

Abstract
Rationale: The clinical features of patients infected with pulmonary nontuberculous mycobacteria (PNTM) are well described, but the genetic components of infection susceptibility are not.
Objectives: To examine genetic variants in patients with PNTM, their unaffected family members, and a control group.
Methods: Whole-exome sequencing was done on 69 white patients with PNTM and 18 of their white unaffected family members. We performed a candidate gene analysis using immune, cystic fibrosis transmembrance conductance regulator (CFTR), cilia, and connective tissue gene sets. The numbers of patients, family members, and control subjects with variants in each category were compared, as was the average number of variants per person.
Measurements and Main Results: A significantly higher number of patients with PNTM than the other subjects had low-frequency, protein-affecting variants in immune, CFTR, cilia, and connective tissue categories (35, 26, 90, and 90%, respectively). Patients with PNTM also had significantly more cilia and connective tissue variants per person than did control subjects (2.47 and 2.55 compared with 1.38 and 1.40, respectively; P = 1.4 × 10−6 and P = 2.7 × 10−8, respectively). Patients with PNTM had an average of 5.26 variants across all categories (1.98 in control subjects; P = 2.8 × 10−17), and they were more likely than control subjects to have variants in multiple categories. We observed similar results for family members without PNTM infection, with the exception of the immune category.
Conclusions: Patients with PNTM have more low-frequency, protein-affecting variants in immune, CFTR, cilia, and connective tissue genes than their unaffected family members and control subjects. We propose that PNTM infection is a multigenic disease in which combinations of variants across gene categories, plus environmental exposures, increase susceptibility to the infection.
Keywords: bronchiectasis, cilia, genetics, immune system diseases, nontuberculous mycobacteria
At a Glance Commentary
Scientific Knowledge on the Subject
Patients who have pulmonary nontuberculous mycobacterial (PNTM) infection with no apparent underlying cause are known to be mostly postmenopausal white women who have a distinct body morphotype, high rates of cystic fibrosis transmembrance conductance regulator (CFTR) heterozygosity, and decreased ciliary beat frequency. However, the genetic underpinnings of these features and the susceptibility to PNTM infection are not understood.
What This Study Adds to the Field
Patients with PNTM have a burden of immune, CFTR, cilia, and connective tissue gene variants higher than that of control subjects, and they are more likely to have variants in multiple gene categories. This suggests that susceptibility to PNTM infection is multigenic, resulting from a combination of variations in genetic categories that mirror the phenotype of the patient.
Nontuberculous mycobacteria (NTM) are ubiquitous in the environment but rarely cause clinical disease. Disseminated nontuberculous mycobacterial infections occur only in the setting of immune compromise, such as genetic or acquired defects of the IFN-γ–IL-12 pathway. Isolated pulmonary nontuberculous mycobacterial (PNTM) infections, by contrast, are associated with disorders of mucociliary clearance, such as cystic fibrosis (CF) or primary ciliary dyskinesia (PCD). PNTM infections also occur in a distinct group of patients in whom there is no apparent underlying cause. These patients tend to be tall and lean white women who are diagnosed with PNTM infection in their sixth decade of life (1). They were found to have higher rates of scoliosis, pectus excavatum (PE), mitral valve prolapse (MVP), and mutations in the cystic fibrosis transmembrance conductance regulator (CFTR) gene than did matched control subjects in the National Health and Nutrition Examination Survey (1). Additionally, these patients may have a significantly decreased ciliary beat frequency compared with healthy control subjects (2). These clinical features, as well as the presence of familial clustering of disease (3), suggest that genetic factors play a role in susceptibility to PNTM disease. To further investigate the genetics of PNTM susceptibility, we performed whole-exome sequencing on 69 white patients with PNTM and 18 of their family members without PNTM infection and compared these data with sequencing data of the appropriate 1000 Genomes Project (1000G) control subjects. Some of the results of these studies have been reported previously in the form of conference abstracts (4, 5).
Methods
A detailed description of the methods can be found in the Materials and Methods section of the online supplement.
Cohort and Sample Collection
Patients were recruited from 2001 to 2013 at the National Institutes of Health (NIH) Clinical Center, Bethesda, MD. All patients and their family members provided informed consent under institutional review board–approved NIH protocols. All patients with PNTM had microbiologic and radiographic evidence of PNTM infection according to the American Thoracic Society criteria for PNTM disease (6). Patients and their family members were also evaluated for evidence of bronchiectasis, scoliosis, PE, MVP, joint hypermobility, the Steinberg thumb sign, and the Walker-Murdock wrist sign (7). DNA was isolated from blood collected from patients with PNTM and their family members seen at the NIH and from saliva (catalog number OG-500, Oragene; DNA Genotek, Ottawa, ON, Canada) of family members who did not travel to the NIH for evaluation. Because of the intrafamilial consistency and the relative reads on the sequences, we believe that the variants reported here reflect germline changes and not somatic ones. Lists of patients and pedigrees are provided in Table E1 and Figure E1, respectively, in the online supplement.
Sequencing and Variant Filtering
Whole-exome sequencing was performed on collected samples, and Illumina sequencing assay reads (Illumina, San Diego, CA) were aligned with Burrows-Wheeler Aligner (8). Single-nucleotide variants (SNVs) and insertions and deletions (indels) were called with GATK (9) and annotated by using SnpEff, ANNOVAR, and CADD (10–12). The sequencing data are publicly available in the database of genomes and phenotypes known as dbGaP (accession number phs000719.v1.p1).
Principal components analysis (PCA) was performed with smartpca in EIGENSTRAT 4.2 (13) using one PNTM-affected proband from each family and from the sporadic cases.
Exome data were filtered for variants present in patients with PNTM (PNTM-associated variants) in VarSifter (14) using a custom query with three parameters: gene name, population frequency less than 2%, and variant consequence (nonsynonymous, splicing, stop-gain, stop-loss, and start-loss variants). The genes examined were in immune, connective tissue, and cilia pathways, as well as CFTR (Table E2). Sequencing files from 1000G were filtered with the same parameters to obtain an overall set of variants not specifically associated with PNTM infection (“PNTM-unassociated variants”). The filtered PNTM-associated variants and PNTM-unassociated variants are outlined in Tables E3 and E4, respectively.
