

Abstract
Rationale: Bronchopulmonary dysplasia (BPD), a prevalent severe lung disease of premature infants, has a strong genetic component. Large-scale genome-wide association studies for common variants have not revealed its genetic basis.
Objectives: Given the historical high mortality rate of extremely preterm infants who now survive and develop BPD, we hypothesized that risk loci underlying this disease are under severe purifying selection during evolution; thus, rare variants likely explain greater risk of the disease.
Methods: We performed exome sequencing on 50 BPD-affected and unaffected twin pairs using DNA isolated from neonatal blood spots and identified genes affected by extremely rare nonsynonymous mutations. Functional genomic approaches were then used to systematically compare these affected genes.
Measurements and Main Results: We identified 258 genes with rare nonsynonymous mutations in patients with BPD. These genes were highly enriched for processes involved in pulmonary structure and function including collagen fibril organization, morphogenesis of embryonic epithelium, and regulation of Wnt signaling pathway; displayed significantly elevated expression in fetal and adult lungs; and were substantially up-regulated in a murine model of BPD. Analyses of mouse mutants revealed their phenotypic enrichment for embryonic development and the cyanosis phenotype, a clinical manifestation of BPD.
Conclusions: Our study supports the role of rare variants in BPD, in contrast with the role of common variants targeted by genome-wide association studies. Overall, our study is the first to sequence BPD exomes from newborn blood spot samples and identify with high confidence genes implicated in BPD, thereby providing important insights into its biology and molecular etiology.
Keywords: exome sequencing, chronic lung disease, bronchopulmonary dysplasia, genetic predisposition to disease, premature
At a Glance Commentary
Scientific Knowledge on the Subject
We identified rare nonsynonymous mutations affecting 258 genes in patients with bronchopulmonary dysplasia, suggesting the role of rare variants in bronchopulmonary dysplasia.
What This Study Adds to the Field
This is the first study to identify genes implicated in bronchopulmonary dysplasia by exome sequencing of newborn blood spot samples.
Bronchopulmonary dysplasia (BPD) is a common chronic lung disease in premature infants, typically leading to significant morbidity and mortality (1). Its prevalence rises with increasing immaturity and occurs in most newborns born between 24 and 26 weeks gestational age, a time when alveolar and distal vascular development is rapidly occurring (2). BPD is phenotypically hallmarked by respiratory impairment accompanied with signs of shallow breathing, retractions, and paradoxical breathing patterns (3). Although BPD symptoms and treatment have been extensively studied, its genetic basis has been largely unclear (4). Twin studies revealed that genetic factors play a significant role in the development of BPD (5, 6). Others have suggested several gene candidates for this disease, including surfactant proteins (7), interleukin-18 (8), and macrophage migration inhibitory factor (9). However, none of these studies has characterized biologic pathways that contribute significantly to BPD, preventing further assessment of their phenotypic association with BPD symptoms. Although genome-wide association studies (GWAS) have been successfully applied to many other diseases, a recent large study from our group did not reveal any single-nucleotide polymorphisms (SNPs) with genome-wide statistical significance (10). When others studied patient samples that had been pooled a limited number of loci, but not genes, were associated with BPD (11).
Given the significant historic high mortality in extremely premature infants who today now survive and develop BPD, genomic loci underlying this disease would likely be under severe selective pressure. As such, severe disease-related alleles should be at low frequency preventing them from reaching high or even medium allele frequencies in the population (12). Because GWAS studies have been primarily designed for genotyping previously known common allelic variants (e.g., HapMap SNPs) with medium to high frequencies, simply genotyping these common variants among patients with BPD may be less likely to capture these disease-associated loci under strong selective pressure. In this study we tested the hypothesis that rare variants contribute to the risk for BPD.
The samples analyzed in this study were extracted from newborn blood spots that are collected in many states for routine screening of inherited disorders. Although widely available, their use for genome and exome sequencing had not been previously assessed. Exome sequencing of DNA extracted from blood spots from BPD-affected and non-BPD revealed rare damaging variants in particular genes in patients with BPD, thereby defining the genetic architecture underlying this disease. Our results help define the biologic pathways and processes implicated in BPD and provide a proof-of-principle that these routinely collected samples from large populations can be effectively used for genomic investigations.
