

Abstract
Rationale: Increased abundance and stiffness of the extracellular matrix, in particular collagens, is a hallmark of idiopathic pulmonary fibrosis (IPF). FK506-binding protein 10 (FKBP10) is a collagen chaperone, mutations of which have been indicated in the reduction of extracellular matrix stiffness (e.g., in osteogenesis imperfecta).
Objectives: To assess the expression and function of FKBP10 in IPF.
Methods: We assessed FKBP10 expression in bleomycin-induced lung fibrosis (using quantitative reverse transcriptase–polymerase chain reaction, Western blot, and immunofluorescence), analyzed microarray data from 99 patients with IPF and 43 control subjects from a U.S. cohort, and performed Western blot analysis from 6 patients with IPF and 5 control subjects from a German cohort. Subcellular localization of FKBP10 was assessed by immunofluorescent stainings. The expression and function of FKBP10, as well as its regulation by endoplasmic reticulum stress or transforming growth factor-β1, was analyzed by small interfering RNA–mediated loss-of-function experiments, quantitative reverse transcriptase–polymerase chain reaction, Western blot, and quantification of secreted collagens in the lung and in primary human lung fibroblasts (phLF). Effects on collagen secretion were compared with those of the drugs nintedanib and pirfenidone, recently approved for IPF.
Measurements and Main Results: FKBP10 expression was up-regulated in bleomycin-induced lung fibrosis and IPF. Immunofluorescent stainings demonstrated localization to interstitial (myo)fibroblasts and CD68+ macrophages. Transforming growth factor-β1, but not endoplasmic reticulum stress, induced FKBP10 expression in phLF. The small interfering RNA–mediated knockdown of FKBP10 attenuated expression of profibrotic mediators and effectors, including collagens I and V and α-smooth muscle actin, on the transcript and protein level. Importantly, loss of FKBP10 expression significantly suppressed collagen secretion by phLF.
Conclusions: FKBP10 might be a novel drug target for IPF.
Keywords: FKBP65, peptidyl-prolyl isomerase, lung fibrosis, collagen cross-linking, extracellular matrix
At a Glance Commentary
Scientific Knowledge on the Subject
Accumulation of extracellular matrix plays an important role in idiopathic pulmonary fibrosis (IPF) disease progression. Deficiency of the chaperone FK506-binding protein 10 (FKBP10) has been reported to attenuate collagen secretion and decrease extracellular collagen cross-linking in, for example, osteogenesis imperfecta.
What This Study Adds to the Field
FKBP10 is specifically up-regulated in interstitial fibroblasts in IPF. Inhibition of FKBP10 in primary IPF fibroblasts attenuates gene expression of various profibrotic genes and decreases collagen secretion.
Idiopathic pulmonary fibrosis (IPF) is the most fatal interstitial lung disease, with a 5-year survival rate of 30 to 50% and few treatment options (1). The etiology of IPF is poorly understood. The current concept involves repeated alveolar injuries of unclear nature, providing signals for fibroblast activation, proliferation, and differentiation to myofibroblasts (2–6). The latter overgrow the delicate alveolar lung tissue and secrete increased amounts of extracellular matrix (ECM) proteins. According to a recently published study, the ECM appears to play a major role not only in the composition but also in the maintenance of the fibrotic phenotype in IPF, contributing to the irreversibility of the disease (7). Therefore, matrix and matrix-processing enzymes will provide promising novel drug targets for IPF. For instance, inhibition of the collagen cross-linking enzyme lysyl oxidase-like 2 (LOXL2) is currently investigated as an IPF treatment option in a phase II trial (8).
Pirfenidone and nintedanib, two drugs recently approved by the U.S. Food and Drug Administration for IPF therapy, decelerate but do not attenuate disease progression in patients with IPF, by decreasing lung function decline. They also show considerable side effects, and their mechanism of action is incompletely understood (9, 10). As such, the need to identify novel drug targets for IPF remains imperative, as we require alternative treatment options for nonresponders, cell-type–specific targeted therapy, and combination therapy options.
The intracellular chaperone FK506-binding protein 10 (FKBP10, also termed FKBP65) has been reported to directly interact with collagen I (11). FKBP10 belongs to the FKBP subfamily of immunophilins, molecular chaperones with peptidyl-prolyl isomerase activity, which bind the immunosuppressive drug FK506 (tacrolimus) (12, 13). Collagen triple helix formation heavily relies on proline isomerization to trans-proline as a prerequisite of linear chain assembly (14). Mutations in FKBP10 lead to collagen-related disorders such as osteogenesis imperfecta, and studies in the last 4 years have suggested an association of these mutations with attenuated collagen secretion and diminished collagen I cross-linking in dermal fibroblasts and bone (15–18). A recent study showed that Fkbp10−/− mouse embryos are postnatally lethal and display reduced collagen cross-linking in calvarial bone (19).
Given that FKBP10 is a collagen-processing enzyme with potential impact on ECM protein secretion and cross-linking, we sought to assess its role in IPF. We analyzed FKBP10 expression in lungs of mice subjected to bleomycin and patients with IPF. Loss-of-function studies were performed in primary human lung fibroblasts (phLF) to investigate the effect of FKBP10 on ECM protein synthesis and secretion.
Methods
For more details on methods, see the online supplement. Statistical analysis was performed in GraphPadPrism 5 (GraphPad Software, San Diego, CA). Results are given as mean ± SEM, and paired t test was used for statistical analysis, if not mentioned otherwise.
