) are shown above bandpass-filtered versions (gray and dark red) of the same EEG recording. The incidence of peaks in the upper 30% by amplitude (maximum negative deflection between two zero crossings; blue line denotes 30th percentile amplitude cutoff) in the filtered signal were counted in epochs scored as waking. I2–6 has been previously validated as a measure of sleep-pressure accumulation in awake mice (Ehlen et al., 2013). Arrowheads indicate detected peaks. I2–6 significantly increased across the active period in Ube3am+/p+ mice (black plot; early night, ZT 12–14, vs late night, ZT 20–22; Student's paired t test, p < 0.05). This measure of sleep pressure (I2–6) failed to significantly accumulate in Ube3am−/p+ mice (red plot; paired t test, p > 0.05).
As a complementary measure of sleep-pressure accumulation in wild-type and Ube3am−/p+ mice, we also examined the incidence of 2–6 Hz waves that occur during epochs of the waking EEG [wave incidence (I2–6)]. I2–6 is a measure of sleep pressure experienced by animals while they are awake, and correlates strongly with other measures of sleep pressure, including waking theta power, but with better temporal resolution (Ehlen et al., 2013). In wild-type mice, I2–6 increased significantly across the night (Fig. 6B; Student–Newman–Keuls post hoc, p < 0.05). In contrast, I2–6 in Ube3am−/p+ mice did not increase over the same period, remaining consistent throughout the night (Fig. 6B; data not shown), further supporting a deficiency in sleep-pressure accumulation during the night in Ube3am−/p+ mice.
The relatively constant levels of sleep, delta power, and I2–6 during the night that occur with maternal Ube3a loss suggest that Ube3am−/p+ mice have a reduced need for a siesta as a consequence of their relative inability to accumulate sleep pressure. However, because Ube3am−/p+ mice tend to sleep slightly more than wild-type mice during the early night (Figs. 4, 5), it is unclear whether these changes in nighttime sleep-pressure accumulation were due to defects in sleep homeostasis or circadian clock outputs that directly regulate sleep–wake patterns. To distinguish between these possibilities, we conducted a standard homeostatic challenge by assessing changes in sleep pressure caused by forced sleep deprivation during the day, when mice sleep the most (Borbély and Neuhaus, 1979). Specifically, we sleep-deprived mice for the first half of the day, ZT 0–6, and focused our analyses on responses occurring during the second half of the day, ZT 6–12. Wild-type mice showed typical significant increases in NREM sleep, delta power, and I2–6 immediately (ZT 6–12) after sleep deprivation, indicating that sleep deprivation substantially increased sleep pressure (Fig. 7). In contrast, sleep deprivation in Ube3am−/p+ mice did not significantly increase NREM sleep, delta power, or I2–6 (Fig. 7), indicating that Ube3am−/p+ mice have a distinct and specific deficit accumulating sleep pressure. During the 18 h recovery period that followed sleep deprivation, wild-type mice exhibited a significantly larger increase in NREM delta power normalized to waking delta power when compared with Ube3am−/p+ mice (wild type, 193 ± 3%; Ube3am−/p+, 121 ± 4%; t(11) = 3.3, p = 0.003, Student's t test). Thus, together, our data suggest that Ube3am−/p+ mice have an impaired sleep homeostatic mechanism that is responsible for the behavioral changes associated with the loss of their siesta.
Figure 7.