Methods
Patient Population and Phenotype Definitions
Infants for study were twin live births identified from the California Perinatal Quality Care Collaborative (CPQCC, http://www.cpqcc.org/) (13) database during the 4 calendar years 2005–2008. The CPQCC tracks more than 90% of all neonatal intensive care unit admissions in California. Infants were included if they had gestational ages 25 weeks to 29 weeks and 6 days, one of the twin pair had birth weight less than 1,500 g, and they both had a minimum of 3 days continuous positive pressure ventilation during their hospitalization period up to 36 weeks postmenstrual age (PMA). We used standard criteria for the diagnosis and classification of BPD (1, 14). Because Lavoie and colleagues (6) demonstrated that heritability was associated with moderate and severe, but not mild cases, we defined BPD cases as infants requiring supplemental oxygen at 36 weeks PMA. The need for supplemental oxygen was determined by the usual practices of the individual neonatal intensive care unit and physiologic assessments (15) were not routinely performed. Infants were excluded if they had major congenital abnormalities, had major surgery, died or left the hospital before 36 week PMA, or if supplemental oxygen status at 36 weeks PMA was not known.
Research Ethic Board Approvals
This study was approved by both the institutional review board of Stanford University School of Medicine and the Committee for the Protection of Human Subjects of the State of California. When institutional review board approvals are obtained newborn screening blood spots may be used for anonymous research studies unless parents specifically request in writing that they not be used.
Whole-Exome Sequencing, SNP Call, and Annotation
See the online supplement for more details.
Statistical Analyses
See the online supplement for more details.
Results
Identifying Rare Nonsynonymous Mutations from Infants with or without BPD by Exome Sequencing
We studied 50 twin pairs born in California, among which 51 individuals had BPD. Genomic DNA was extracted from their newborn screening blood spots (16) and subjected to exome sequencing and single nucleotide variant calling and standardized filtering (see Methods). With the identified variants, we were able to distinguish the 21 monozygotic twin pairs (four pairs with discordant BPD status) from the 29 dizygotic twin pairs, thereby serving as a useful quality control for the samples. In total we identified 57,535 high-confidence nonsynonymous variants (missense or loss-of-function) (see Table E1 in the online supplement) among the 100 individuals. To reveal the population differentiation of these sequenced individuals (17), we performed principal component analysis (PCA) for these nonsynonymous sites and observed clear population stratification within these samples, which was largely consistent with their self-reported ethnic background (P < 2.2 × 10−16) (Figure 1A), except one twin pair that coclustered with the African American population. Notably, the Hispanic individuals (the western U.S. Hispanic descents) formed a separate cluster closer to the non-Hispanic whites than to other populations, demonstrating the overall quality of the identified variants that enabled us to assess population structure in a finer resolution.
Figure 1.

Principal component analysis for population stratification. This analysis was for all the nonsynonymous sites (A), the rare nonsynonymous sites (B), and the extremely conserved rare nonsynonymous sites (C) identified from the 100 study participants. It is clear that the population structure has been lost when only considering the rare nonsynonymous sites, and has reached almost zero differentiation when using the extremely conserved rare nonsynonymous sites. BPD = bronchopulmonary dysplasia.
Because most samples were phenotypically concordant or dizygotic twin pairs (only four phenotypically discordant monozygotic twin pairs), we decided not to directly use the twin relationship between the samples, but to directly assess the gene sets differentially affected between subjects with and without BPD. We hypothesized that rare variants likely play a more important role in this disease, and thus only considered nonsynonymous variants identified in study participants, but not observed in the 1,000 Genome dataset. As shown in Figure 1B, PCA analysis for this set of rare nonsynonymous variants revealed a clear loss of population structure. To exclude the possibility that these rare variants were mere artifacts from our variant calls for the exome sequencing data, we performed a comparative genomic analysis to examine the selective pressure on these low-frequency loci. We speculated that if these variants are truly underrepresented within human population, they are more likely to be under strong purifying selection during the course of evolution, and such would strongly inhibit an increase in their minor allele frequencies (12, 18).
