

Abstract
Genetically engineered mice provide powerful tools for understanding mammalian gene function. These models traditionally rely on gene overexpression from transgenes or targeted, irreversible gene mutation. By adapting the tetracycline (tet)-responsive system previously used for gene overexpression, we have developed a simple transgenic system to reversibly control endogenous gene expression using RNA interference (RNAi) in mice. Transgenic mice harboring a tet-responsive RNA polymerase II promoter driving a microRNA-based short hairpin RNA targeting the tumor suppressor Trp53 reversibly express short hairpin RNA when crossed with existing mouse strains expressing general or tissue-specific ‘tet-on’ or ‘tet-off’ transactivators. Reversible Trp53 knockdown can be achieved in several tissues, and restoring Trp53 expression in lymphomas whose development is promoted by Trp53 knockdown leads to tumor regression. By leaving the target gene unaltered, this approach permits tissue-specific, reversible regulation of endogenous gene expression in vivo, with potential broad application in basic biology and drug target validation.
Mouse models are powerful tools for studying gene function in mammalian systems. Through transgenesis and homologous recombination, the mouse genome can be manipulated to bring about heritable, tissue-specific and reversible overexpression of protein-coding cDNAs, disruption of single genes and knock-in of mutant alleles1. Conditional gene inactivation can also be achieved using site-specific recombination systems, including Cre/loxP and Flp/FRT2. As recombinase activity in these transgenic systems can be controlled temporally and spatially, they have been particularly useful for studying the adult function of genes that are essential for mouse development3. Furthermore, acute disruption of gene function can circumvent genetic compensatory mechanisms inherent to germline mutant models4.
Despite their utility, recombination-based systems for conditional gene inactivation in mice have limitations: the crossing strategies to generate these models are time consuming, recombinase expression can be genotoxic5 and, most notably, recombination disrupts the target locus irreversibly. In many experimental contexts, such as studying gene function during mouse development or mimicking effects of potential targeted therapeutics in mouse models of human disease6, transient and/or tissue-specific inhibition of endogenous genes is desirable.
Tet-regulated gene overexpression systems were initially developed for use in cultured cells7, but they are also effective in transgenic mice8. Regulated transcription from the tet-responsive TRE promoter relies on the activity of a tet transactivator protein, which can be either activated (‘tet-on’) or repressed (‘tet-off’) by tetracycline or its analog doxycycline. Expressing tet transactivators from tissue-specific promoters9 allows spatial and temporal control of gene overexpression, a strategy widely used to study mouse embryonic and postnatal development10,11 and oncogene addiction12. Hence, many mouse lines have been developed that express the tTA (tet-off) or rtTA (tet-on) transactivators in different cell types.
RNAi holds great promise as an experimental tool for loss-of-function genetics. In many cell-based systems, short hairpin RNAs (shRNAs) have been expressed from tet-responsive or Cre/loxP-regulated promoters, allowing reversible gene inhibition13. Recent studies indicate that ubiquitous14 or tissue-specific15 RNA polymerase II promoters can be used to express microRNA-based shRNAs in transgenic animals, and other transgenic models have used Cre/loxP-mediated excision or activation of RNA polymerase III promoters to control shRNA expression unidirectionally16–19. However, these approaches do not fully exploit a major experimental advantage of RNAi: in principle, its effects can be tissue specific and reversible.
The aim of this study was to determine whether RNAi can be used as a tool to reversibly regulate expression of an endogenous gene in mice. We and others recently showed that RNA polymerase II promoters, including the tet-responsive TRE promoter, can be used to express shRNAs based on microRNA precursors20,21. This raised the possibility that the tet-regulated expression system, used thus far for cDNA overexpression in mice, could be exploited to reversibly inhibit gene expression in vivo. Here we create a transgenic mouse line harboring a tet-regulated shRNA cassette whose expression can be regulated in a tissue-specific manner when crossed with existing tet transactivator mouse strains. Using this system, we demonstrate reversible Trp53 knockdown in several adult tissues, and we examine the impact of restoring Trp53 expression in tumors driven by attenuated Trp53 function. Our results establish proof of principle for a transgenic RNAi technology that may eventually be used to spatially, temporally and reversibly regulate the expression of any endogenous gene.