The indel dataset was filtered by gene name and impact factor (“moderate” or “high”). Filtered indels are given in Table E5.
Targeted Sanger sequencing was done for variants interesting to the authors. For that sequencing, custom primers for the relevant genes were used (Table E3). Primer sequences are available upon request. We observed a false-positive rate of 0% (n = 0 of 39).
Statistical Analysis
Statistical testing was performed in R (15). The number of samples with variants was compared with a two-tailed Fisher’s exact test. The average numbers of variants per person were compared by unpaired two-tailed t testing with Welch’s correction. P values of 0.05 or less were considered statistically significant.
A C-α burden test was performed on the quality-controlled SNV dataset in plinkseq; the results are given in Table E6.
Results
Cohort Description
We performed whole-exome sequencing on 102 individuals (20 patients with PNTM, 25 unaffected family members spread across 12 families, and 57 patients with sporadic PNTM infection). After quality control, three PNTM-affected samples and two PNTM-unaffected relatives’ samples were removed. To determine the appropriate control population based on the genetic ancestry of the PNTM infection cohort, PCA was performed on the remaining family probands and sporadic cases. Two families and one sporadic case were grouped with the CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) East Asian populations, and the other nine families and sporadic cases were grouped with the CEU (Utah Residents with Northern and Western European Ancestry) and TSI (Toscani in Italia) European populations (Figure 1). We continued analysis with only the European samples (15 patients with PNTM, 18 unaffected family members spread across 9 families, and 54 patients with sporadic PNTM infection) (Figure 2A). Features associated with PNTM disease, such as bronchiectasis or scoliosis (1), were present in some family members without PNTM infection. The 1000G Phase I European samples were used as controls. This control dataset is composed of 53% women and 47% men, with no available age or phenotype information.
Figure 1.
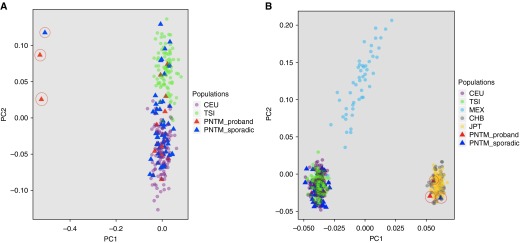
Principal components analysis of patients with pulmonary nontuberculous mycobacterial (PNTM) infection. (A) The majority of patients with PNTM group with the 1000 Genomes TSI (Toscani in Italia) and CEU (Utah residents with Northern and Western European Ancestry) European populations. (B) Three samples (circled in A and B) group with CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) populations, indicating that they are of Asian descent. Those patients and their family members were excluded from further analysis. Also note that, though several patients with PNTM self-identified as Hispanic, none group with the MEX (Mexican ancestry from Los Angeles, CA) population. PC1 and PC2 = principal components 1 and 2; PNTM_proband = one patient with PNTM infection from each family; PNTM_sporadic = patients with sporadic PNTM infection.
Figure 2.
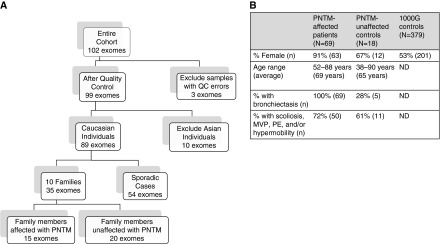
The structure and characteristics of the pulmonary nontuberculous mycobacterial (PNTM) infection whole-exome cohort. (A) A schematic depiction of how the original dataset was winnowed down to the samples used for analysis in this study. (B) The final breakdown of patients with PNTM infection, their unaffected family members, and 1000 Genomes Project (1000G) control samples used in candidate gene set analysis, including sex, age, incidence of bronchiectasis, and proportion with at least one connective tissue feature. In this case, connective tissue features included scoliosis, mitral valve prolapse (MVP), pectus excavatum (PE), and joint hypermobility. ND = not determined; QC = quality control.
Gene Filtering
Our initial approach to gene discovery was to look for variants that tracked with PNTM disease in each of the families and then to check for their presence in sporadic cases. This approach was limited by family structures that were not conducive to effective variant filtering, and we did not identify any obvious candidate variants. Next, we performed burden testing across the whole exome in plinkseq, which also did not yield any specific unifying genes. Therefore, we pursued a broad candidate gene approach using the previously identified gene sets as recognized through the study of well-characterized diseases (e.g., CF, PCD, inherited immune disorders, and inherited disorders of connective tissue). Using the final breakdown into PNTM-affected, PNTM-unaffected, and 1000G control groups (Figure 2B), we present a gene set analysis in each of these four genetic domains (i.e., immune, CFTR, cilia, and connective tissue). We also discuss variants found in each category that seemed particularly intriguing, details on which can be found in the online supplement.
Immune Genes
The genes involved in the synthesis of and response to IFN-γ are essential for the control of mycobacteria, as clearly exemplified by Mendelian defects in these genes in cases of disseminated bacillus Calmette-Guérin and NTM infections, sometimes referred to as “Mendelian susceptibility to mycobacterial disease” (MSMD) (16). Because these genes are so clearly implicated in mycobacterial susceptibility outside the lung, we explored them in the setting of isolated lung NTM disease. For the immune gene set, we included the 10 genes often considered part of MSMD (IFNGR1, IFNGR2, IL12RB1, IL12B, STAT1, IKBKG, CYBB, ISG15, IRF8, and GATA2). We also included other genes that might have an overlap with MSMD genes in terms of convergent pathways or reported phenocopies (RAG1, RAG2, IL12A, IL12RB2, MPEG1, CARD9, and CLEC4D). Interrogation of these 17 genes in our cohort identified 19 PNTM-associated variants that passed the filters (Table E3). None of the previously recognized MSMD mutations were identified; the heterozygous changes seen in STAT1, IRF8, CARD9, CLEC4D, and MPEG1 have not previously been associated with disseminated NTM. Thirty-five percent of patients with PNTM had variants in one of these immune pathway genes, significantly more than the 11% observed in the control population and 6% observed in the unaffected family members (P = 2.5 × 10−6 and P = 0.02, respectively) (Table 1). Those who had variants typically only had one, regardless of sample group (Table 1).