The GERP++ algorithm (19) was designed to compare the variability of each nucleotide in the human genome against orthologous positions across 33 other mammalian genomes and quantify the evolutionary constraints over each genomic locus in the human genome thereby providing a cross-species comparison and a complementary and independent test for the population genetic data within a single species. As expected, we observed that these rare nonsynonymous loci were overall assigned with substantially higher GERP++ scores relative to the nonsynonymous variants identified from the 1,000 Genome Project (P < 1 ×10−3; Wilcoxon rank sum test). This demonstrates that the loss of population structure (Figure 1B) is likely not explained by the technical artifacts, but is a result of an elevated selection force on these loci.
To exclude stochastic mutations with low frequencies in a given population among these rare variants, we further analyzed the mutations affecting the extremely conserved genomic loci (GERP++ score ≥ 5.83, <95% of the nonsynonymous sites in the 1,000 Genome dataset) and thus considered 315 and 234 rare nonsynonymous variants identified in 51 individuals with BPD and 49 individuals without BPD, respectively. As shown in Figure 1C, PCA analysis on this set of variants revealed that most of the individuals (96%) were clustered around the center with little population differentiation, excluding two twin pairs (4%) far away from the center. Because individuals from the same populations with the outliers (Asian/Pacific Islanders and non-Hispanic whites) were coclustered with other study participants, the outlier status of the two twin pairs thus should not be explained by their population stratification, but by their highly distinct mutation patterns associated with these conserved rare nonsynonymous sites. Interestingly, these four outliers were all patients with BPD, so it is likely that genes affected by these variants are implicated in BPD.
To confirm that the complete loss of population structure (Figure 1C) was not caused by undersampling of the conserved rare nonsynonymous variants from the mutation pool of all the individuals, we performed a set of permutation tests where in each simulation we randomly sampled the same number of nonsynonymous sites and repeated the previous PCA analysis. These randomly sampled sites clearly recapitulated (P < 1 × 10−10; permutation test) the population structure in Figure 1A, demonstrating that the lack of population stratification in Figure 1C cannot be explained by insufficient sampling, but by the strong selective pressure against population differentiation. Because such a signal of negative selection typically suggests critical biologic functions during evolution, we next closely examined the genes affected by these conserved rare nonsynonymous mutations.
When we excluded the genes that were affected in both cases and control subjects we found conserved rare nonsynonymous variants affecting 258 and 182 nonoverlapping genes in the subject with BPD and without BPD, respectively. We next examined whether the affected genes in the patients with BPD shared some genetic properties that were absent from the genes identified from the siblings without BPD. Importantly, because the variant calls were masked to whether infants had or did not have BPD, our observation is not affected by a systematic bias toward infants with BPD, or unequal gene length or GC content. Furthermore, because the overall population differentiation for this set of variants was close to zero (Figure 1C), our analysis should not be affected by population stratification. Finally, because we are comparing functional differences for the nonoverlapping gene sets between individuals with and without BPD, this comparison is not affected by the genetic relatedness between the twin pairs.
Functional Enrichment of the Affected Genes in Individuals with BPD
The inheritance of BPD does not exhibit a classic Mendelian pattern and many of the identified variants in our exome screen are in heterozygous states. We therefore hypothesized that BPD, at least in part, is a dominant or semidominant trait, where its associated loci are dosage sensitive and thus a single copy loss contributes to the disease etiology. In the genetic context, the heterozygous effect or the dosage sensitivity is quantified by the degree of haploinsufficiency for a single gene, and particular in the human genome, haploinsufficiency for most of the genes has been accurately predicted by a recent study (20). To test our hypothesis, we examined the predicted haploinsufficiency scores for genes affected in individuals with and without BPD, and indeed observed that the affected genes in patients with BPD were substantially more haploinsufficient than all the protein-coding genes in the human genome (P = 1.86 × 10−6; Wilcoxon rank sum test) (Figure 2), whereas this trend was absent from the genes identified from the non-BPD control group (P = 0.14 compared with all the protein-coding genes in the human genome; Wilcoxon rank sum test) (Figure 2). This comparison supports our hypothesis and suggests that the genes implicated in BPD are likely dosage sensitive.