Induction and Measurement of Murine Pulmonary Fibrosis
Pulmonary fibrosis was induced in female C57BL/6 mice (10–12 wk old) by a single intratracheal instillation of 50 μl of bleomycin (3 U/kg; Sigma Aldrich, Taufkirchen, Germany) dissolved in sterile saline and applied using the MicroSprayer Aerosolizer, Model IA-1C (Penn-Century, Wyndmoor, PA). Control mice were instilled with 50 μl of saline. After instillation, mice were kept for 14, 28, and 56 days. Before death, mice were anesthetized with ketamine/xylazine followed by lung function measurement and tissue harvesting as previously described (20). Fibrosis was further assessed by bronchoalveolar lavage cell counts and histology evaluation. All animal experiments were conducted under strict governmental and international guidelines and were approved by the local government for the administrative region of Upper Bavaria, Germany.
Gene Expression Data
Data for FKBP10 in IPF lungs (n = 99) and normal histology control lungs (n = 43) was extracted from the gene expression microarray data generated by us on lung samples obtained from the National Lung, Heart, and Blood Institute–funded Tissue Resource Consortium, as described previously (21, 22). Gene expression microarray data (Agilent Technologies, Santa Clara, CA), and associated clinical data are available on the Lung Genomics Research Consortium website (https://www.lung-genomics.org/research/) as well as on accession number GSE47460 or the Lung Tissue Research Consortium website (http://www.ltrcpublic.com). Significance was calculated using t statistics, and multiple testing was controlled by the false discovery rate method at 5% (23).
Human Material
Resected human lung tissue and lung explant material were obtained from the Asklepios biobank for lung diseases at the Comprehensive Pneumology Center. Biopsies were obtained from six patients with IPF (usual interstitial pneumonia pattern, mean age 54 ± 9 yr, five men, one woman). All participants gave written informed consent, and the study was approved by the local ethics committee of Ludwig-Maximilians University of Munich, Germany.
Isolation and Culture of phLF
For FKBP10 knockdown, phLF were isolated from IPF biopsies (n = 4), the adjacent normal region of a lung tumor resection (n = 2), and donor tissue (n = 3). For more details, see the online supplement.
Treatment of phLF with Transforming Growth Factor-β1, Nintedanib, or Pirfenidone
Cells were seeded at a density of 20,000 to 25,000 cells/cm2, starved for 24 hours in Dulbecco’s modified Eagle medium/F12 with 0.5% fetal bovine serum and 0.1 mM 2-phospho-l-ascorbic acid, followed by treatment with the indicated concentrations of transforming growth factor (TGF)-β1 (R&D Systems, Minneapolis, MN), nintedanib, and pirfenidone (both Selleck, Houston, TX) in starvation medium for the indicated time points. For more details, see the online supplement.
Transfection of phLF
Cells were reverse transfected either with human FKBP10 small interfering RNA (siRNA) (s34171; Life Technologies, Carlsbad, CA) or negative control siRNA No. 1 (Life Technologies). Twenty-four hours after transfection, cells were starved for another 24 hours in Dulbecco’s modified Eagle medium/F-12 including 0.5% fetal bovine serum and 0.1 mM 2-phospho-l-ascorbic acid, followed by treatment with 2 ng/ml TGF-β1 in starvation medium. Twenty-four and 48 hours after beginning the TGF-β1 treatment, cells and cell culture supernatants were harvested for RNA and protein analysis. In total, eight completely independent knockdowns were performed in eight different human primary fibroblast lines.
After 24 hours of siRNA transfection, forward transfection with the mothers against decapentaplegic homolog (SMAD) signaling luciferase reporter plasmid was performed in three different cell lines with pGL3-CAGA(9)-luc (24) or pGL3 control vector (Promega, Madison, WI). After incubation for 6 hours, cells were starved for 18 hours, followed by treatment with 2 ng/ml TGF-β1. Luminescence was recorded in a TriStar LB 941 Multimode Reader (Berthold Technologies, Bad Wildbad, Germany), and results were normalized to pGL3 control luciferase activity.
For more details, see the online supplement.
Real-Time Quantitative Reverse Transcriptase Polymerase Chain Reaction Analysis
Relative transcript abundance of a gene is expressed as −ΔCt values (ΔCt = Cttarget − Ctreference) or as fold change derived from the relevant ΔΔCt values, using 2−(ΔΔCt). For specific gene amplification, primers listed in Table E1 in the online supplement were used. Glyceraldehyde phosphate dehydrogenase (GAPDH) and hypoxanthine-guanine phosphoribosyltransferase (HPRT) were used as endogenous controls for standardization of relative mRNA expression in mice and phLF, respectively. For more technical details on RNA isolation and quantitative reverse transcriptase polymerase chain reaction, see the online supplement.
Protein Isolation and Western Blot Analysis
See online supplement.
Quantification of Secreted Collagen
For quantification of total secreted collagen, the Sircol assay was performed according to manufacturer’s instructions (Biocolor, Carrickfergus, UK).
For specific quantification of secreted collagen I, collagens were precipitated from cell culture supernatant as follows: Proteins were precipitated with 0.2 g/ml ammonium sulfate and incubated on ice for 30 minutes, followed by centrifugation at 20,000 × g at 4°C for 30 minutes. The pellet was dissolved in 0.1 M acetic acid containing 0.1 mg/ml pepsin (Thermo Fisher Scientific, Waltham, MA) and incubated overnight at 4°C. Then, 5 M NaCl was added to yield a final concentration of 0.7 M, followed by incubation on ice for 30 minutes and centrifugation at 20,000 × g at 4°C for 30 minutes. The pellet containing collagen I was resuspended in 0.1 M acetic acid and analyzed by immunoblotting.
Immunofluorescent and Masson Trichrome Stainings
Cultured phLF were seeded on coverslips, and immunostaining was performed as described previously (25). For staining of tissue sections, human and murine lung tissue was fixed in 10% formalin before paraffin embedding. Three-micron sections were prepared and mounted on slides, followed by deparaffinization and immunofluorescent staining or Masson trichrome staining according to a standard protocol. For more details, see the online supplement.