Table 1.
Immune Gene Results
| Immune Variants | PNTM Affected (n) | PNTM Unaffected (n) | 1000G Control (n) |
|---|---|---|---|
| Samples with at least one PNTM-associated variant | 24 | 1 | 41 |
| Samples without a PNTM-associated variant* | 45 | 17 | 338 |
| Average number of variants per person† | 1.17 | 1.00 | 1.07 |
Definition of abbreviations: 1000G = 1000 Genomes Project; ND = not done, because there were too few samples in the unaffected family members of patients with pulmonary nontuberculous mycobacterial infection; PNTM = pulmonary nontuberculous mycobacterial infection.
PNTM affected versus PNTM unaffected, P = 0.02; PNTM unaffected versus 1000G control, not significant (P > 0.05); PNTM affected versus 1000G control, P = 2.5 × 10−6.
PNTM affected versus PNTM unaffected, ND; PNTM unaffected versus 1000G control, ND; PNTM affected versus 1000G control, not significant (P > 0.05).
We found the following variants of particular interest. Signal transducer and activator of transcription 1 (STAT1) c.796G>A; p.V266I, heterozygous in two unrelated patients with PNTM infection, is a reported gain-of-function mutation originally described in an immune dysregulation–polyendocrinopathy–enteropathy, X-linked–like syndrome (17). IRF8 c.287C>T, p.T96M, heterozygous in two unrelated patients with PNTM, is in the DNA binding domain of the protein, similar to the other mutations reported in this gene (18). Preliminary results show that introduction of this mutation into mouse bone marrow derived macrophages decreased the expression of Irf8 target genes compared with wild-type protein (E. P. Szymanski and S. Togi, unpublished results). Three patients with PNTM had heterozygous changes in macrophage-expressed gene 1, perforin-2 (MPEG1): c.217A>G, p.T73A; c.946C>T, p.P316S; c.1192C>T, and p.Q398X. MPEG1 changes have not previously been reported in humans, but preliminary functional studies of these patients’ cells showed decreased killing of intracellular Mycobacterium avium (R. McCormack, unpublished results). Six patients with PNTM, including a sibling pair, had heterozygous variants in caspase recruitment domain family 9 (CARD9): c.1434+1G>C (splice variant), E249K, R315H, and V385L. CARD9 biallelic loss-of-function mutations lead to Candida meningitis, chronic mucocutaneous candidiasis, and other disseminated fungal infections (19–23). The C-type lectin receptors that signal through CARD9 recognize mycobacteria in addition to fungi, and pulmonary mycobacterial disease in Card9−/− mice cannot be controlled (24, 25). There are no reports of the clinical effects of heterozygous CARD9 mutations in humans, but Card9+/− mice show a modest increase in susceptibility to fungal infection (R. Drummond and M. Lionakis, unpublished results).
The lung is a major point of entry for environmental NTM infection, where even a slight defect in mycobacterial handling might permit persistence of mycobacteria and other organisms. Therefore, late-onset, milder clinical disease localized to the lung may be more likely to be associated with apparently mild heterozygous changes in immune response genes. These immune variants that we have identified in patients with PNTM and not their family members are distinct from the known MSMD mutations.
CFTR Variants
Cystic fibrosis is a critical paradigm for understanding PNTM disease. PNTM infection is associated with bronchiectasis; it is limited to the lung; and its incidence increases with age (26, 27). Previous studies have identified increased rates of heterozygous CFTR variants in patients with PNTM disease (1, 28). Therefore, we examined CFTR variants in our patients with PNTM and their unaffected family members. Sixteen CFTR PNTM-associated variants passed our filters (Table E3) and were found in 23% of patients with PNTM infection and 44% of unaffected family members, with the prevalence in both groups being significantly different from the 6% prevalence observed in control subjects (P = 2.8 × 10−5 and P = 1.1 × 10−5, respectively) (Table 2). Two CFTR deletions were also noted in two additional patients with PNTM, for an overall CFTR variation rate of 26% in patients with PNTM (Table E5). This percentage would increase to 32% if we included four additional patients with PNTM who had intronic CFTR variants that may contribute to PNTM disease (see online supplement). All 18 changes were subsequently classified by the mutation report in the CFTR2 database (The Clinical and Functional Translation of CFTR [CFTR2]; available at http://cftr2.org): 6% are known to cause CF (n = 1); 44% are parts of complex alleles but do not cause CF on their own (n = 8); 17% are associated with elevated sweat chloride and may cause CFTR dysfunction (n = 3); 6% are associated with normal sweat chloride and do not cause CFTR dysfunction (n = 1); and 28% are rare variants of unknown effect (n = 5). Of the 26 members of the total PNTM cohort (i.e., patients with PNTM and their unaffected family members) with changes in CFTR, 69% had just one change (either variant or indel) in CFTR (n = 18) and 31% had two changes (n = 8). Of note, the only two unaffected family members with two changes in CFTR had bronchiectasis with no infection. The average number of CFTR variants per person was not significantly different relative to that of control subjects (Table 2). Of the 18 patients with PNTM with a CFTR change, 17 had at least one variant in another candidate gene category.
Table 2.
CFTR Results
| CFTR Variants | PNTM Affected (n) | PNTM Unaffected (n) | 1000G Control (n) |
|---|---|---|---|
| Samples with at least one PNTM-associated variant | 16 | 8 | 22 |
| Samples without a PNTM-associated variant* | 53 | 10 | 357 |
| Average number of variants per person† | 1.19 | 1.25 | 1.32 |
Definition of abbreviations: 1000G = 1000 Genomes Project; CFTR = cystic fibrosis transmembrance conductance regulator; PNTM = pulmonary nontuberculous mycobacterial infection.
PNTM affected versus PNTM unaffected, not significant (P > 0.05); PNTM unaffected versus 1000G control, P = 1.1 × 10−5; PNTM affected versus 1000G control, P = 2.8 × 10−5.
All comparisons, not significant (P > 0.05).