Figure 2.

Increased haploinsufficiency score for bronchopulmonary dysplasia (BPD) candidate genes. Predicted haploinsufficiency scores were assigned with genes in the genome background (all the protein-coding genes), genes with rare nonsynonymous variants in the non-BPD individuals, and genes identified from the individuals with BPD. All P values were derived from a comparison against the genome background, and the reference line (dashed horizontal red line) indicates the median haploinsufficiency of the genes identified from the subjects with BPD. The bottom and top of the boxes denote the first and third quartiles, respectively. The whiskers indicate the minimal value within 1.5 interquartile range (IQR) of the lower quartile and the maximum value within 1.5 IQR of the upper quartile.
We next performed gene ontology (GO) analysis (GO biologic processes, excluding the IEA terms that are inferred from electronic annotations) for the identified genes to determine their involvement of specific biologic processes. As shown in Figure 3, genes specifically affected among individuals with BPD were enriched for a broad category of functions. Importantly, as referenced by the affected genes in the infants with no BPD, our analysis further revealed that several pathways were specifically enriched for the gene set only identified from patients with BPD, but not in the affected genes in subjects without BPD (Figure 3), including collagen fibril organization (Benjamini–Hochberg false-discovery rate [FDR] = 0.026), morphogenesis of embryonic epithelium (FDR = 0.035), and regulation of Wnt signaling pathway (FDR = 0.03). Perturbations of these pathways in BPD have been suggested previously, but not shown, to predispose prematurely born infants to develop BPD (21). Thus, our pathway analysis of genes identified via exome sequencing has identified biologic pathways involved in BPD.
Figure 3.
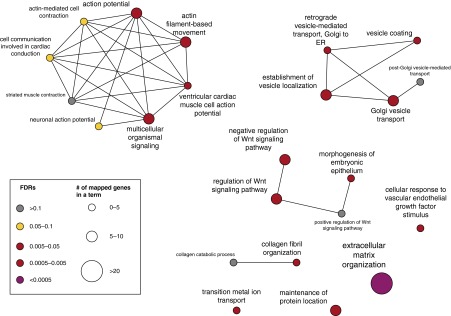
Differential enrichment for gene ontology (GO) biologic processes for bronchopulmonary dysplasia candidate genes. Each node represents a GO term, where larger size of the nodes indicate strong statistical significance and the darker color indicates the increased fold change of the number of genes assigned with each term. Connections between GO terms represent term–term associations, and significant terms were indicated by their false-discovery rates (FDRs). ER = endoplasmic reticulum; Wnt = wingless-type MMTV integration site family.
Expression Analysis of the Identified BPD Candidate Genes
We postulated that if the identified gene candidates were relevant to lung pathophysiology in the prematurely born, an increased expression in the developing fetal human lung would be expected. We therefore examined gene expression profiling data from multiple human tissues (22) and specifically investigated the expression profiles of the identified genes in lungs. As shown in Figure 4, expression of the BPD candidate genes displayed a marked elevation relative to all other genes in the genome (P = 0.04; Wilcoxon rank sum test), whereas the identified genes from the individuals without BPD exhibited similar expression levels with the transcriptome background (P = 0.20; Wilcoxon rank sum test). Taken together, this comparison indicates the strong candidacy of the identified genes in BPD, and suggests that these BPD candidate genes are important in lung pathophysiology.
Figure 4.