Results
Mimicking strategies used previously for tet-regulated gene overexpression in mice, we generated a transgene comprising the tet-responsive TRE-CMV promoter7 upstream of a shRNA targeting mouse Trp53 referred to as p53.1224 (ref. 20) (Fig. 1a). We generated several founder lines harboring the TRE-p53.1224 transgene using standard pronuclear injection procedures22. To test each line for its ability to express the Trp53 shRNA from the TRE promoter, we isolated transgenic mouse embryonic fibroblasts (MEFs) and infected them with a retrovirus expressing tTA (tet-off), which transactivates the TRE promoter in the absence of doxycycline. Of nine TRE-p53.1224 founder lines tested, two (A and D) showed marked, tTA-dependent Trp53 knockdown in MEFs (Fig. 1b,c) that was fully reversible upon addition of doxycycline (Fig. 1d). Unless otherwise indicated, we present results obtained using founder line A; however, line D behaved similarly in each context tested.
Figure 1.
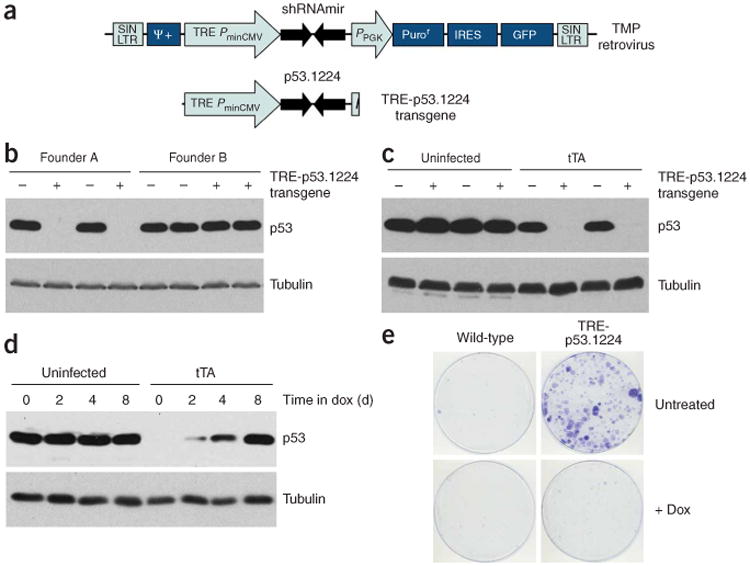
Germline transmission of a functional TRE-p53.1224 cassette. (a) Diagram of the TRE-p53.1224 transgene, derived from the TMP-p53.1224 retroviral vector20. (b) Trp53 protein blot of MEFs of indicated TRE-p53.1224 genotype (+ indicates transgenic) isolated from two different TRE-p53.1224 founder lines (A and B), infected with tTA. Note that Trp53 knockdown is only observed in line A. (c) Trp53 protein blot of MEFs of indicated TRE-p53.1224 genotype, either infected with tTA (right) or uninfected (left). (d) Trp53 protein blot of TRE-p53.1224 transgenic MEFs infected with tTA and cultured in doxycycline (Dox) for various periods. Uninfected controls are shown. (e) Colony formation assay for TRE-p53.1224 transgenic and nontransgenic littermate MEFs infected with tTA, plated at low density and cultured with or without doxycycline.
Consistent with the known effects of Trp53 deficiency, transgenic MEFs expressing tTA formed colonies when plated at low density but grew similarly to wild-type MEFs in the presence of doxycycline (Fig. 1e). Thus, in these founder lines, the TRE-p53.1224 transgenes are integrated at genomic locations conducive to reversible regulation by a tet transactivator. We observed similar tTA-dependent gene knockdown in MEFs isolated from a single transgenic line (of four tested) harboring a TRE-p16/p19.478 transgene (Supplementary Fig. 1 online). In this case, the shRNA is designed to target the transcript that encodes both p19ARF and p16INK4a, verifying that genes other than Trp53 can be targeted with this approach.
The rtTA (tet-on) transactivator is expressed in MEFs derived from Rosa26-rtTA transgenic embryos11. To test whether the amount of rtTA produced by the Rosa26-rtTA transgene was sufficient to drive effective shRNA expression, we cultured MEFs derived from a cross between TRE-p53.1224 and Rosa26-rtTA mice. We observed substantial Trp53 knockdown upon doxycycline treatment of Rosa26-rtTA/TRE-p53.1224 ‘double-transgenic’ MEFs (Fig. 2a), and Trp53 expression was restored with similar kinetics after doxycycline withdrawal (Fig. 2b). We observed functional Trp53 knockdown in Rosa26-rtTA/TRE-p53.1224 MEFs isolated using founder lines A and D (Fig. 2c–e) and in double-transgenic MEFs isolated from a cross between TRE-p53.1224 and a different general tet-on mouse strain, CMV-rtTA23 (Fig. 2f,g). These results demonstrate that effective and reversible knockdown of an endogenous gene can be achieved in an entirely transgenic setting.