Cilia Genes
An increasing number of genes are recognized to control cilia formation, structure, and function. We filtered for the 30 genes that have been clearly implicated in human PCD (29), plus an additional 10 genes with roles in ciliogenesis or ciliary beat frequency; 105 PNTM-associated variants passed those filters (Table E3). We identified cilia-related gene variants in 90% of patients with PNTM, significantly more than 45% in control subjects (P = 4.3 × 10−13) (Table 3). Patients with PNTM had an average of 2.47 cilia variants per patient, significantly more than the 1.38 cilia variants per control subject (P = 1.4 × 10−6, Table 3). The number of unaffected family members with cilia variants was not significantly different from control subjects, but the average number of variants per person was significantly higher than that of control subjects (2.63) (Table 3). Four deletions—in GAS2L2, DNAH5, and RPGR—also passed our filters, but they were not included in the statistical analysis (Table E5).
Table 3.
Cilia Gene Results
| Cilia Variants | PNTM Affected (n) | PNTM Unaffected (n) | 1000G Control (n) |
|---|---|---|---|
| Samples with at least one PNTM-associated variant | 62 | 11 | 170 |
| Samples without a PNTM-associated variant* | 7 | 7 | 209 |
| Average number of variants per person† | 2.47 | 2.63 | 1.38 |
Definition of abbreviations: 1000G = 1000 Genomes Project; PNTM = pulmonary nontuberculous mycobacterial infection.
PNTM affected versus PNTM unaffected, P = 0.007; PNTM unaffected versus 1000G control, not significant (P > 0.05); PNTM affected versus 1000G control, P = 4.3 × 10−13.
PNTM affected versus PNTM unaffected, not significant (P > 0.05); PNTM unaffected versus 1000G control, P = 0.03; PNTM affected versus 1000G control, P = 1.4 × 10−6.
We found the following variants of particular interest. Two siblings with PNTM infection had homozygous splice mutations in radial spoke head 1 homolog (RSPH1) that caused mild PCD (30). Each of three patients with PNTM had different heterozygous variants in macrophage-stimulating 1 receptor (MST1R), which is not known to be a disease-causing gene, but has been implicated in bronchiectasis and ciliary beat frequency (31, 32).
Connective Tissue Genes
Connective tissue features have long been recognized as part of the clinical phenotype of PNTM disease, sometimes eponymized as “Lady Windermere syndrome” (33). These features include tallness, leanness, scoliosis, pectus abnormalities, and MVP, all of which occur at increased rates compared with both general and demographically similar control populations (1, 34, 35). Therefore, we included 24 connective tissue genes in our survey; 99 PNTM-associated variants passed our filters (Table E3). Ninety percent of patients with PNTM had at least one of these variants, significantly more than the 57% frequency in control subjects (P = 3.0 × 10−8) (Table 4). Patients with PNTM had an average of 2.55 connective tissue variants per patient, significantly more than the 1.40 connective tissue variants per control subject (P = 2.7 × 10−8) (Table 4). This significance held also when we compared connective tissue variants in patients without PNTM with those of control subjects (Table 4). Of interest, 78% of unaffected family members had bronchiectasis, scoliosis, MVP, PE, and/or joint hypermobility, all features that may be influenced by the wide variation in connective tissue genes within this group. Three indels also passed our filters, although they were not included in our statistical comparison. Each of three patients with PNTM had apparently deleterious indels in FBN2, TGFBR1, and SMAD6 (Table E5).
Table 4.
Connective Tissue Gene Results
| Connective Tissue Variants | PNTM Affected (n) | PNTM Unaffected (n) | 1000G Control (n) |
|---|---|---|---|
| Samples with at least one PNTM-associated variant | 62 | 18 | 214 |
| Samples without a PNTM-associated variant* | 7 | 0 | 165 |
| Average number of variants per person† | 2.55 | 2.5 | 1.40 |
Definition of abbreviations: 1000G = 1000 Genomes Project; PNTM = pulmonary nontuberculous mycobacterial infection.
PNTM affected versus PNTM unaffected, not significant (P > 0.05); PNTM unaffected versus 1000G control, P = 7.9 × 10−5; PNTM affected versus 1000G control, P = 3.0 × 10−8.
PNTM affected versus PNTM unaffected, not significant (P > 0.05); PNTM unaffected versus 1000G control, P = 0.0008; PNTM affected versus 1000G control, P = 2.7 × 10−8.
We found the following variants of particular interest. One patient with PNTM infection had heterozygous FBN2 c.2260G>A, p.G754S, a variant that likely causes congenital contractural arachnodactyly (36), as well as heterozygous COL5A1 (collagen type V, alpha 1) c.1588G>A, p.G530S mutation, which causes mild Ehlers-Danlos syndrome (EDS) in the homozygous state (37–39). The latter mutation was found in 12% of the PNTM infection cohort compared with 4% in European 1000G control subjects and with other reported control frequencies of 2–6% (37–39). Six patients with PNTM with the G530S mutation had bronchiectasis and four had scoliosis, joint hypermobility, and/or the wrist and thumb signs. Of the five unaffected family members with the G530S mutation, four had scoliosis, PE, or wrist and thumb signs. The G530S mutation is known to be associated with mild collagen defects and could possibly affect body morphotype and lung structure and healing in this PNTM infection cohort (37–39).
Multiple Categories
Table 5 summarizes the findings of this candidate gene set approach. More patients with PNTM had at least two cilia or connective tissue variants than did control subjects (P = 4.4 × 10−16 and P = 6.3 × 10−14, respectively). This significance held true, but to a lesser extent, when we compared unaffected family members with control subjects.
Table 5.