Subcellular Fractionation
Protein fractionation was performed with a Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Fisher Scientific) according to the manufacturer's instructions. Purity of the fractions was assessed using the following marker proteins for Western blot analysis of the obtained fractions: calreticulin and protein disulfide isomerase 3 (PDIA3) for the membrane extract, glyceraldehyde phosphate dehydrogenase (GAPDH) for the cytosolic extract, and lamin A/C for the nuclear and chromatin-bound extract.
Results
FKBP10 Expression Is Increased in Lung Fibrosis
We first studied FKBP10 expression in the mouse model of bleomycin-induced lung fibrosis. Bleomycin instillation led to increased ECM deposition, as demonstrated by Masson trichrome staining, and a significant decrease in lung compliance at Day 14, which was completely restored after 56 days (Figures 1A and 1B). Western blot analysis showed a clear increase of FKBP10 protein levels in total lung lysates at Day 14 after instillation of bleomycin (Figure 1D). After resolution of fibrosis, at Day 56, FKBP10 expression had returned to baseline levels (cf. Figures 1A, 1B, and 1E). With a fold change of 3.6, up-regulation of FKBP10 at Day 14 occurred in part on the transcriptional level (Figure 1C).
Figure 1.

FK506-binding protein 10 (FKBP10) is up-regulated in a mouse model of bleomycin-induced lung fibrosis. (A) Representative Masson trichrome staining demonstrating that increased collagen deposition peaked at Day 14 and was gradually lost afterward. Bottom row shows magnification of the boxed areas in the top row. Upper scale bar = 1,000 μm; lower scale bar = 100 μm. (B) Bleomycin instillation led to a significant decrease in lung compliance at Day 14, which was completely restored after 56 days. (C) FKBP10 gene expression was up-regulated at Day 14 after bleomycin instillation. (D) Representative Western blot showing that FKBP10 levels in total lung homogenate were significantly increased at Day 14 after bleomycin instillation; HeLa lysate was used as positive control. (E) Quantification by densitometric analysis demonstrating that FKBP10 levels returned to baseline at Day 28 and Day 56. Data shown are mean ± SEM, and a two-tailed Mann-Whitney test was used for statistical analysis for comparison between bleomycin groups (Bleo) at different time points and phosphate-buffered saline (PBS) control vs. bleomycin for each time point. Actb = β-actin as loading control. Gapdh = glyceraldehyde phosphate dehydrogenase. **P < 0.01, ***P < 0.001.
Next, we studied FKBP10 gene expression in IPF. Analysis of microarray data of 99 IPF samples and 43 normal histology control samples revealed significant up-regulation of FKBP10 (fold change, 1.7; false discovery rate < 5%; P < 1 × 10−7) (Figures 2A and 2B). Up-regulation of FKBP10 was confirmed on the protein level using lung homogenates from patients with IPF of an independent cohort: FKBP10 was highly increased in comparison with donor samples (Figures 2C and 2D). Moreover, FKBP10 levels appeared to correlate with levels of α-smooth muscle actin (α-SMA), a marker of myofibroblasts (Figure 2C).
Figure 2.
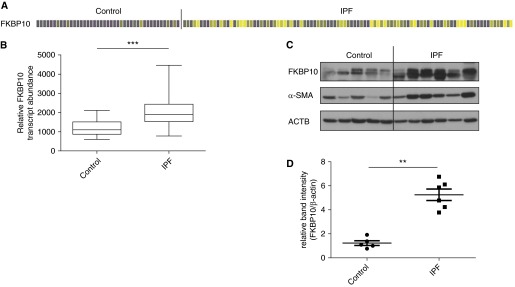
FK506-binding protein 10 (FKBP10) is up-regulated in idiopathic pulmonary fibrosis (IPF). (A) Heat map of FKBP10 gene expression, extracted from microarray data of normal histology control (n = 43) and samples from patients with IPF (n = 99). Every column corresponds to a patient. Increases and decreases are denoted in increasing shades of yellow and purple, respectively, and gray is unchanged (false discovery rate < 5%). (B) Box-and-whisker plot for FKBP10 gene expression data. The difference is highly significant (***P < 1 × 10−7). (C) Western blot analysis of total lung tissue homogenate showed up-regulation of FKBP10 in patients with IPF relative to donor samples in an independent cohort. (D) Densitometric analysis of the Western blot showed that FKBP10 up-regulation in IPF is highly significant. Data shown are mean ± SEM, and a two-tailed Mann-Whitney test was used for statistical analysis (**P < 0.01). α-SMA = α-smooth muscle actin; ACTB = β-actin as loading control.
FKBP10 Is Expressed in Interstitial Fibroblasts, Including Myofibroblasts
Immunofluorescent stainings of lung tissue sections from the bleomycin-treated mice and patients with IPF confirmed increased expression of FKBP10 in fibrotic lungs, with little staining of FKBP10 in phosphate-buffered saline–instilled control mice or donor lungs. FKBP10 expression was predominantly localized to interstitial fibroblasts, as evidenced by colocalization with α-SMA (Figures 3 and 4A) or desmin (Figure 4A), both markers of myofibroblasts. In addition, a fraction of FKBP10-expressing cells were found to be interstitial macrophages (evidenced by CD68+ staining; Figure 4B). In contrast, FKBP10 did not colocalize with T1α (AT1 cells) or TTF1 (AT2 cells) (Figures 3 and 4A).
Figure 3.

FK506-binding protein 10 (FKBP10) is expressed in interstitial fibroblasts, including myofibroblasts, in a mouse model of bleomycin-induced lung fibrosis. Immunofluorescent stainings of paraffin sections from control (phosphate-buffered saline [PBS], left panels) and bleomycin-treated (Bleo, right panels) mice at Day 14 after bleomycin instillation. Representative Fkbp10 immunostaining is shown in red, α-smooth muscle actin (α-SMA), podoplanin (T1α), or thyroid transcription factor 1 (TTF1) in green, and 4′,6-diamidino-2-phenylindole (DAPI) in blue, as indicated on the right side. White squares in the Bleo stainings are shown enlarged to the right. Sections shown are representative stainings from three PBS- and three bleomycin-treated mice.