Figure 2.
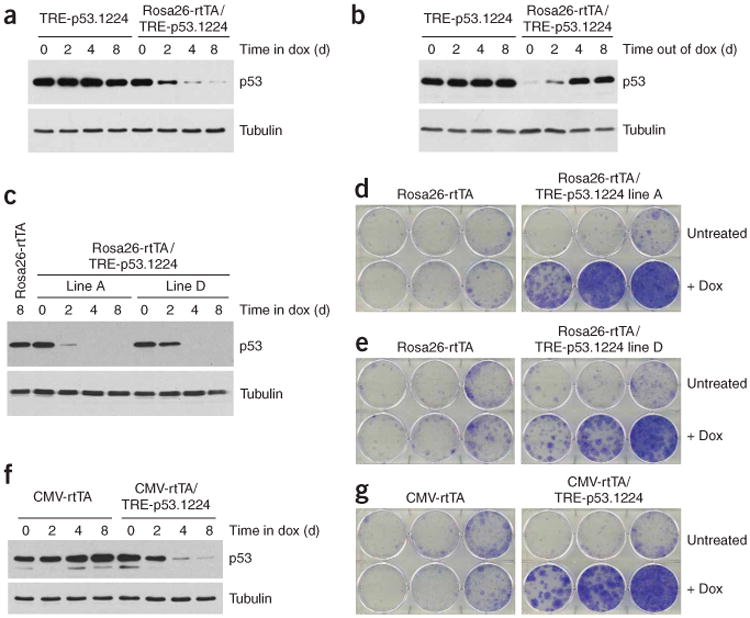
Transgenic tet-transactivators can drive Trp53 knockdown in MEFs. (a) Time course of Trp53 knockdown in Rosa26-rtTA/TRE-p53.1224 double-transgenic MEFs in response to doxycycline (Dox) treatment. (b) Time course of Trp53 re-expression in Rosa26-rtTA/TRE-p53.1224 double-transgenic MEFs that were cultured in doxycycline for 8 d and then shifted to normal medium at day 0. (c) Time course of Trp53 knockdown in Rosa26-rtTA/TRE-p53.1224 (from TRE-p53.1224 founder lines A and D) double-transgenic MEFs in response to doxycycline treatment. Littermate control doxycycline-treated Rosa26-rtTA MEFs are also shown. (d) Colony formation assay for Rosa26-rtTA/TRE-p53.1224 (founder line A) MEFs plated at low density and cultured with or without doxycycline. Littermate control Rosa26-rtTA MEFs are also shown. (e) As in d, but for TRE-p53.1224 founder line D. (f) Tme course of Trp53 knockdown in CMV-rtTA/TRE-p53.1224 double-transgenic MEFs in response to doxycycline treatment. Littermate control doxycycline-treated CMV-rtTA MEFs are also shown. (g) Colony formation assay for CMV-rtTA/TRE-p53.1224 MEFs plated at low density and cultured with or without doxycycline. Littermate control CMV-rtTA MEFs are also shown.
The Rosa26-rtTA and CMV-rtTA mouse lines have been used previously to regulate tet-on cDNA overexpression in tissues of adult mice11,23. To test whether they could also drive shRNA expression in these tissues, we measured p53.1224 short interfering RNA (siRNA) production in adult organs of doxycycline-treated Rosa26-rtTA/TRE-p53.1224 and CMV-rtTA/TRE-p53.1224 mice. Though we were unable to detect siRNA production in Rosa26-rtTA/TRE-p53.1224 tissues (data not shown), several tissues from CMV-rtTA/TRE-p53.1224 mice contained abundant siRNA, consistent with the known expression pattern of the CMV promoter (Fig. 3a). p53.1224 siRNA production was doxycycline dependent (Fig. 3b) and, in tissues such as muscle and skin, was of similar abundance as in positive control tumor samples in which Trp53 knockdown is required for tumor maintenance (Fig. 3c).
Figure 3.