Variants in PNTM-affected, PNTM-unaffected, and 1000G Samples across All Candidate Genes Tested
| PNTM Affected [n (%)] | PNTM Unaffected [n (%)] | 1000G Control [n (%)] | P Value* | |
|---|---|---|---|---|
| Number of people with variants in each category | ||||
| Immune | ||||
| 0 immune variants | 45 (65%) | 17 (94%) | 338 (89%) | |
| 1 immune variant | 21 (30%) | 1 (6%) | 38 (10%) | |
| ≥2 immune variants | 3 (4%) | 0 (0%) | 3 (1%) | |
| CFTR | ||||
| 0 CFTR variants | 53 (77%) | 10 (56%) | 357 (94%) | |
| 1 CFTR variant | 13 (19%) | 6 (33%) | 16 (4%) | |
| ≥2 CFTR variants | 3 (4%) | 2 (11%) | 6 (2%) | |
| Cilia | ||||
| 0 cilia variants | 7 (10%) | 7 (39%) | 209 (55%) | |
| 1 cilia variant | 22 (32%) | 3 (17%) | 121 (32%) | |
| ≥2 cilia variants | 40 (58%) | 8 (44%) | 49 (13%) | |
| Connective Tissue | ||||
| 0 connective tissue variants | 7 (10%) | 0 (0%) | 165 (44%) | |
| 1 connective tissue variant | 17 (25%) | 3 (17%) | 142 (37%) | |
| ≥2 connective tissue variants | 45 (65%) | 15 (83%) | 72 (19%) | |
| Variants in all candidate genes | ||||
| Average number of variants per person† | 5.26 | 4.7 | 1.98 | |
| Number of people with variants | ||||
| At least 1 hit | 63 (91%) | 17 (94%) | 156 (41%) | 2.2 × 10−15 |
| At least 2 hits | 44 (64%) | 12 (67%) | 28 (7%) | 2.7 × 10−24 |
| At least 3 hits | 17 (25%) | 3 (17%) | 0 (0%) | 2.4 × 10−15 |
| 4 hits | 1 (1%) | 0 (0%) | 0 (0%) | NS |
Definition of abbreviations: 1000G = 1000 Genomes Project; CFTR = cystic fibrosis transmembrance conductance regulator; NS = not significant (P > 0.05); PNTM = pulmonary nontuberculous mycobacterial infection.
P values were derived by using Fisher’s exact test to compare the number of patients with PNTM with the number of 1000G control subjects.
PNTM affected versus PNTM unaffected, NS; PNTM unaffected versus 1000G control, P = 0.0001; PNTM affected versus 1000G control, P = 2.8 × 10−17.
Patients with PNTM and their unaffected family members had strikingly more variants in any category per person than did control subjects (5.26, 4.7, and 1.98 variants, respectively; P = 2.8 × 10−17) (Table 5). When we separated those variants by category, a significantly higher number of patients with PNTM and their unaffected family members had one, two, or three category “hits,” where a hit was defined as one or more variants in the immune or CFTR categories but two or more variants in cilia or connective tissue genes (Table 5). Two or more variants were used for cilia and connective tissue because of the higher burden of variants per person in those categories (Tables 3–5). The number of variants in each category is summarized by sample in Table E6. The number of genes in which SNVs and indels were found for each category are summarized in Figures 3A and 3B, respectively.
Figure 3.

Number of genes in which variants or indels were found for each category. (A) The number of genes in which a single nucleotide variant (SNV) was found for each category. Nine genes of 17 were found in the immune category, 1 of 1 in cystic fibrosis transmembrance conductance regulator (CFTR), 29 of 40 in cilia, and 23 of 28 in connective tissue. (B) The number of genes in which an indel was found for each category. None of 17 genes were found in the immune category, 1 of 1 for CFTR, 3 of 40 for cilia, and 3 of 28 for connective tissue. Dark gray bar areas indicate the number of genes with a variant or indel, and the entire bar for each category indicates the total number of genes tested.
plinkseq Burden Test
Because we had resorted to candidate gene assessment, we wanted to explore whether there were any overlooked or unrecognized genes or pathways that might help explain PNTM disease. Therefore, we applied a broad exome-wide search using plinkseq to assess genetic burden using an unbiased approach. The C-α test was used because it performs well when there is a mixture of protective and detrimental variants in the dataset. With this test, 89 unique genes reached significance (Table E7). The list was assessed for gene function and pathways in Ingenuity Pathway Analysis (IPA; QIAGEN, Redwood City, CA), some of which were consistent with our candidate gene categories (Table E8). We also noted genes involved in lipid or estrogen pathways, possibly relevant to the typically tall, slender body habitus and female predominance of patients with PNTM. Fifty-three percent of burden genes fell under one or more candidate categories, and the other 47% were in other noncandidate categories (Table 6). Interestingly, one of the genes significant by burden testing is SCNN1B, which codes for the β-subunit of ENaC, a gene implicated in nonclassical CF (40). The only probable pathogenic variant in this gene (c.245C>G, p.S82C) was identified in PNTM-affected patient CJF_38.
Table 6.
Significant Genes from the C-α Burden Test by Candidate Category
| Category | Number of Genes (%) |
|---|---|
| Candidate categories | |
| Immune | 12 (13%) |
| Respiratory | 1 (1%) |
| Connective tissue | 19 (21%) |
| Additional metabolism genes | |
| Lipid | 1 (1%) |
| Estrogen | 1 (1%) |
| Multiple candidate categories* | 13 (15%) |
| Total candidate categories | 47 (53%) |
| All significant burden genes | 89 |
Genes that were categorized in multiple categories (i.e., immune, respiratory, connective tissue, lipid, and/or estrogen).
Discussion
Mendelian genetics has transformed our understanding of disease and the approach to medicine itself. Whereas genetic techniques have proven highly effective in rare diseases with clear patterns of inheritance and high penetrance, the genetic dissection of common or complex disease has been, as expected, much more difficult. By their very nature, common, complex diseases are likely to be a summation of multiple genetic and environmental influences. This is likely to be especially true in diseases that are late in onset, which thereby allows many environmental, degenerative, or somatic mutational events to occur. The disease under study here presents in the fifth to sixth decade of life; has sex, morphologic, and strong geographic associations (41); and is associated with pulmonary mycobacterial and other infections. These characteristics make it a particularly compelling example of these multiple confounding features. Therefore, despite the presence of familial clusters, it seems most likely that the genetic basis of PNTM disease is complex and the result of multiple convergent pathways’ leading to full phenotypic disease expression.