Figure 4.

FK506-binding protein 10 (FKBP10) is expressed in interstitial fibroblasts, including myofibroblasts, and interstitial CD68+ macrophages in human idiopathic pulmonary fibrosis (IPF). Representative immunofluorescent stainings of paraffin sections from donor (left panels) and IPF tissue (right panels). Representative FKBP10 immunostaining is always shown in red and 4′,6-diamidino-2-phenylindole (DAPI) in blue. (A) Representative immunofluorescent costainings with α-smooth muscle actin (α-SMA), desmin, podoplanin (T1α), and thyroid transcription factor 1 (TTF1) in green show FKBP10 expression in interstitial fibroblasts but not in alveolar epithelial cells. White squares in the IPF stainings are shown enlarged to the right. Scale bar = 100 μm. (B) Representative immunofluorescent costainings with CD68 show FKBP10 expression in interstitial macrophages (indicated by white arrows). Sections shown are representative stainings from three donors and three patients with IPF. Scale bar = 50 μm.
FKBP10 Is an Endoplasmic Reticulum Resident Protein but Not Up-regulated by ER Stress in Primary Human Lung Fibroblasts
In immunofluorescent stainings of phLF, FKBP10 localized mainly to the cytoplasm and showed colocalization with PDIA3, an endoplasmic reticulum (ER)-resident protein (Figure 5A). In some cells, positive FKBP10 staining was also observed in the nucleus. In contrast, costaining with a marker for the Golgi apparatus, Golgin subfamily A member 1 (GOLGA1) showed no colocalization (Figure 5B). Subcellular fractionation of phLF resulted in a clear enrichment of FKBP10 in the ER/membrane extract (Figure 5C). Additionally, FKBP10 was also weakly detected in the cytosolic and the nuclear extract.
Figure 5.

FK506-binding protein 10 (FKBP10) is mainly an endoplasmic reticulum (ER)-resident protein and does not localize to the Golgi apparatus. (A) Immunostaining of FKBP10 and the ER-marker protein disulfide isomerase A3 (PDIA3) is shown in red and green, respectively. 4′,6-diamidino-2-phenylindole (DAPI) staining is shown in blue. The white square was chosen as the region of interest and enlarged to the right. Arrows in the region of interest depict examples for colocalization of FKBP10 and PDIA3. While the staining shown to the left is an extended-focus picture generated from a z-stack, the enlarged figure to the right shows one focal plane from the z-stack only. (B) Immunostaining of FKBP10 and the Golgi-marker protein Golgin subfamily A member 1 is shown in red and green, respectively. DAPI staining is shown in blue. The presented staining is an extended-focus picture generated from a z-stack. (C) Western blot analysis of subcellular fractionation of primary human lung fibroblasts. FKBP10 is enriched in the membrane extract (ME), similar to the ER-resident proteins calreticulin (CALR) and PDIA3. FKBP10 was additionally found in the cytosolic extract (CE) and the nuclear extract (NE) but not in the chromatin-bound fraction (CB). Lamin A/C and glyceraldehyde phosphate dehydrogenase (GAPDH) were used as marker proteins for NE/CB and CE, respectively.
Although tunicamycin and thapsigargin up-regulated the ER stress markers BiP, ATF4, and CHOP, FKBP10 levels were drastically decreased by both (see Figure E1 in the online supplement). FKBP10 usually migrated at approximately 70 kD, but in the presence of tunicamycin an additional FKBP10 band appeared at approximately 65 kD, in agreement with the theoretical size of the unmodified protein (cf. Figure E1A).
TGF-β1 Up-regulates FKBP10 Expression in phLF
TGF-β1 induced FKBP10 expression in a dose-dependent manner, with a clearly visible up-regulation for concentrations of 1 ng/ml and higher after 48 hours of treatment (Figure 6A). Efficacy of TGF-β1 treatment was confirmed by Western blot analysis of phosphorylated and total SMAD3 (Figure 6A). From these initial experiments, an effective concentration of 2 ng/ml TGF-β1 was derived and used in the following FKBP10 knockdown experiments, where the TGF-β1 effect on FKBP10 expression was also quantified (Figure 6B). Results in Figure 6B are based on results with control siRNA from the loss-of-function experiments (cf. Figure 6C for a representative Western blot). Induction of FKBP10 expression by TGF-β1 was already observed at transcript level (Figure E2).
Figure 6.

FK506-binding protein 10 (FKBP10) is up-regulated by transforming growth factor (TGF)-β1 in primary human lung fibroblasts (phLF), and knockdown of FKBP10 attenuates synthesis of collagen I, collagen V, and α-smooth muscle actin (α-SMA). (A) Western blot analysis of phLF treated with increasing concentrations of TGF-β1 (0.1, 0.2, 1.0, 2.0, and 5.0 ng/ml) shows up-regulation of FKBP10 at 48 hours in the presence of 1.0 ng/ml and higher TGF-β1 concentrations. Efficacy of TGF-β1 treatment was confirmed by monitoring phosphorylation of mothers against decapentaplegic homolog 3 (SMAD3, depicted as pSMAD3) in comparison with total SMAD3 levels (SMAD3). (B) 2.0 ng/ml TGF-β1 significantly increased FKBP10 in phLF after 48 h, as quantified by densitometric analysis from Western blot analysis (cf. Figure 7C for representative Western blot, lanes with scrambled [−] small interfering RNA [siRNA] only). The effect of the knockdown was always significant (P < 0.01 or P < 0.001) but is not specified in the interest of clarity. (C) FKBP10 knockdown effects in phLF in combination with 24 and 48 hours of TGF-β1 treatment (2.0 ng/ml). A representative Western blot analysis for detection of FKBP10, collagen I, collagen V, fibronectin, and α-SMA is shown. (D) Densitometric quantification of collagen I, collagen V, fibronectin, and α-SMA protein levels. β-actin (ACTB) was used as loading control. Scrambled siRNA was used as control. Data are based on eight completely independent experiments and are given as mean ± SEM. Statistical analysis for comparison of FKBP10 siRNA vs. scrambled siRNA control was performed by paired two-tailed t test. *P < 0.05, **P < 0.01. The well-known effect of TGF-β1 on these proteins was mostly significant but is not specified in the interest of clarity. ctrl = control.