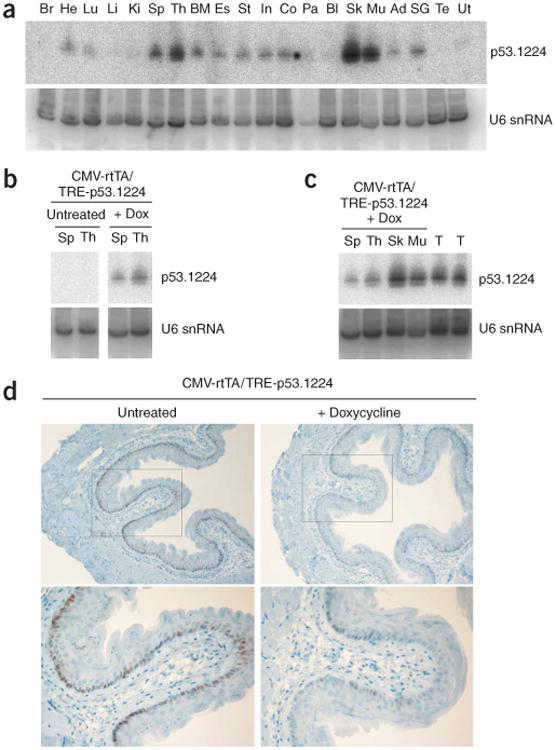
CMV-rtTA drives widespread and functional p53.1224 shRNA expression. (a) Small RNA blot showing expression of p53.1224 siRNA in tissues of doxycycline (Dox)-treated CMV-rtTA/TRE-p53.1224 double-transgenic mice. Br, brain; He, heart; Lu, lung; Li, liver; Ki, kidney; Sp, spleen; Th, thymus; BM, bone marrow; Es, esophagus; St, stomach; In, small intestine; Co, colon; Pa, pancreas; Bl, bladder; Sk, skin; Mu, muscle; Ad, adipose tissue; SG, salivary gland; Te, testis; Ut, uterus. U6 small nuclear (sn) RNA is shown as a loading control. (b) Small RNA blot showing induction of p53.1224 siRNA by doxycycline in spleen and thymus of CMV-rtTA/TRE-p53.1224 double-transgenic mice. Left and right panels are derived from the same blot. (c) Small RNA blot comparing p53.1224 siRNA expression in tissues of doxycycline-treated CMV-rtTA/TRE-p53.1224 double-transgenic mice with control tumor samples (T) in which Trp53 knockdown is known to drive tumor growth20. (d) Trp53 immunohistochemistry of esophagi isolated from untreated (left) or doxycycline-treated (right) CMV-rtTA/TRE-p53.1224 double-transgenic mice, 6 h after 6 Gy of whole-body ionizing radiation. Note attenuated Trp53 expression in cells of the basal layer of the esophageal epithelium in doxycycline-treated double-transgenic mice.
To test Trp53 knockdown in adult tissues, we subjected doxycycline-treated CMV-rtTA/TRE-p53.1224 mice to whole-body ionizing radiation (which induces Trp53 in some tissues24) and examined Trp53 levels by immunohistochemistry or immunoblotting of cell lysates. Radiation-induced Trp53 expression was substantially reduced in the basal layer of the esophageal epithelium of doxycycline-treated mice (Fig. 3d). Furthermore, we observed a marked reduction in radiation-induced apoptosis in the thymus of doxycycline-treated CMV-rtTA/TRE-p53.1224 mice compared with their single transgenic littermates (Fig. 4a) and also in cultured thymocytes derived from these mice, in which Trp53 protein level was clearly reduced after radiation treatment (Fig. 4b,c and Supplementary Fig. 2 online). Together, these results indicate that doxycycline-dependent Trp53 shRNA expression in thymocytes of adult CMV-rtTA/TRE-p53.1224 mice is sufficient to protect them from an apoptotic stimulus in vivo.
Figure 4.