Increased gene expression of the bronchopulmonary dysplasia (BPD) candidate genes in the human lung. Normalized messenger RNA (mRNA) abundance in the human lung was profiled by microarray for genes in the transcriptome background (all the genes profiled), genes identified in the non-BPD subjects, and genes identified in the individuals with BPD. All P values were derived from a comparison against the transcriptome background, and the reference line (dashed horizontal red line) indicates the median mRNA abundance of the genes identified from the subjects with BPD. The bottom and top of the boxes denote the first and third quartiles, respectively. The whiskers indicate the minimal value within 1.5 interquartile range (IQR) of the lower quartile and the maximum value within 1.5 IQR of the upper quartile. The plus symbols represent outliers. The black dashed lines indicate the lower and upper limits of the regions with regular scale. Outliers outside of the black dashed lines are visualized with compressed scale in the regions surrounded by gray lines for better visualization.
Because BPD in the post surfactant era is characterized by alveolar simplification (23), and comparable pathologic findings can be induced in newborn rat pups exposed to a hyperoxic environment (24), we evaluated the BPD gene candidates identified in our screen by examining their expression pattern in the hyperoxic newborn model that mimics BPD (25). Microarrays were used to measure the amount of mRNA in the lungs of hyperoxia-exposed newborns and their room air littermates (wild-type) at postnatal Days 14 and 29 (P14 and P29, respectively). We reanalyzed the transcriptomic expression data (see Methods) and mapped the identified human genes onto their unambiguous one-to-one mouse orthologs. At both P14 and P29 the candidate genes we identified in our BPD-affected infants displayed a significant increase in their expression in hyperoxia-exposed lungs relative to levels in the transcriptome background (P = 0.046 and 8.2 × 10−3 for P14 and P29, Wilcoxon rank sum test, respectively) (Figures 5A and 5B). No increase was seen in the genes identified in the infants without BPD (P > 0.1 in both P14 and P29). These comparisons thus suggest that genes identified in our exome sequencing screen were up-regulated in the lung in response to increased oxygen concentration. It is also important to note that despite the overall trend, some individual genes implicated in BPD might also exhibit down-regulation on oxygen exposure, representing different mechanisms of action.
Figure 5.

Dynamic response of the identified bronchopulmonary dysplasia (BPD) candidate in a murine BPD model. Lungs from hyperoxia-exposed animals at postnatal Day 14 (A) and 29 (B) demonstrated a log2-fold increase in the messenger RNA (mRNA) abundance of BPD-related genes relative to the transcriptome background (all the genes profiled) or genes identified in the non-BPD subjects. All P values were derived from a comparison against the transcriptome background, and the reference line (dashed horizontal red line) indicates the median mRNA abundance of the genes identified in patients with BPD. The bottom and top of the boxes denote the first and third quartiles, respectively. The whiskers indicate the minimal value within 1.5 interquartile range (IQR) of the lower quartile and the maximum value within 1.5 IQR of the upper quartile. The plus symbols represent outliers. The black dashed lines indicate the lower and upper limits of the regions with regular scale. Outliers outside of the black dashed lines are visualized with compressed scale in the regions surrounded by gray lines for better visualization.
Phenotypic Implications of the Identified BPD Candidate Genes
Lastly, we attempted to systematically determine the overall phenotypic consequences in mouse mutants of the genes that were affected in BPD. We first performed our analysis using MamPhEA (26) and observed a significant enrichment of the BPD candidate genes for growth/size phenotype (MP [Mammalian Phenotype identifier in the Mouse Genome Informatics (MGI) database]: 0005378; Bonferroni, P = 0.018), abnormal embryogenesis and development (MP: 0001672; Bonferroni, P = 0.03), and preweaning lethality (MP: 0010770; Bonferroni, P = 0.024), whereas there was no overall enrichment for the genes affected in infants without BPD (Bonferroni, P > 0.05). Because the MamPhEA system computed the Bonferroni P values from comparisons with every phenotype term predefined in MGI, and most of these were presumably irrelevant to BPD symptoms (e.g., eye segment morphology, pigmentation, and skin morphology), we examined a set of several key phenotypes most relevant to BPD including respiratory distress, cyanosis, abnormal lung size, and abnormal breathing pattern. We found that 10% of the affected genes in patients with BPD were involved in the term “cyanosis” compared with 3% in the genome background (P = 7.85 × 10−4; Fisher exact test). The fraction was only 1.37% in the genes affected in the individuals without BPD.