Loss of FKBP10 in IPF Fibroblasts Attenuates ECM Protein Synthesis and Decreases Levels of Secreted Collagen Independent of TGF-β Signaling
The siRNA-mediated knockdown of FKBP10 in IPF fibroblasts showed significant down-regulation of collagen I, collagen V, and fibronectin protein levels in crude cell lysates after 48 hours of TGF-β1 treatment (Figures 6C and 6D). Interestingly, TGF-β1 partly counteracted this effect for collagen I, but not for collagen V or fibronectin (Figure 6D). Moreover, levels of the myofibroblast marker α-SMA were also significantly decreased by knockdown of FKBP10 in the presence of TGF-β1 (Figure 6D, bottom right panel). The effects observed for collagen I and V manifested at transcript level already and also transcription of the TGF-β target gene PAI1 were decreased by knockdown of FKBP10, suggestive of an effect on overall TGF-β signaling (Figure 7A). However, the corresponding TGF-β–induced transcript fold changes did not indicate attenuation of TGF-β signaling in the FKBP10 knockdown (Figure 7B), and neither a Smad signaling luciferase reporter assay (Figure 7C) nor analysis of SMAD 3 phosphorylation or protein levels (Figure 7D and Figure E3) showed an appreciable effect of FKBP10 knockdown on canonical TGF-β signaling.
Figure 7.

Transforming growth factor (TGF)-β1 induces FK506-binding protein 10 (FKBP10) expression, and knockdown of FKBP10 in primary human lung fibroblasts (phLF) significantly decreases expression of collagen α-1(I) chain (COL1A1), collagen α-1(V) chain (COL5A1), and plasminogen activator inhibitor 1 (PAI1) on the transcriptional level independent of the canonical TGF-β signaling pathway. (A) Quantitative reverse transcriptase–polymerase chain reaction analysis of transcript levels of COL1A1 and COL5A1 (encoding the α1-chain of collagen I and V, respectively) and PAI1 as a representative TGF-β1–transduced gene after FKBP10 knockdown in combination with 24 and 48 hours of TGF-β1 treatment (2.0 ng/ml). Data are depicted as mean ± SEM from eight independent experiments, and a paired two-tailed t test was used for statistical analysis for comparison of FKBP10 small interfering RNA (siRNA) vs. scrambled siRNA control. The well-known effect of TGF-β1 on these transcripts was always highly significant (P < 0.01 or P < 0.001) but is not specified in the interest of clarity. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) was used as endogenous control. (B) TGF-β–induced transcript fold changes obtained from the same data do not show attenuation of TGF-β signaling. (C) Transfection of phLF with a mothers against decapentaplegic homolog (SMAD) signaling luciferase reporter plasmid during FKBP10 knockdown showed no effect on canonical TGF-β signaling (n = 3). (D) Western blot analysis (n = 3) demonstrated that FKBP10 knockdown did not affect levels of total and phosphorylated SMAD3. β-actin (ACTB) was used as loading control. For quantification of these data, see Figure E3 in the online supplement. *P < 0.05, **P < 0.01, ***P < 0.001. ctrl = control.
Finally, we assessed levels of secreted collagen in these experiments by two approaches. First, specifically collagen type I levels were semiquantified using Western blot analysis followed by densitometry (Figures 8A and 8B); second, total secreted collagen was quantified using the Sircol assay (Figure 8C). Both approaches showed significant decreases of secreted collagen in the FKBP10 knockdown in absence and presence of TGF-β1.
Figure 8.

Knockdown of FK506-binding protein 10 (FKBP10) attenuates collagen secretion. (A) Western blot analysis of secreted collagen I. Collagen I was precipitated from cell culture supernatant after FKBP10 knockdown in combination with 24 and 48 hours of transforming growth factor (TGF)-β1 treatment (2.0 ng/ml) and analyzed by Western blot analysis. (B) Densitometric analysis of secreted collagen I as detected in A. Data shown are based on seven independent experiments and given as mean ± SEM. (C) Results of a Sircol assay for assessment of total secreted collagen in cell culture supernatant after FKBP10 knockdown in combination with 24 and 48 hours of TGF-β1 treatment. Data shown are based on eight independent experiments and given as mean ± SEM. Statistical analysis was performed using paired two-tailed t test for comparison of FKBP10 siRNA vs. scrambled siRNA control. ctrl = control; siRNA = small interfering RNA. *P < 0.05, **P < 0.01.
In our efforts to interpret the relevance of FKBP10 knockdown in IPF fibroblasts in the context of currently approved IPF therapeutics, we used the same experimental setup to address inhibition of total collagen secretion in IPF fibroblasts by nintedanib and pirfenidone. Importantly, FKBP10 knockdown significantly decreased total collagen secretion by about 20%, in an amount similar to the effects observed with nintedanib, whereas we did not observe any effect of pirfenidone on total collagen secretion (Figure 9).
Figure 9.