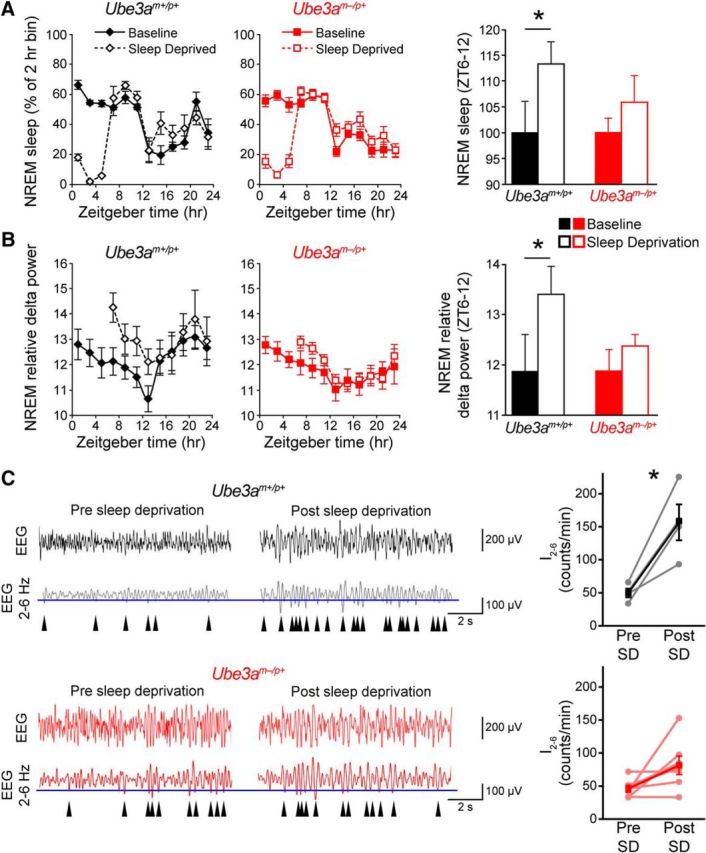
AS model mice exhibit blunted recovery responses from sleep deprivation. A, Line plots, NREM sleep responses to sleep deprivation (SD) during ZT 0–6. Baseline sleep data are replotted from Figure 5D for comparison. Bar graph, Comparison of the effect of sleep deprivation on NREM sleep amounts immediately after the sleep deprivation period (ZT 6–12). Data are plotted as a percentage of the baseline NREM sleep amount. Only wild-type mice (Ube3am+/p+) displayed a significant increase in NREM sleep following sleep deprivation. Asterisk indicates significant effect of sleep deprivation (p < 0.05, Student's paired one-tailed t test; n = 4 Ube3am+/p+ or n = 8 Ube3am−/p+ mice). B, Line plots, NREM relative delta power (power 0.5–4 Hz/total EEG power) during baseline sleep (replotted from Fig. 6A) and in response to sleep deprivation during ZT 0–6. Bar graph, Comparisons of the effect on average relative delta power between genotypes immediately following sleep deprivation (ZT 6–12). Only wild-type mice (Ube3am+/p+) displayed a significant increase in delta power following sleep deprivation. Asterisk indicates significant effect of sleep deprivation (p < 0.05, Student's paired one-tailed t test; n = 4 Ube3am+/p+ or n = 8 Ube3am−/p+ mice). C, Left, Representative EEG recordings of I2–6, as described in Figure 6, shown before and after the 6 h sleep-deprivation period. Note the increase in I2–6 (arrowheads) in Ube3am+/p+ mice in response to sleep deprivation. Right, I2–6 before and after sleep deprivation. I2–6 significantly increased across the sleep-deprivation period in Ube3am+/p+ mice (black plot; presleep deprivation, ZT 0–1, vs postsleep deprivation, ZT 6–7; paired t test, p < 0.05), but not in Ube3am−/p+ mice (red plot; paired t test, p > 0.05).
Discussion
We found that loss of maternal Ube3a produces a penetrant reorganization of sleep–wake architecture across the night, without substantially disrupting circadian rhythms in locomotor activity or molecular clockwork. Our findings suggest a role for maternal Ube3a in regulating the accumulation of sleep pressure during wakefulness, and identify Ube3a as a novel sleep-regulation gene.
We observed a consistently timed rest period (siesta) in wild-type mice during the latter part of the active phase, as previously reported in C57BL/6 mice (Valentinuzzi et al., 1997; Challet et al., 2001). In wild-type mice, this rest period was coincident with a peak in sleep propensity, suggesting that it may be a product of sleep-pressure accumulation during a long bout of wakefulness. This timed rest period was absent in Ube3am−/p+ mice. Our study is the first to link sleep-pressure accumulation and the occurrence of a timed rest period, and suggests that Ube3am−/p+ mice may serve as a novel model for understanding the mechanisms that control the timed rest period during the active phase.