We also performed the mammalian phenotypic enrichment test using EnrichR (27), which computes enrichment signals for phenotypic terms at the same ontology hierarchies, allowing us to increase detection sensitivity. As expected, the BPD candidate genes were highly enriched for the terms of abnormal respiratory systems morphology/physiology (MP: 0002132 and 0002133, with adjusted P = 9.01 × 10−3 and 0.026, respectively), whereas the genes affected in infants without BPD did not show enrichment for both terms (adjusted P ≥ 0.1). Collectively these analyses support the role of the identified candidate genes in BPD risk, and revealed potential functions in lung development and associations with cyanosis.
Discussion
Previous GWAS studies were primarily designed on the basis of the “the common disease–common variant” hypothesis, where common variants in the population are expected to account for a large fraction of disease risk. Although many common variants have been identified through GWAS for various complex diseases (28), the limited success in BPD motivated us to reexamine the common disease–common variant hypothesis, and hypothesized that rare variants could underlie the risk of BPD. It is important to note that “rare” single nucleotide variations are actually very common in any one human’s individual genome (29, 30). By exome sequencing of DNAs extracted from neonatal blood spots, we identified genes that could be involved in the heritability of BPD and also provide functional implications through downstream molecular characterizations.
Through our screen we prioritized 258 genes as candidates for BPD, indicating a polygenic nature of this disease. We observed that these candidate genes were more likely to be haploinsufficient, suggesting dosage sensitivity of these genes, whereby their heterozygous perturbations are likely linked with BPD, and harboring a heterozygous mutation in these genes is likely to increase BPD susceptibility, suggesting phenotypic dominance in this disease.
The identified mutations were highly heterogeneous among infants with BPD; however, our genomic analyses clearly revealed a convergence of these seemingly heterogeneous mutations onto common genetic pathways relevant to BPD etiologies. We showed that the affected genes were significantly enriched for collagen fibril organization, which supports the findings from a previous study (31). These enriched pathways, together with the significantly up-regulated gene expression in lung of BPD candidate genes, further elucidated pathways associated with this disease, and suggested that collagen fibril organization, morphogenesis of embryonic epithelium, and regulation of the Wnt signaling pathway were heavily involved in the development of BPD.
These findings were consistent with the observation that infants with BPD showed abnormal alveolarization and decreased pulmonary microvascular development in the lung (23). Notably, the identified BPD candidate genes also exhibited strong functional enrichment for extracellular matrix organization (Figure 3). By carefully examining the 14 affected genes annotated under this term (ADAMTS3, COL12A1, COL5A2, COL6A3, DMP1, FN1, ITGA1, ITGA2, ITGA9, ITGB6, KDR, LAMB2, NID2, and SDC2), we observed that most of these proteins are collagens (COL12A1, COL5A2, and COL6A3) or mediate collagen binding, including the integrins ITGA1, ITGA2, and ITGA9. Interestingly, the ADAMTS3 (ADAM metallopeptidase with thrombospondin type 1 motif, 3) cleaves the propeptides of type II collagen prior to fibril assembly (UniProtKB annotation), and a deficiency of this protein causes defects of connective tissues (annotated by RefSeq). Similarly, FN1 (fibronectin 1) and NID2 (nidogen 2, osteonidogen) interact with collagens at the cell surface, but the latter also binds to laminin and is involved in maintaining the structure of the basement membrane (annotated by RefSeq).
Another major constituent of the basement membrane was also identified, where LAMB2 interacts with integrins and modulates the organization of cells into tissues during embryonic development. Among these genes, ITGB6 is a prominent example, where its mouse mutant exhibited abnormal respiratory systems morphology and physiology (MGI annotation). Earlier studies have suggested the protein’s involvement in the development of pulmonary emphysema, which was caused by the overproduction of elastolytic matrix metalloproteinase-12 resulting from integrin-mediated transforming growth factor-β activation (32). Taken together, despite the seemingly heterogeneous mutations identified in different individuals, functional analyses of the affected genes suggest convergent pathways underlying BPD centered on the integrin and collagen proteins.