In idiopathic pulmonary fibrosis fibroblasts, FK506-binding protein 10 (FKBP10) knockdown inhibits collagen secretion with similar efficiency as nintedanib, whereas pirfenidone shows no effect. (A–C) Normalized levels of secreted collagen in response to (A) FKBP10 knockdown, (B) nintedanib, and (C) pirfenidone treatment at varying concentrations. In contrast to pirfenidone, nintedanib shows a dose-dependent effect. In comparison, FKBP10 knockdown performs with similar efficiency as submicromolar concentrations of nintedanib. For FKBP10 knockdown, scrambled small interfering RNA (siRNA) was used as control. Data shown are based on four independent experiments and given as mean ± SEM. Statistical analysis was performed using paired two-tailed t test. *P < 0.05, **P < 0.01. ctrl = control; TGF-β = transforming growth factor-β.
Discussion
In the present study, we show that FKBP10 is up-regulated in bleomycin-induced lung fibrosis and IPF. FKBP10 is predominantly expressed in interstitial (myo)fibroblasts and CD68+ macrophages. TGF-β1, but not ER stress, induced FKBP10 expression in phLF. Loss of FKBP10 in phLF led to attenuation of collagen synthesis and secretion and reduced the expression of α-SMA and PAI1 independent of the TGF-β signaling pathway. Finally, FKBP10 knockdown inhibited collagen secretion with an efficiency similar to that of nintedanib.
FKBP10 was up-regulated in the mouse model of bleomycin-induced lung fibrosis and returned to baseline with resolution of the disease (Figure 1). In agreement, a previous study has indicated up-regulation of FKBP10 mRNA during bleomycin-induced lung injury (26). In this study, however, analysis was restricted to Day 10 after bleomycin administration, and up-regulation was merely assessed at transcript level. We clarify that FKBP10 overexpression occurs on the protein level and correlated with the fibrotic phase and lung function, both aspects of importance for the translation of these results to IPF, a state of presumably irreversible fibrosis. Most importantly, we show for the first time that FKBP10 expression is increased in IPF (Figure 2). This appears to be a universal phenomenon, as we discovered this in two completely independent cohorts, one in the United States and one in Europe, strongly arguing for an important function of FKBP10 in IPF. Immunofluorescent stainings of fibrotic mouse and IPF tissue sections revealed strong FKBP10 expression by interstitial fibroblasts, including myofibroblasts, the major ECM-producing cell type in fibrotic lung disease (Figures 3 and 4), indicating that FKBP10 might have a function in ECM protein synthesis in phLF. Notably, many myofibroblasts, both in the mouse model as well as in the IPF sections, were FKBP10-negative, highlighting the phenotypic diversity of interstitial mesenchymal cell types in fibrosis.
The subcellular localization provided an important clue for FKBP10 function in fibroblasts, as FKBP10 substrates proposed in the literature include not only ECM protein precursors, such as procollagen I or tropoelastin (11, 27), but also cytosolic proteins, such as HSP90 and c-Raf-1 (28). Here, we demonstrate that FKBP10 localized mainly to the ER in phLF (Figure 5). No colocalization was observed with the Golgi-specific protein GOLGA1 (29), indicating that FKBP10 is not associated with transport processes across the trans-Golgi network to the extracellular space. This suggests that FKBP10 mainly functions as an ER-resident chaperone and/or foldase in phLF, which agrees with previous studies in chondroblasts and embryonic fibroblasts (19, 27).
As there is evidence that ER stress contributes to IPF pathology (30, 31), we assessed whether FKBP10 is induced by ER stress as a possible explanation for its increased expression in IPF. Interestingly, we observed a drastic decrease in FKBP10 levels (band at 70 kD; cf. Figure E1A) in response to tunicamycin, an inhibitor of N-protein glycosylation (32). This decrease was accompanied by the appearance of a lower band at the height of the predicted size of the unmodified protein (65 kD; cf. Figure E1A), in line with the previous identification of FKBP10 as an N-glycosylated protein (33). This is in agreement with a previous study showing that FKBP10 is degraded by the proteasome in response to ER stress (34). As the observed decrease of FKBP10 might reflect deglycosylation of the protein rather than an overall decrease in expression levels, a mechanistically independent ER stress inducer, thapsigargin, was used to confirm FKBP10 down-regulation in ER stress (Figure E1B). Hence, it is unlikely that the unfolded protein response contributes to FKBP10 up-regulation in IPF.
In contrast, TGF-β1 significantly induced FKBP10 expression in phLF on the mRNA and protein level (cf. Figure E2, Figure 6B). This effect has previously only been observed in fetal lung fibroblasts (26) and implies that adult IPF phLF have retained the capacity to up-regulate FKBP10 under fibrotic conditions, supporting the use of this cell culture system for the functional analysis of FKBP10 in vitro. In agreement with a collagen chaperone function, inhibition of FKBP10 by an siRNA-mediated approach in this model resulted in a consistent decrease of collagen I and collagen V protein levels (cf. Figure 6C and 6D) as well as in decreased secreted collagen (Figure 8). In line with our results, deficiency of FKBP10 in osteogenesis imperfecta has been shown to attenuate collagen secretion and, moreover, decrease the extent of lysyl hydroxylation and extracellular collagen cross-linking and increase protease sensitivity of extracellular collagen I in dermal fibroblasts from patients with osteogenesis imperfecta (15, 17, 18).
Surprisingly, loss of FKBP10 affected collagen levels not only on protein but also on transcript levels (Figure 7A). This is unexpected, as FKBP10—as a chaperone and peptidyl-prolyl isomerase—is mainly ascribed a post-transcriptional role in procollagen I processing (11, 14, 15, 17, 18). Interestingly, loss of FKBP10 also attenuated the expression of the TGF-β–responsive genes PAI1 and α-SMA (Figures 6D and 7A). However, this was not due to inhibition of the canonical TGF-β signaling pathway (Figure 7B–7D). Hence, it appears that FKBP10 exerts a previously unappreciated function in signal transduction processes for the transcriptional regulation of profibrotic genes. Nevertheless, the data also show an increase in TGF-β–induced collagen expression and secretion, even in the context of FKBP10 knockdown, indicating that alternative, FKBP10-independent, profibrotic mechanisms contribute to overall ECM deposition.