Given the distinct and widely reported phenotypes associated with PNTM infection, a combinatorial genetic approach to studying PNTM disease seemed reasonable to pursue. Compared with 1000G control subjects, more patients with PNTM and their unaffected family members had low frequency, protein-affecting variants across all categories, as well as more variants per person. However, the variants in the immune category genes were almost exclusively limited to the patients with PNTM infection, suggesting that these variants may be critical distinguishing factors between patients and their unaffected relatives. However, 23 of 24 patients with PNTM who had an immune variant had variants in at least one other category, implying that these immune changes alone are not sufficient to cause PNTM disease. Overall, members of the PNTM infection cohort were more likely to have variants in multiple candidate categories. This observation still held true when we specifically considered patients in whom we identified Mendelian disease variants (i.e., STAT1 gain of function, RSPH1 causing PCD, COL5A1 causing mild EDS, and CFTR changes). Multiple family members affected by PNTM shared specific variants in a few other instances (Table E3); however, there was not sufficient evidence for causality in those cases. Further familial genetic studies may provide more power for analysis, provided the study is carefully designed to account for the nuances of familial PNTM disease.
A parallel analysis of PNTM infection–unassociated variants showed extremely similar results (data not shown). This method included filtered variants that were found in the control dataset, but not in the PNTM infection dataset, providing additional assurance that our categorical findings are valid.
Because there is always a risk of bias in candidate searches, we undertook an unbiased, exome-wide C-α burden test in plinkseq, which identified a high proportion of the gene categories we had previously selected. Similarly, in a recent study of the molecular basis of asthma, investigators identified immunity- and cilia-related categories in microarray gene expression data (42). Both of these findings offer further support to the relevance of the categories we chose. As a whole, our categorical data provide genetic evidence to support the “susceptible persons” model of PNTM infection (43, 44), whereby an increased burden of variants in candidate categories conspire to alter patients’ skeletal structure, epithelial function, mucociliary clearance, and local immune function, thus enabling infection.
Digenic inheritance (alterations in two different genes within the same pathway combining to cause clinical disease) has been described in many disorders, including familial hemophagocytic lymphohistiocytosis, retinitis pigmentosa, Hirschsprung disease, and facioscapulohumeral muscular dystrophy (45, 46). The model of polygenic disease that we suggest for PNTM disease carries this mechanism to the next logical level (Figure 4). We hypothesize that variants in a handful of different genes within a few relevant pathways collude to cause PNTM disease. The variants alone are not enough to cause clinical disease, but together they make disease more likely. On top of these complex genetic factors, the clear influence of environmental factors such as high water vapor pressure and geographic variation in exposure to NTM (47) illustrates the multiple ways in which susceptible people can be further affected by a conducive environment (Figure 3). The late onset of PNTM disease allows ample time for genetic, environmental, and other factors to accumulate.
Figure 4.

A Venn diagram of our suggested model for multiple-category “hits” leading to infection. A susceptible person has low-frequency, protein-affecting variants in multiple gene categories, leading to mild abnormalities in the phenotypes related to those categories. This genetic predisposition also acts within a conducive environment and likely interacts with other components as well (e.g., age, hormones). Pulmonary nontuberculous mycobacterial (PNTM) disease occurs at the intersection of multiple underlying factors. CFTR = cystic fibrosis transmembrance conductance regulator.
We found it difficult to pinpoint a single, specific genetic basis of PNTM infection. Patients with PNTM and their family members shared a higher burden of variants in several categories, except immune genes. This overlap is not entirely surprising, as 75% of PNTM infection family members in our cohort had at least one body characteristic associated with PNTM disease, which is reflected in a larger study of families affected by PNTM infection (3). Because of these similarities, there is the chance that unaffected family members have not developed PNTM disease yet. We have tried to minimize this possibility by including unaffected family members of similar ages to the patients with PNTM (Figure 2B). The genetic burdens we report in CFTR, ciliary function, and connective tissue may be associated more with susceptibility to bronchiectasis than with PNTM infection itself: 35% of family members of patients with PNTM had bronchiectasis without PNTM infection. Given that immune variants were almost exclusively found in patients with PNTM, these immune genes might be critical discriminating genetic factors for PNTM disease. The involvement of immune function in PNTM infection susceptibility is reiterated by the associations of oral prednisone, anti–TNF-α therapy, other immunomodulatory drugs, AIDS, and MSMD with NTM infection (43, 44, 48, 49). Interestingly, the variants in immune genes we identified (STAT1, IRF8, MPEG1, and CARD9) are not MSMD-type mutations and do not appear to be associated with NTM disease outside the lung. Finally, although we recruited patients on the basis of NTM isolation, the vast majority had concomitant pulmonary fungal and bacterial infections, making the underlying infectious driver hard to discern. Therefore, it may be best to regard these genetic associations as being characterized by, but not limited to, NTM infection.
We are aware that our study is only moderately sized and used 1000G reference data for the control exomes. Still, this is the largest whole-exome database of patients with PNTM to date. There are undoubtedly other pathways to be identified that are relevant to this disease. This study is also important for what we did not find: The majority of the genes relevant in MSMD were not identified, confirming that the factors underlying disseminated and pulmonary disease segregate at the genetic level as they do clinically. These findings may provide insight into the ongoing quest to understand this complex but relatively common disease.
Acknowledgments
Acknowledgment
The authors thank Paul J. Gallins of the University of North Carolina School of Medicine for assistance with running the plinkseq burden test, as well as all of the patients and their families for their continued participation in our studies.
Footnotes
Supported by Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), and the Division of Intramural Research, NHLBI, National Institutes of Health (NIH); for exome sequencing and analyses, supported in part with federal funds to the Institute for Genome Sciences (C.M.F., principal investigator) from the NIAID, NIH, U.S. Department of Health and Human Services, under contract number HHSN272200900009C; and also supported by NIH grants 5R01 HL071798 and U54 HL096458 (M.K.). The Genetic Disorders of Mucociliary Clearance (U54 HL096458) is a part of the National Center for Advancing Translational Studies (NCATS) Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research, NCATS, funded through a collaboration between NCATS and NHLBI.