By depriving mice of sleep during the day, when mice sleep the most, we revealed that the effect of Ube3a loss on sleep pressure is not limited to the active period. Ube3am−/p+ mice have blunted sleep drive following sleep deprivation compared with littermate controls. Thus, Ube3am−/p+ mice fail to accumulate sleep pressure while they are awake, regardless of time of day. That Ube3am−/p+ mice exhibit NREM delta power levels that are comparable to wild-type mice during the early daytime (Fig. 6A, ZT 1) suggests that Ube3am−/p+ mice are capable of typical levels of sleep pressure, but their failure to accumulate sleep pressure in response to wakefulness suggests the possibility that the early-morning sleep pressure may develop independent of wakefulness. Ube3a may provide an entry point for deciphering the circuitry and molecular mechanisms underlying sleep homeostasis across both the active and inactive phases of the day.
The effect of maternal Ube3a loss on sleep homeostasis appears to be independent of a substantial disruption in circadian clock function in the SCN. Another group (Shi et al., 2015) recently reported that the same AS model mouse line (Jiang et al., 1998) exhibits a modest lengthening of circadian period (>0.5 h change) in constant darkness and faster entrainment to experimental jet lag. Our data trend in the same direction for both measures; however, the effects did not reach significance in our hands. We believe the discrepancy between studies highlights the subtlety of the effect of maternal Ube3a deletion on circadian behavior. We also found that Ube3a imprinting is uniquely relaxed in neurons within the SCN. Thus, it remains possible that homozygous Ube3a deletion could produce a stronger circadian phenotype. However, RNAi-mediated Ube3a knockdown in human U2OS cells produced a similarly small (∼0.6 h) period lengthening, suggesting that even homozygous Ube3a deletion is unlikely to markedly impair circadian clock function.
Observational studies of AS individuals, obtained from surveys of parents and caregivers, indicate that AS individuals have decreased nighttime sleep amounts and difficulty falling and staying asleep (Bruni et al., 2004; Didden et al., 2004; Goldman et al., 2012; Thibert et al., 2013; Larson et al., 2015). These studies have led to the idea that individuals with AS generally have less need for nighttime sleep. EEG/EMG recordings of nighttime sleep support these observations, and further suggest that children with AS also have reduced levels of REM sleep and decreased sleep efficiency (Miano et al., 2004). EEG sleep studies in individuals with AS, however, were limited to the 7–9 h corresponding to bedtime, and did not include the daytime/active period. Our results suggest that nighttime sleep deficits that affect individuals with AS may be due, at least in part, to specific changes in sleep-pressure accumulation that are most prominent during the daytime. Thus, EEG sleep studies in individuals with AS during the day may reveal broader differences in sleep homeostasis. Accordingly, our data raise the possibility that interventions that affect sleep regulation during the daytime may substantially improve the overall nighttime sleep profile of individuals with AS.
Footnotes
This work was supported by grants from the Angelman Syndrome Foundation (to B.D.P.); Simons Foundation Grant SFARI #274426 (to B.D.P.); National Institutes of Health (NIH) Grants R01NS085093 (to B.D.P.), R01MH093372 (to B.D.P. and M.J.Z.), DP1ES024088 (to M.J.Z.), 8G12MD007602 (director: V.C. Bond; project principal investigators: K.N.P., J.P.D., and J.C.E.), U54NS083932 (director: P.R. MacLeish; project coprincipal investigators: K.N.P. and J.P.D.), R01NS039444 (to R.J.W.), R01NS078410 (to K.N.P.), and SC1GM109861 (to J.P.D.). A.J.B. was supported by NIH Grant F32 HL116077. K.A.J. was supported by an NIH T32 postdoctoral training grant through the Carolina Institute for Developmental Disabilities (NIH T32HD40127). The University of North Carolina Confocal and Multiphoton Imaging Core is funded by NIH Grants P30NS045892 and U54HD079124. We thank Vladimir Ghukasyan at the University of North Carolina Confocal and Multiphoton Imaging Core for technical assistance.
The authors declare no competing financial interests.