To understand the molecular functions of the identified genes, we explored expression dynamics of these identified BPD candidate genes with a hyperoxic mouse model. We observed increased expression of these genes on high-level oxygen exposure in the postnatal stages, and these observations were concordant with their overall phenotypic enrichment for the abnormal respiratory systems morphology and physiology in their respective mouse mutants. However, it is also important to note that although hyperoxia is one of the most frequently used models both because of its clinical relevance and ability to reproduce the pathologic changes, the molecular etiology of BPD might involve many other mechanisms of action, and thus molecular functions of the identified genes should be studied in other relevant experimental contexts in future. Moreover, because mouse models might not fully recapitulate the molecular causes of human disease, the generality of our observations on humans requires further investigation in future.
This study, for the first time, analyzed exomes from DNA extracted from dried neonatal blood spots of infants with or without BPD to unravel the heritability of BPD and to identify candidate genes for further validation. These more focused genetic investigations on rare variants based on high-throughput sequencing substantially extend the genetic knowledge of BPD and opens a new approach for examining the genetics of diseases that afflict infants and children.
Acknowledgments
Acknowledgment
The authors express their appreciation to Drs. Fred Lorey and Shabbir Ahmad for so aptly directing efforts to make newborn blood specimens available for analyses, to Allan Santos for his detailed efforts in finding and processing blood spots, and to the many individuals associated with the California Perinatal Quality Care Collaborative for their efforts to create such an important database. The authors thank the anonymous reviewers for all the constructive comments.
Footnotes
Supported by Banting Postdoctoral Fellowship administered by the Government of Canada (J.L.), Winston Chen Stanford Graduate Fellowship (K.-H.Y.), and National Institutes of Health grants 5U01HL10739304, 1P50HG00773501, and 5P50HG00773502 (M.S.). This work was funded by National Institutes of Health/NHLBI grant RC2 HL101748.
Author Contributions: J.L. and K.-H.Y. designed and conducted the analysis, interpreted the results, and drafted the manuscript. M.S., G.M.S., H.M.O’B., and D.K.S. interpreted the data and revised the manuscript. J.O., L.L.J.-P., and J.B.G. acquired the data.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201501-0168OC on June 1, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163:1723–1729. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 2.Coalson JJ. Pathology of bronchopulmonary dysplasia. Semin Perinatol. 2006;30:179–184. doi: 10.1053/j.semperi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Kinsella JP, Greenough A, Abman SH. Bronchopulmonary dysplasia. Lancet. 2006;367:1421–1431. doi: 10.1016/S0140-6736(06)68615-7. [DOI] [PubMed] [Google Scholar]
- 4.Shaw GM, O’Brodovich HM. Progress in understanding the genetics of bronchopulmonary dysplasia. Semin Perinatol. 2013;37:85–93. doi: 10.1053/j.semperi.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhandari V, Bizzarro MJ, Shetty A, Zhong X, Page GP, Zhang H, Ment LR, Gruen JR Neonatal Genetics Study Group. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics. 2006;117:1901–1906. doi: 10.1542/peds.2005-1414. [DOI] [PubMed] [Google Scholar]
- 6.Lavoie PM, Pham C, Jang KL. Heritability of bronchopulmonary dysplasia, defined according to the consensus statement of the national institutes of health. Pediatrics. 2008;122:479–485. doi: 10.1542/peds.2007-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryckman KK, Dagle JM, Kelsey K, Momany AM, Murray JC. Genetic associations of surfactant protein D and angiotensin-converting enzyme with lung disease in preterm neonates. J Perinatol. 2012;32:349–355. doi: 10.1038/jp.