Importantly, the effects of FKBP10 knockdown on TGF-β–induced collagen secretion in phLF were comparable to those of submicromolar concentrations of nintedanib (Figures 9A and 9B). In contrast, we did not observe any effect of pirfenidone in this context. Previous studies in human lung fibroblasts and A549 cells have shown moderate reduction of collagen I expression only at concentrations higher than those used in this study (1.0 mM vs. 1.6 and 2.7 mM pirfenidone, respectively [35, 36]). The effect of pirfenidone on TGF-β–induced collagen secretion in vitro has, to our knowledge, only been studied in trabecular meshwork cells, where collagen secretion was significantly reduced at a concentration of 2.5 mM (37). These concentrations are hardly physiologically relevant, indicating that the well-known antifibrotic effects of pirfenidone in vivo (38) differ substantially in underlying mechanisms or involve metabolic activation.
The ability to bind the immunosuppressive drug FK506 (tacrolimus) via at least one peptidyl-prolyl isomerase domain is characteristic for all FKBPs, including FKBP10 (11, 39). Interestingly, immunosuppressive peptidyl-prolyl isomerase inhibitors like FK506 have been ascribed potent antifibrotic effects in lung fibrosis (40–43). FK506 suppresses collagen synthesis and expression of the TGF-β1 receptor in dermal and lung fibroblasts, but the underlying mechanisms are largely unclear (41, 44, 45). Our findings put forward the possibility that inhibition of FKBP10 might contribute to the antifibrotic effects of FK506.
In conclusion, we show that FKBP10 is overexpressed in bleomycin-induced lung fibrosis and IPF. Up-regulation of FKBP10 expression is at least partly mediated by TGF-β1. Loss of FKBP10 in phLF derived from patients with IPF or control patients attenuated ECM protein expression and secretion and suppressed fibroblast-to-myofibroblast differentiation/activation. Although FKBP10 has been suggested to be important for normal lung development, with little expression in normal adult tissues, its expression appears to be reactivated during lung fibrosis (26, 27). Considering the impact of FKBP10 on collagen cross-linking (15, 17, 18) and the effects described herein on collagen secretion, FKBP10 might provide a very specific and effective drug target for treatment of IPF.
Acknowledgments
Acknowledgment
The authors thank Daniela Dietel for excellent technical assistance, Mona Dotzler for initial contributions to this project, and Melanie Königshoff and Kathrin Mutze for valuable help and discussion.
Footnotes
Supported by the Helmholtz Association, the German Center for Lung Research, and National Institutes of Health grants RO1HL108642 and RC2HL101715 (N.K.).
Author Contributions: Conception and design: C.A.S.-W., I.E.F., K.H., J.B., N.K., and O.E. Experimental work, analysis, and interpretation: C.A.S.-W., I.E.F., L.K., J.M., K.H., B.M.J.-G., E.H., G.P., H.W., C.N., R.H., M.L., N.K., and O.E. Drafting the manuscript and intellectual content: C.A.S.-W., I.E.F., L.K., K.H., N.K., and O.E.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201412-2233OC on June 3, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:285–292. doi: 10.1513/pats.200601-005TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selman M, King TE, Pardo A American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 3.Renzoni E, Srihari V, Sestini P. Pathogenesis of idiopathic pulmonary fibrosis: review of recent findings. F1000Prime Rep. 2014;6:69. doi: 10.12703/P6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–179. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, Bar-Joseph Z, Bitterman P, Blackburn MR, Bradford W, et al. Future directions in idiopathic pulmonary fibrosis research: an NHLBI workshop report. Am J Respir Crit Care Med. 2014;189:214–222. doi: 10.1164/rccm.201306-1141WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. 2014;189:1161–1172. doi: 10.1164/rccm.201312-2221PP. [DOI] [PubMed] [Google Scholar]
- 7.Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, Connett JE, Henke CA, Larsson O, Bitterman PB. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis: aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190:867–878. doi: 10.1164/rccm.201403-0509PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 10.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa Y, Vranka J, Wirz J, Nagata K, Bächinger HP. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J Biol Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- 12.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 13.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16:318–325. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- 14.Ishikawa Y, Bächinger HP. A molecular ensemble in the rER for procollagen maturation. Biochim Biophys Acta. 2013;1833:2479–2491. doi: 10.1016/j.bbamcr.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley BP, Malfait F, Bonafe L, Baldridge D, Homan E, Symoens S, Willaert A, Elcioglu N, Van Maldergem L, Verellen-Dumoulin C, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res. 2011;26:666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes AM, Cabral WA, Weis M, Makareeva E, Mertz EL, Leikin S, Eyre D, Trujillo C, Marini JC. Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum Mutat. 2012;33:1589–1598. doi: 10.1002/humu.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarze U, Cundy T, Pyott SM, Christiansen HE, Hegde MR, Bank RA, Pals G, Ankala A, Conneely K, Seaver L, et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet. 2013;22:1–17. doi: 10.1093/hmg/dds371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lietman CD, Rajagopal A, Homan EP, Munivez E, Jiang MM, Bertin TK, Chen Y, Hicks J, Weis M, Eyre D, et al. Connective tissue alterations in Fkbp10-/- mice. Hum Mol Genet. 