References
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- Anand SN, Maywood ES, Chesham JE, Joynson G, Banks GT, Hastings MH, Nolan PM. Distinct and separable roles for endogenous CRY1 and CRY2 within the circadian molecular clockwork of the suprachiasmatic nucleus, as revealed by the Fbxl3(Afh) mutation. J Neurosci. 2013;33:7145–7153. doi: 10.1523/JNEUROSCI.4950-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggs JE, Price TS, DiTacchio L, Panda S, Fitzgerald GA, Hogenesch JB. Network features of the mammalian circadian clock. PLoS Biol. 2009;7:e52. doi: 10.1371/journal.pbio.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- Borbély A, Neuhaus H. Sleep-deprivation: effects on sleep and EEG in the rat. J Comp Physiol. 1979;133:71–87. doi: 10.1007/BF00663111. [DOI] [Google Scholar]
- Braam W, Didden R, Smits MG, Curfs LM. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol. 2008;23:649–654. doi: 10.1177/0883073808314153. [DOI] [PubMed] [Google Scholar]
- Bruni O, Ferri R, D'Agostino G, Miano S, Roccella M, Elia M. Sleep disturbances in Angelman syndrome: a questionnaire study. Brain Dev. 2004;26:233–240. doi: 10.1016/S0387-7604(03)00160-8. [DOI] [PubMed] [Google Scholar]
- Challet E, Turek FW, Laute M, Van Reeth O. Sleep deprivation decreases phase-shift responses of circadian rhythms to light in the mouse: role of serotonergic and metabolic signals. Brain Res. 2001;909:81–91. doi: 10.1016/S0006-8993(01)02625-7. [DOI] [PubMed] [Google Scholar]
- Colas D, Wagstaff J, Fort P, Salvert D, Sarda N. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol Dis. 2005;20:471–478. doi: 10.1016/j.nbd.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Dan B. Angelman syndrome: current understanding and research prospects. Epilepsia. 2009;50:2331–2339. doi: 10.1111/j.1528-1167.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- DeBruyne JP, Noton E, Lambert CM, Maywood ES, Weaver DR, Reppert SM. A clock shock: mouse CLOCK is not required for circadian oscillator function. Neuron. 2006;50:465–477. doi: 10.1016/j.neuron.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Didden R, Korzilius H, Smits MG, Curfs LM. Sleep problems in individuals with Angelman syndrome. Am J Mental Retard. 2004;109:275–284. doi: 10.1352/0895-8017(2004)109<275:SPIIWS>2.0.CO%3B2. [DOI] [PubMed] [Google Scholar]
- DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, Panda S. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333:1881–1885. doi: 10.1126/science.1206022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, McKnight SL. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science. 2003;301:379–383. doi: 10.1126/science.1082795. [DOI] [PubMed] [Google Scholar]
- Egawa K, Kitagawa K, Inoue K, Takayama M, Takayama C, Saitoh S, Kishino T, Kitagawa M, Fukuda A. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci Transl Med. 2012;4:163ra157. doi: 10.1126/scitranslmed.3004655. [DOI] [PubMed] [Google Scholar]
- Ehlen JC, Jefferson F, Brager AJ, Benveniste M, Paul KN. Period-amplitude analysis reveals wake-dependent changes in the electroencephalogram during sleep deprivation. Sleep. 2013;36:1723–1735. doi: 10.5665/sleep.3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JA, Leise TL, Castanon-Cervantes O, Davidson AJ. Dynamic interactions mediated by nonredundant signaling mechanisms couple circadian clock neurons. Neuron. 2013;80:973–983. doi: 10.1016/j.neuron.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SE, Bichell TJ, Surdyka K, Malow BA. Sleep in children and adolescents with Angelman syndrome: association with parent sleep and stress. J Intellect Disabil Res. 2012;56:600–608. doi: 10.1111/j.1365-2788.2011.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, Glossop N, Zhang YQ, Fukada Y, Meng QJ. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014;42:5765–5775. doi: 10.1093/nar/gku225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39:283–291. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Burns AJ, Nonneman RJ, Baker LK, Riddick NV, Nikolova VD, Riday TT, Yashiro K, Philpot BD, Moy SS. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–90. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Deboer T, Tobler I. Effects of sleep deprivation on sleep and sleep EEG in three mouse strains: empirical data and simulations. Brain Res. 2000;857:8–19. doi: 10.1016/S0006-8993(99)02248-9. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/S0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol. 2014;522:1874–1896. doi: 10.1002/cne.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64:947–960. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM, Shinnick JE, Shaaya EA, Thiele EA, Thibert RL. Angelman syndrome in adulthood. Am J Med Genet A. 2015;167A:331–344. doi: 10.1002/ajmg.a.36864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Judson MC, Zylka MF, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- Miano S, Bruni O, Leuzzi V, Elia M, Verrillo E, Ferri R. Sleep polygraphy in Angelman syndrome. Clin Neurophysiol. 2004;115:938–945. doi: 10.1016/j.clinph.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Miura K, Kishino T, Li E, Webber H, Dikkes P, Holmes GL, Wagstaff J. Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol Dis. 2002;9:149–159. doi: 10.1006/nbdi.2001.0463. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul KN, Laposky AD, Turek FW. Reproductive hormone replacement alters sleep in mice. Neurosci Lett. 2009;463:239–243. doi: 10.1016/j.neulet.2009.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008;9:434–441. doi: 10.1016/j.sleep.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- Shi SQ, Bichell TJ, Ihrie RA, Johnson CH. Ube3a imprinting impairs circadian robustness in angelman syndrome models. Curr biol. 2015;25:537–545. doi: 10.1016/j.cub.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Jiang YH, Galijaard RJ, Matsuura T, Fang P, Kubota T, Christian SL, Bressler J, Cattanach B, Ledbetter DH, Beaudet AL. The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 1997;7:368–377. doi: 10.1101/gr.7.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaesu Y, Komada Y, Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med. 2012;13:1164–1170. doi: 10.1016/j.sleep.2012.06.015. [DOI] [PubMed] [Google Scholar]
- Thibert RL, Larson AM, Hsieh DT, Raby AR, Thiele EA. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013;48:271–279. doi: 10.1016/j.pediatrneurol.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Valentinuzzi VS, Scarbrough K, Takahashi JS, Turek FW. Effects of aging on the circadian rhythm of wheel-running activity in C57BL/6 mice. Am J Physiol. 1997;273:R1957–R1964. doi: 10.1152/ajpregu.1997.273.6.R1957. [DOI] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- Walz NC, Beebe D, Byars K. Sleep in individuals with Angelman syndrome: parent perceptions of patterns and problems. Am J Ment Retard. 2005;110:243–252. doi: 10.1352/0895-8017(2005)110[243:SIIWAS]2.0.CO%3B2. [DOI] [PubMed] [Google Scholar]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12:385–395. doi: 10.1097/GIM.0b013e3181def138. [DOI] [PubMed] [Google Scholar]
- Wisor JP, Pasumarthi RK, Gerashchenko D, Thompson CL, Pathak S, Sancar A, Franken P, Lein ES, Kilduff TS. Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J Neurosci. 2008;28:7193–7201. doi: 10.1523/JNEUROSCI.1150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yu G, Parks H, Hebert T, Goh BC, Dietrich MA, Pelled G, Izadpanah R, Gazit D, Bunnell BA, Gimble JM. Circadian mechanisms in murine and human bone marrow mesenchymal stem cells following dexamethasone exposure. Bone. 2008;42:861–870. doi: 10.1016/j.bone.2007.12.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–847. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Takahashi JS. Real-time luminescence reporting of circadian gene expression in mammals. Methods Enzymol. 2005;393:288–301. doi: 10.1016/S0076-6879(05)93012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, Liu X, Atwood A, Huss JW, 3rd, Janes J, Su AI, Hogenesch JB, Kay SA. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Shearman LP, Weaver DR, Reppert SM. Three period homologs in mammals: different light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of brain. Neuron. 1998;20:1103–1110. doi: 10.1016/S0896-6273(00)80492-4. [DOI] [PubMed] [Google Scholar]