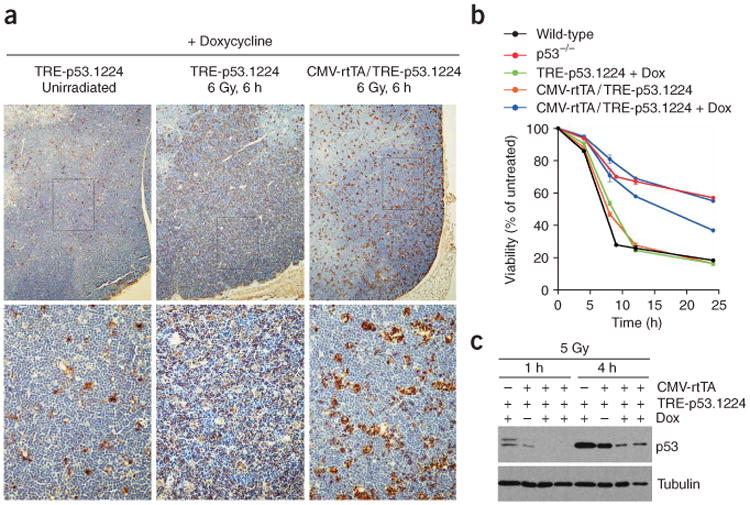
Trp53 knockdown protects thymocytes from radiation-induced apoptosis in vivo. (a) Representative TUNEL staining of thymi isolated from doxycycline (Dox)-treated mice, 6 h after 6 Gy of whole-body ionizing radiation. Note widespread apoptosis in the thymus of a control TRE-p53.1224 mouse (center) compared with a CMV-rtTA/TRE-p53.1224 double-transgenic thymus (right). A control, unirradiated TRE-p53.1224 thymus (left) is also shown. (b) Time course showing viability of cultured thymocytes after 5 Gy of ionizing radiation.Each line represents average viability ± s.e.m.from triplicate samples from a single mouse, normalized to unirradiated samples from the same animal. Black, wild-type; red, Trp53-null; green, doxycycline (Dox)-treated TRE-p53.1224 mice; orange, untreated CMV-rtTA/TRE-p53.1224 mice; blue, doxycycline-treated CMV-rtTA/TRE-p53.1224 mice. (c) Trp53 protein blot of cultured thymocytes after 5 Gy of ionizing radiation. Note substantial Trp53 knockdown in thymocytes derived from two doxycycline-treated CMV-rtTA/TRE-p53.1224 mice.
Tissue-specific control of transgene expression using tetracycline-based systems has proven to be effective for studying the impact of gene overexpression in particular tissues. To test whether p53.1224 siRNA expression could be similarly restricted to a particular tissue in adult mice, we crossed TRE-p53.1224 mice with LAP-tTA transgenic mice9. In these mice, the liver-enriched transcriptional activator protein (LAP; also known as C/EBPβ) promoter drives expression of the tTA (tet-off) protein specifically in differentiated hepatocytes. As predicted, we detected Trp53 siRNA only in the liver of adult LAP-tTA/TRE-p53.1224 mice (Fig. 5a). siRNA production was inhibited by addition of doxycycline to the drinking water, with no effect on the levels of the endogenous liver-specific microRNA miR-122 (ref. 25) (Fig. 5b). We observed similar tissue-specific siRNA expression in LAP-tTA/TRE-p53.1224 line D mice (Supplementary Fig. 3 online). To test whether this level of siRNA production was sufficient to affect Trp53 expression, we injected mice with recombinant adenovirus, which is known to localize to the liver and induce Trp53 protein26 (an alternative to radiation, which does not induce Trp53 in the liver24). Immunofluorescence showed a substantial reduction in Trp53 levels in livers of LAP-tTA/TRE-p53.1224 mice but not in doxycycline-treated animals (Fig. 5c). Thus, tet transactivator strains previously designed for tissue-specific gene overexpression can also drive reversible gene knockdown in specific organs.
Figure 5.