2014;23:4822–4831. doi: 10.1093/hmg/ddu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.John G, Kohse K, Orasche J, Reda A, Schnelle-Kreis J, Zimmermann R, Schmid O, Eickelberg O, Yildirim AO. The composition of cigarette smoke determines inflammatory cell recruitment to the lung in COPD mouse models. Clin Sci (Lond) 2014;126:207–221. doi: 10.1042/CS20130117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer Y, Tedrow J, de Bernard S, Birker-Robaczewska M, Gibson KF, Juan Guardela B, Hess P, Klenk A, Lindell KO, Poirey S, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2015;52:217–231. doi: 10.1165/rcmb.2013-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang IV, Pedersen BS, Rabinovich E, Hennessy CE, Davidson EJ, Murphy E, Guardela BJ, Tedrow JR, Zhang Y, Singh MK, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:1263–1272. doi: 10.1164/rccm.201408-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma SF, Tseng GC, Feingold E, Juan-Guardela BM, Richards TJ, Lussier Y, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5:205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mise N, Savai R, Yu H, Schwarz J, Kaminski N, Eickelberg O. Zyxin is a transforming growth factor-β (TGF-β)/Smad3 target gene that regulates lung cancer cell motility via integrin α5β1. J Biol Chem. 2012;287:31393–31405. doi: 10.1074/jbc.M112.357624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patterson CE, Abrams WR, Wolter NE, Rosenbloom J, Davis EC. Developmental regulation and coordinate reexpression of FKBP65 with extracellular matrix proteins after lung injury suggest a specialized function for this endoplasmic reticulum immunophilin. Cell Stress Chaperones. 2005;10:285–295. doi: 10.1379/CSC-118R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patterson CE, Schaub T, Coleman EJ, Davis EC. Developmental regulation of FKBP65: an ER-localized extracellular matrix binding-protein. Mol Biol Cell. 2000;11:3925–3935. doi: 10.1091/mbc.11.11.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coss MC, Stephens RM, Morrison DK, Winterstein D, Smith LM, Simek SL. The immunophilin FKBP65 forms an association with the serine/threonine kinase c-Raf-1. Cell Growth Differ. 1998;9:41–48. [PubMed] [Google Scholar]
- 29.Munro S. The golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb Perspect Biol. 2011;3:6. doi: 10.1101/cshperspect.a005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elbein AD. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit Rev Biochem. 1984;16:21–49. doi: 10.3109/10409238409102805. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 34.Murphy LA, Ramirez EA, Trinh VT, Herman AM, Anderson VC, Brewster JL. Endoplasmic reticulum stress or mutation of an EF-hand Ca(2+)-binding domain directs the FKBP65 rotamase to an ERAD-based proteolysis. Cell Stress Chaperones. 2011;16:607–619. doi: 10.1007/s12192-011-0270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. doi: 10.1016/j.ejps.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 36.Hisatomi K, Mukae H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S, Fujita H, Nakamichi S, Oku H, Urata Y, et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm Med. 2012;12:24. doi: 10.1186/1471-2466-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattabiraman PP, Maddala R, Rao PV. Regulation of plasticity and fibrogenic activity of trabecular meshwork cells by Rho GTPase signaling. J Cell Physiol. 2014;229:927–942. doi: 10.1002/jcp.24524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20:85–97. doi: 10.1183/09059180.00001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coss MC, Winterstein D, Sowder RC, II, Simek SL. Molecular cloning, DNA sequence analysis, and biochemical characterization of a novel 65-kDa FK506-binding protein (FKBP65) J Biol Chem. 1995;270:29336–29341. doi: 10.1074/jbc.270.49.29336. [DOI] [PubMed] [Google Scholar]
- 40.Horita N, Akahane M, Okada Y, Kobayashi Y, Arai T, Amano I, Takezawa T, To M, To Y. Tacrolimus and steroid treatment for acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2011;50:189–195. doi: 10.2169/internalmedicine.50.4327. [DOI] [PubMed] [Google Scholar]
- 41.Nagano J, Iyonaga K, Kawamura K, Yamashita A, Ichiyasu H, Okamoto T, Suga M, Sasaki Y, Kohrogi H. Use of tacrolimus, a potent antifibrotic agent, in bleomycin-induced lung fibrosis. Eur Respir J. 2006;27:460–469. doi: 10.1183/09031936.06.00070705. [DOI] [PubMed] [Google Scholar]
- 42.Correale M, Totaro A, Lacedonia D, Montrone D, Di Biase M, Barbaro Foschino MP, Brunetti ND. Novelty in treatment of pulmonary fibrosis: pulmonary hypertension drugs and others. Cardiovasc Hematol Agents Med Chem. 2013;11:169–178. doi: 10.2174/187152571131100086. [DOI] [PubMed] [Google Scholar]
- 43.Inase N, Sawada M, Ohtani Y, Miyake S, Isogai S, Sakashita H, Miyazaki Y, Yoshizawa Y. Cyclosporin A followed by the treatment of acute exacerbation of idiopathic pulmonary fibrosis with corticosteroid. Intern Med. 2003;42:565–570. doi: 10.2169/internalmedicine.42.565. [DOI] [PubMed] [Google Scholar]
- 44.Lan CC, Fang AH, Wu PH, Wu CS. Tacrolimus abrogates TGF-beta1-induced type I collagen production in normal human fibroblasts through suppressing p38MAPK signalling pathway: implications on treatment of chronic atopic dermatitis lesions. J Eur Acad Dermatol Venereol. 2014;28:204–215. doi: 10.1111/jdv.12086. [DOI] [PubMed] [Google Scholar]
- 45.Wu CS, Wu PH, Fang AH, Lan CC. FK506 inhibits the enhancing effects of transforming growth factor (TGF)-β1 on collagen expression and TGF-β/Smad signalling in keloid fibroblasts: implication for new therapeutic approach. Br J Dermatol. 2012;167:532–541. doi: 10.1111/j.1365-2133.2012.11023.x. [DOI] [PubMed] [Google Scholar]