Author Contributions: E.P.S.: designed and performed the genetic analysis, performed Sanger validation, and wrote the manuscript; J.M.L., C.J.F., and C.H.: collected samples and phenotyped and treated patients; A.P.H.: performed Sanger validation and provided critical input throughout the study; F.C. and P.D.: assisted with quality control of the sequencing data, performed the principal components analysis, and provided valuable statistical expertise; A.J.O.: provided crucial bioinformatics and computing expertise; R.M. and E.P.: did preliminary studies on MPEG1; R.A.D. and M.S.L.: did preliminary studies on CARD9; S.K.B.: treated patients; D.R.P.: provided statistical expertise; M.K.: provided expertise in cilia genes and provided critical input; G.C.: provided expertise in CFTR and provided critical input; X.L., S.E.D., C.M.F., and H.T.: did exome sequencing, alignment, and variant calling; K.N.O.: treated patients, helped conceive the study, and provided critical input; S.M.H.: conceived the project and revised the manuscript. All authors had a chance to comment on the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201502-0387OC on June 3, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Kim RD, Greenberg DE, Ehrmantraut ME, Guide SV, Ding L, Shea Y, Brown MR, Chernick M, Steagall WK, Glasgow CG, et al. Pulmonary nontuberculous mycobacterial disease: prospective study of a distinct preexisting syndrome. Am J Respir Crit Care Med. 2008;178:1066–1074. doi: 10.1164/rccm.200805-686OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fowler CJ, Olivier KN, Leung JM, Smith CC, Huth AG, Root H, Kuhns DB, Logun C, Zelazny A, Frein CA, et al. Abnormal nasal nitric oxide production, ciliary beat frequency, and Toll-like receptor response in pulmonary nontuberculous mycobacterial disease epithelium. Am J Respir Crit Care Med. 2013;187:1374–1381. doi: 10.1164/rccm.201212-2197OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung JM, Fowler C, Smith C, Adjemian J, Frein C, Claypool RJ, Holland SM, Prevots RD, Olivier K. A familial syndrome of pulmonary nontuberculous mycobacteria infections. Am J Respir Crit Care Med. 2013;188:1373–1376. doi: 10.1164/rccm.201306-1059LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szymanski EP, Leung JM, Fowler CJ, Liu X, Devine SE, Fraser CM, Tettelin H, Browne SK, Olivier KN, Holland SM. STAT1 gain of function mutations are infrequent in pulmonary nontuberculous mycobacterial infection [abstract] J Clin Immunol. 2014;35:362. [Google Scholar]
- 5.Szymanski EP, Leung JM, Fowler CJ, Liu X, Devine SE, Fraser CM, Tettelin H, Hsu AP, Browne SK, Olivier KN, et al. Pulmonary nontuberculous mycobacterial infection: a multisystem multigenic disease [abstract] J Clin Immunol. 2015;35:309. doi: 10.1164/rccm.201502-0387OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, et al. ATS Mycobacterial Diseases Subcommittee An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases Am J Respir Crit Care Med 2007175367–416.[Published erratum appears in Am J Respir Crit Care Med 2007;175:744–745.] [DOI] [PubMed] [Google Scholar]
- 7.Dean JC. Management of Marfan syndrome. Heart. 2002;88:97–103. doi: 10.1136/heart.88.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D.Principal components analysis corrects for stratification in genome-wide association studies Nat Genet 200638904–909 [DOI] [PubMed] [Google Scholar]
- 14.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.RC Team. Vienna, Austria: R Foundation for Statistical Computing; 2013. A language and environment for statistical computing. [Google Scholar]
- 16.Al-Muhsen S, Casanova JL. The genetic heterogeneity of Mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol. 2008;122:1043–1051, quiz 1052–1053. doi: 10.1016/j.jaci.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 17.Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, Noel RJ, Verbsky JW, Freeman AF, Janssen E, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation–polyendocrinopathy–enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. 2013;131:1611–1623. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365:127–138. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, van Leeuwen EM, Roos D, Scalais E, de Beaufort C, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. 2013;121:2385–2392. doi: 10.1182/blood-2012-08-450551. [DOI] [PubMed] [Google Scholar]
- 20.Glocker EO, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Wang W, Lin Z, Wang X, Li T, Yu J, Liu W, Tong Z, Xu Y, Zhang J, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol. 2014;133:905–908, e3. doi: 10.1016/j.jaci.2013.09.033. [DOI] [PubMed] [Google Scholar]
- 22.Lanternier F, Barbati E, Meinzer U, Liu L, Pedergnana V, Migaud M, Héritier S, Chomton M, Frémond ML, Gonzales E, et al. Inherited CARD9 deficiency in 2 unrelated patients with invasive Exophiala infection. J Infect Dis. 2015;211:1241–1250. doi: 10.1093/infdis/jiu412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, Migaud M, Taibi L, Ammar-Khodja A, Boudghene Stambouli O, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. 2013;369:1704–1714. doi: 10.1056/NEJMoa1208487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz LX, Schroder K. Novel insights into the innate immune response to non-tuberculous Mycobacteria. Immunol Cell Biol. 2012;90:568–570. doi: 10.1038/icb.2011.86. [DOI] [PubMed] [Google Scholar]
- 25.Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, Hanke K, Gross O, Ruland J, Kaufmann SH. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med. 2010;207:777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olivier KN, Weber DJ, Lee JH, Handler A, Tudor G, Molina PL, Tomashefski J, Knowles MR Nontuberculous Mycobacteria in Cystic Fibrosis Study Group. Nontuberculous mycobacteria. II: nested-cohort study of impact on cystic fibrosis lung disease. Am J Respir Crit Care Med. 2003;167:835–840. doi: 10.1164/rccm.200207-679OC. [DOI] [PubMed] [Google Scholar]