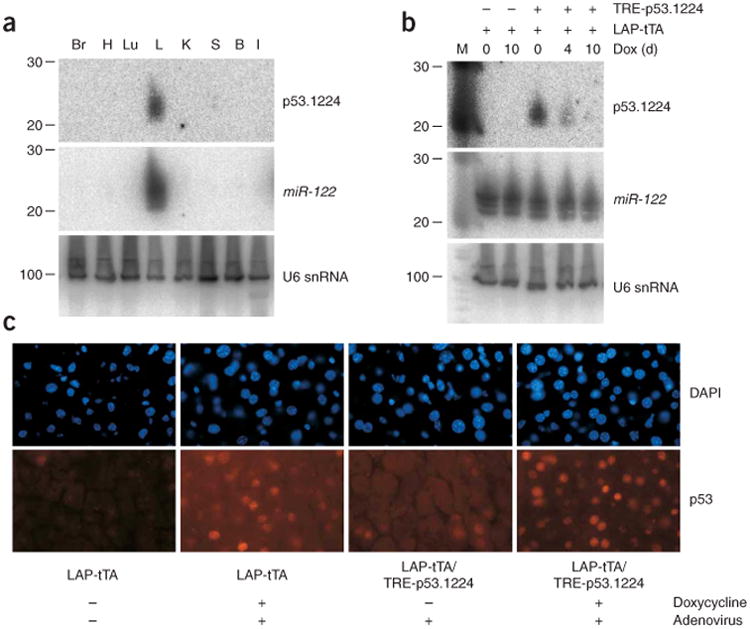
Liver-specific Trp53 knockdown in LAP-tTA/TRE-p53.1224 mice. (a) p53.1224 small RNA blot for various tissues isolated from LAP-tTA/TRE-p53.1224 double-transgenic mice. The blot was stripped and reprobed for the liver-specific microRNA miR-122. U6 snRNA is shown as a loading control. Br, brain; H, heart; Lu, lung; L, liver; K, kidney; S, spleen; B, bone marrow; I, small intestine. (b) p53.1224 small RNA blot of livers isolated from LAP-tTA/TRE-p53.1224 double-transgenic mice and LAP-tTA single transgenic controls. Mice were given doxycycline (Dox) for the indicated amounts of time before livers were harvested. miR-122 and U6 snRNA abundance is also shown. (c) Trp53 immuno-fluorescence (IF) in livers isolated from untreated or doxycycline-treated LAP-tTA/TRE-p53.1224 double-transgenic mice and LAP-tTA single transgenic controls. Trp53 was induced in the liver by injection of adenovirus into the tail vein, as indicated.
We also tested whether tissue-specific expression of the p53.1224 shRNA from the transgenic TRE promoter was sufficient to inhibit Trp53 function in vivo. To this end, we used a well-characterized model in which Trp53 loss promotes tumorigenesis: the Eμ-Myc transgenic mouse. These animals express the Myc oncogene in B cells and develop pre-B or B cell lymphoma27. Eμ-Myc lymphoma-genesis is markedly accelerated by Trp53 deficiency28,29 or after reconstitution of lethally irradiated mice with Eμ-Myc hematopoietic stem cells transduced with Trp53 shRNAs30. To test whether shRNA expression from a germline TRE-p53.1224 transgene was similarly functional, we generated ‘triple transgenic’ Eμ-Myc/Eμ-tTA/TRE-p53.1224 mice. Previous studies have shown that tet-regulated Myc overexpression driven by the Eμ-tTA transgene (Eμ-SRα control element) causes an immature T cell disease31, suggesting a slightly different transgene expression pattern compared with the Eμ-Myc transgene (Eμ-VH control element). Despite this, we predicted that Trp53 knockdown and Myc overexpression would overlap in a subset of hematopoietic cells of Eμ-Myc/Eμ-tTA/TRE-p53.1224 mice and that these animals would more rapidly develop lymphoma.
Indeed, the latency of tumorigenesis in Eμ-Myc/Eμ-tTA/TRE-p53.1224 mice compared with their collective Eμ-Myc single and double-transgenic littermates was significantly reduced (Fig. 6a). Tumor latency was similar for the three control genotypes: Eμ-Myc alone, Eμ-Myc/Eμ-tTA double-transgenic and Eμ-Myc/TRE-p53.1224 double-transgenic (Supplementary Fig. 4 online), suggesting that shRNA expression from the TRE-p53.l224 transgene is tTA dependent and not leaky. In contrast to the canonical grossly enlarged lymph nodes observed in their Eμ-Myc littermates27 (Fig. 6b), many triple transgenic mice became moribund without a large peripheral tumor burden (Fig. 6b and Table 1). Of nine triple-transgenic mice that succumbed with relatively small peripheral lymph nodes, six also had partial hind leg paralysis. This phenotype has previously been associated with lymphoma in mice32, although it is infrequently observed in control Eμ-Myc single- or double-transgenic mice. In principle, these distinct phenotypes could result from the coexpression of Myc and the p53.1224 shRNA in a particular B cell type targeted by both the Eμ-VH and Eμ-SRα control elements or from a specific hypomorphic state produced by incomplete suppression of Trp53, as has been described30.
Figure 6.