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Floros J, Londono D, Gordon D, Silveyra P, Diangelo SL, Viscardi RM, Worthen GS, Shenberger J, Wang G, Lin Z, et al. IL-18R1 and IL-18RAP SNPs may be associated with bronchopulmonary dysplasia in African-American infants. Pediatr Res. 2012;71:107–114. doi: 10.1038/pr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prencipe G, Auriti C, Inglese R, Devito R, Ronchetti MP, Seganti G, Ravà L, Orzalesi M, De Benedetti F. A polymorphism in the macrophage migration inhibitory factor promoter is associated with bronchopulmonary dysplasia. Pediatr Res. 2011;69:142–147. doi: 10.1203/PDR.0b013e3182042496. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, St Julien KR, Stevenson DK, Hoffmann TJ, Witte JS, Lazzeroni LC, Krasnow MA, Quaintance CC, Oehlert JW, Jelliffe-Pawlowski LL, et al. A genome-wide association study (GWAS) for bronchopulmonary dysplasia. Pediatrics. 2013;132:290–297. doi: 10.1542/peds.2013-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadchouel A, Durrmeyer X, Bouzigon E, Incitti R, Huusko J, Jarreau PH, Lenclen R, Demenais F, Franco-Montoya ML, Layouni I, et al. Identification of SPOCK2 as a susceptibility gene for bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2011;184:1164–1170. doi: 10.1164/rccm.201103-0548OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartl DL, Clark AG. Principles of population genetics. Sunderland, MA: Sinauer Associates; 2007. [Google Scholar]
- 13.Gould JB. The role of regional collaboratives: the California Perinatal Quality Care Collaborative model. Clin Perinatol. 2010;37:71–86. doi: 10.1016/j.clp.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Walsh MC, Szefler S, Davis J, Allen M, Van Marter L, Abman S, Blackmon L, Jobe A. Summary proceedings from the bronchopulmonary dysplasia group. Pediatrics. 2006;117:S52–S56. doi: 10.1542/peds.2005-0620I. [DOI] [PubMed] [Google Scholar]
- 15.Walsh MC, Yao Q, Gettner P, Hale E, Collins M, Hensman A, Everette R, Peters N, Miller N, Muran G, et al. National Institute of Child Health and Human Development Neonatal Research Network. Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics. 2004;114:1305–1311. doi: 10.1542/peds.2004-0204. [DOI] [PubMed] [Google Scholar]
- 16.St Julien KR, Jelliffe-Pawlowski LL, Shaw GM, Stevenson DK, O’Brodovich HM, Krasnow MA Stanford BPD Study Group. High quality genome-wide genotyping from archived dried blood spots without DNA amplification. PLoS One. 2013;8:e64710. doi: 10.1371/journal.pone.0064710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat Genet. 2008;40:340–345. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 19.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLOS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. 2007;357:1946–1955. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- 22.She X, Rohl CA, Castle JC, Kulkarni AV, Johnson JM, Chen R. Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genomics. 2009;10:269. doi: 10.1186/1471-2164-10-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol. 1998;29:710–717. doi: 10.1016/s0046-8177(98)90280-5. [DOI] [PubMed] [Google Scholar]
- 24.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, Kourembanas S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med. 2009;180:1122–1130. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong J, Carey WA, Abel S, Collura C, Jiang G, Tomaszek S, Sutor S, Roden AC, Asmann YW, Prakash YS, et al. MicroRNA-mRNA interactions in a murine model of hyperoxia-induced bronchopulmonary dysplasia. BMC Genomics. 2012;13:204. doi: 10.1186/1471-2164-13-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weng MP, Liao BY. MamPhEA: a web tool for mammalian phenotype enrichment analysis. Bioinformatics. 2010;26:2212–2213. doi: 10.1093/bioinformatics/btq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 29.Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. Broad GO; Seattle GO; NHLBI Exome Sequencing Project. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sweet DG, McMahon KJ, Curley AE, O’Connor CM, Halliday HL. Type I collagenases in bronchoalveolar lavage fluid from preterm babies at risk of developing chronic lung disease. Arch Dis Child Fetal Neonatal Ed. 2001;84:F168–F171. doi: 10.1136/fn.84.3.F168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature. 2003;422:169–173. doi: 10.1038/nature01413. [DOI] [PubMed] [Google Scholar]