- 27.Olivier KN, Weber DJ, Wallace RJ, Jr, Faiz AR, Lee JH, Zhang Y, Brown-Elliot BA, Handler A, Wilson RW, Schechter MS, et al. Nontuberculous Mycobacteria in Cystic Fibrosis Study Group. Nontuberculous mycobacteria. I: Multicenter prevalence study in cystic fibrosis. Am J Respir Crit Care Med. 2003;167:828–834. doi: 10.1164/rccm.200207-678OC. [DOI] [PubMed] [Google Scholar]
- 28.Ziedalski TM, Kao PN, Henig NR, Jacobs SS, Ruoss SJ. Prospective analysis of cystic fibrosis transmembrane regulator mutations in adults with bronchiectasis or pulmonary nontuberculous mycobacterial infection. Chest. 2006;130:995–1002. doi: 10.1378/chest.130.4.995. [DOI] [PubMed] [Google Scholar]
- 29.Kurkowiak M, Ziętkiewicz E, Witt M. Recent advances in primary ciliary dyskinesia genetics. J Med Genet. 2015;52:1–9. doi: 10.1136/jmedgenet-2014-102755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knowles MR, Ostrowski LE, Leigh MW, Sears PR, Davis SD, Wolf WE, Hazucha MJ, Carson JL, Olivier KN, Sagel SD, et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am J Respir Crit Care Med. 2014;189:707–717. doi: 10.1164/rccm.201311-2047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakamoto O, Iwama A, Amitani R, Takehara T, Yamaguchi N, Yamamoto T, Masuyama K, Yamanaka T, Ando M, Suda T. Role of macrophage-stimulating protein and its receptor, RON tyrosine kinase, in ciliary motility. J Clin Invest. 1997;99:701–709. doi: 10.1172/JCI119214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takano Y, Sakamoto O, Suga M, Suda T, Ando M. Elevated levels of macrophage-stimulating protein in induced sputum of patients with bronchiectasis. Respir Med. 2000;94:784–790. doi: 10.1053/rmed.2000.0822. [DOI] [PubMed] [Google Scholar]
- 33.Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern: the Lady Windermere syndrome. Chest. 1992;101:1605–1609. doi: 10.1378/chest.101.6.1605. [DOI] [PubMed] [Google Scholar]
- 34.Iseman MD, Buschman DL, Ackerson LM. Pectus excavatum and scoliosis: thoracic anomalies associated with pulmonary disease caused by Mycobacterium avium complex. Am Rev Respir Dis. 1991;144:914–916. doi: 10.1164/ajrccm/144.4.914. [DOI] [PubMed] [Google Scholar]
- 35.Kartalija M, Ovrutsky AR, Bryan CL, Pott GB, Fantuzzi G, Thomas J, Strand MJ, Bai X, Ramamoorthy P, Rothman MS, et al. Patients with nontuberculous mycobacterial lung disease exhibit unique body and immune phenotypes. Am J Respir Crit Care Med. 2013;187:197–205. doi: 10.1164/rccm.201206-1035OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callewaert BL, Loeys BL, Ficcadenti A, Vermeer S, Landgren M, Kroes HY, Yaron Y, Pope M, Foulds N, Boute O, et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Hum Mutat. 2009;30:334–341. doi: 10.1002/humu.20854. [DOI] [PubMed] [Google Scholar]
- 37.Giunta C, Nuytinck L, Raghunath M, Hausser I, De Paepe A, Steinmann B. Homozygous Gly530Ser substitution in COL5A1 causes mild classical Ehlers-Danlos syndrome. Am J Med Genet. 2002;109:284–290. doi: 10.1002/ajmg.10373. [DOI] [PubMed] [Google Scholar]
- 38.Giunta C, Steinmann B. Compound heterozygosity for a disease-causing G1489E [corrected] and disease-modifying G530S substitution in COL5A1 of a patient with the classical type of Ehlers-Danlos syndrome: an explanation of intrafamilial variability? Am J Med Genet. 2000;90:72–79. doi: 10.1002/(sici)1096-8628(20000103)90:1<72::aid-ajmg13>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 39.Malfait F, Coucke P, Symoens S, Loeys B, Nuytinck L, De Paepe A. The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum Mutat. 2005;25:28–37. doi: 10.1002/humu.20107. [DOI] [PubMed] [Google Scholar]
- 40.Sheridan MB, Fong P, Groman JD, Conrad C, Flume P, Diaz R, Harris C, Knowles M, Cutting GR. Mutations in the beta-subunit of the epithelial Na+ channel in patients with a cystic fibrosis-like syndrome. Hum Mol Genet. 2005;14:3493–3498. doi: 10.1093/hmg/ddi374. [DOI] [PubMed] [Google Scholar]
- 41.Adjemian J, Olivier KN, Seitz AE, Falkinham JO, III, Holland SM, Prevots DR. Spatial clusters of nontuberculous mycobacterial lung disease in the United States. Am J Respir Crit Care Med. 2012;186:553–558. doi: 10.1164/rccm.201205-0913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Modena BD, Tedrow JR, Milosevic J, Bleecker ER, Meyers DA, Wu W, Bar-Joseph Z, Erzurum SC, Gaston BM, Busse WW, et al. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am J Respir Crit Care Med. 2014;190:1363–1372. doi: 10.1164/rccm.201406-1099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dirac MA, Horan KL, Doody DR, Meschke JS, Park DR, Jackson LA, Weiss NS, Winthrop KL, Cangelosi GA. Environment or host?: A case–control study of risk factors for Mycobacterium avium complex lung disease. Am J Respir Crit Care Med. 2012;186:684–691. doi: 10.1164/rccm.201205-0825OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marras TK. Host susceptibility or environmental exposure in Mycobacterium avium complex lung disease: it takes two to tango. Am J Respir Crit Care Med. 2012;186:585–586. doi: 10.1164/rccm.201208-1432ED. [DOI] [PubMed] [Google Scholar]
- 45.Schäffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013;50:641–652. doi: 10.1136/jmedgenet-2013-101713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, Johnson JA, Filipovich AH. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. 2014;124:1331–1334. doi: 10.1182/blood-2014-05-573105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prevots DR, Adjemian J, Fernandez AG, Knowles MR, Olivier KN. Environmental risks for nontuberculous mycobacteria: individual exposures and climatic factors in the cystic fibrosis population. Ann Am Thorac Soc. 2014;11:1032–1038. doi: 10.1513/AnnalsATS.201404-184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan ED, Iseman MD. Underlying host risk factors for nontuberculous mycobacterial lung disease. Semin Respir Crit Care Med. 2013;34:110–123. doi: 10.1055/s-0033-1333573. [DOI] [PubMed] [Google Scholar]
- 49.Winthrop KL, Chang E, Yamashita S, Iademarco MF, LoBue PA. Nontuberculous mycobacteria infections and anti-tumor necrosis factor-α therapy. Emerg Infect Dis. 2009;15:1556–1561. doi: 10.3201/eid1510.090310. [DOI] [PMC free article] [PubMed] [Google Scholar]