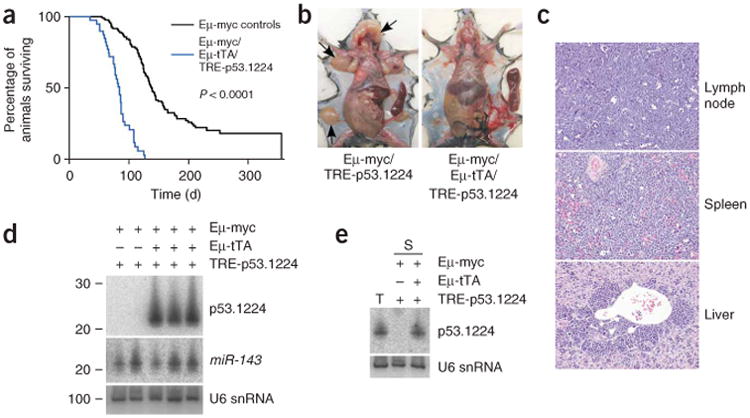
Tissue-specific Trp53 knockdown accelerates Eμ-Myc lymphomagenesis. (a) Kaplan-Meier curves showing decreased survival of Eμ-Myc/Eμ-tTA/TRE-p53.1224 triple-transgenic mice (blue; median survival 82 d) compared with their collective double transgenic (Eμ-Myc/Eμ-tTA and Eμ-Myc/TRE-p53.1224) and single Eμ-Myc transgenic littermates (black; median survival 125 d), referred to as Eμ-Myc controls. (b) Gross anatomy of a representative Eμ-Myc/Eμ-tTA/TRE-p53.1224 triple-transgenic mouse (right) alongside an Eμ-Myc/TRE-p53.1224 control (left). Spleens have been dissected out for visualization. In the control (left), note massive lymph node enlargement (arrows) characteristic of spontaneously arising tumors in Eμ-Myc mice, compared with the grossly enlarged spleen but relatively small peripheral lymph nodes in the Eμ-Myc/Eμ-tTA/TRE-p53.1224 mouse. (c) Histopathology of lymph node, spleen and liver harvested from a representative moribund Eμ-Myc/Eμ-tTA/TRE-p53.1224 mouse. Sections are stained with hematoxylin and eosin (H&E). Note similar appearance of lymph node, spleen and perivascular areas of the liver, all packed with lymphoma cells. (d) p53.1224 small RNA blot of spleens isolated from moribund Eμ-Myc/Eμ-tTA/TRE-1224 triple-transgenic mice, with double-transgenic controls. (e) Small RNA blot showing similar p53.1224 siRNA abundance in lymphoma-laden triple-transgenic spleen (S) compared with a positive control tumor sample (T), derived from transformed MEFs in which Trp53 knockdown is known to drive tumor growth20.
Table 1. Pathology of transgenic mice of various genotypes.
| Mouse | Genotype | Peripheral lymph nodes | Immunotype | Spleen p53.1224 expression |
|---|---|---|---|---|
| 3.5 | Eμ-Myc | Large | B | ND |
| C12 | Eμ-Myc | Large | Pre-B | No |
| 2.5 | Eμ-Myc/TRE-p53.1224 | Large | T | No |
| 4.1 | Eμ-Myc/TRE-p53.1224 | Large | B | No |
| 6.4 | Eμ-Myc/TRE-p53.1224 | Large | B | No |
| 1 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Small | Pre-B | Yes |
| 2 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Small | Pre-B | Yes |
| 2.2 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Small | Pre-B | Yes |
| 1.2 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Small | Pre-B | Yes |
| C11 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Small | Pre-B | Yes |
| C21 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Small | Pre-B | Yes |
| 2.4 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Large | B | Yes |
| S31 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Large | Pre-B | Yes |
| S23 | Eμ-Myc/Eμ-tTA/TRE-p53.1224 | Large | B | No |
ND, not determined.
Moribund triple-transgenic mice with relatively small peripheral lymph nodes also showed a grossly enlarged spleen taken over by B220+ IgM− (pre-B) tumor cells (Fig. 6c and Table 1), with lymphoma dissemination into the lymph nodes and liver (Fig. 6c). Of nine such Eμ-Myc/Eμ-tTA/TRE-p53.1224 mice examined, all except one had highly abundant p53.1224 siRNA in spleen and lymph nodes (Fig. 6d and Table 1). We made similar observations in triple-transgenic mice harboring the TRE-p53.1224 line D transgene (Supplementary Fig. 5 online). Notably, we did not observe any change in the endogenous microRNA miR-143 (Fig. 6d), suggesting that expression of the p53.1224 shRNA was not disrupting natural pre-microRNA processing. We did not detect p53.1224 expression in whole spleen of Eμ-tTA/TRE-p53.1224 double-transgenic mice (Supplementary Fig. 6 online), presumably because expression from the Eμ promoter is highest in active B cells that are present only at low levels in normal spleen. Notably, p53.1224 siRNA was present in diseased Eμ-Myc/Eμ-tTA/TRE-p53.1224 spleens at levels known to promote Trp53 knockdown and tumor progression in other contexts20 (Fig. 6d,e). Thus, Trp53 shRNA can be abundantly expressed from a cell type–specific, tet-regulated transgene in vivo, thereby compromising the tumor suppressive functions of Trp53.
To determine whether knockdown of Trp53 was reversible, we examined the biological impact of doxycycline addition in lymphoma-bearing triple-transgenic mice. Initially, three primary Eμ-Myc/Eμ-tTA/TRE-p53.1224 mice were treated with doxycycline in their drinking water at the onset of paralysis. Within 2 d, each mouse regained full movement and lived for a further 24, 25 and 64 d, respectively, before tumors eventually relapsed. In contrast, untreated mice progressed to full paralysis within a few days. Magnetic resonance imaging (MRI) of one mouse showed enlarged spleen and lymph nodes at the time of paralysis, which markedly regressed after 7 d of doxycycline treatment (Fig. 7a). The spleen of the treated mouse also appeared darker by MRI, consistent with tumor cell clearance (Fig. 7a).
Figure 7.
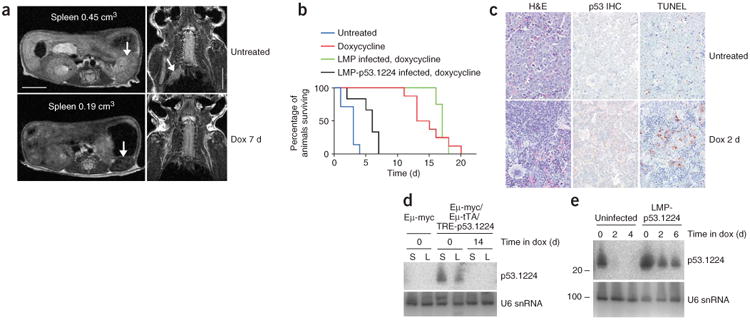
Restoring Trp53 expression in tumors causes apoptosis and regression. (a) MRI of a primary Eμ-Myc/Eμ-tTA/TRE-p53.1224 triple-transgenic mouse, before and after 7 d of doxycycline (Dox) treatment. Left: transverse sections through abdomen (arrows indicate spleen). Right: coronal sections through upper thoracic region (arrow indicates enlarged mediastinal lymph nodes). Scale bars represent 0.5 cm. (b) Kaplan-Meier survival curve for mice transplanted with Eμ-Myc/Eμ-tTA/TRE-p53.1224 lymphoma cells. For all mice, time zero was when disease first became evident (partial hind leg paralysis); mice were killed when moribund. Blue, untreated. Red, doxycycline-treated. Green, mice transplanted with triple-transgenic cells infected with LMP empty vector and treated with doxycycline. Black, mice transplanted with triple-transgenic cells infected with LMP-p53.1224 and treated with doxycycline. (c) H&E staining, Trp53 immunohistochemistry (IHC) and apoptosis in spleens from Eμ-Myc/Eμ-tTA/TRE-p53.1224 lymphoma cell transplant recipients, either without treatment or after 2 d on doxycycline. (d) p53.1224 small RNA blot analysis of spleens (S) and lymph nodes (L) from Eμ-Myc/Eμ-tTA/TRE-p53.1224 lymphoma cell transplant recipients, either without treatment or after 14 d on doxycycline. (e) p53.1224 small RNA blot analysis of spleens from mice receiving transplants of Eμ-Myc/Eμ-tTA/TRE-p53.1224 lymphoma cells, either uninfected or infected with LMP-p53.1224, and treated with doxycycline for various times.
To examine the performance of the tet-regulated system more systematically, we transplanted lymphoma cells harvested from Eμ-Myc/Eμ-tTA/TRE-p53.1224 mice into a cohort of immunocompromised mice. Approximately two weeks after tail vein injection, all mice presented with partial hind leg paralysis similar to that observed in many of the primary triple-transgenic mice. If left untreated, these animals progressed to complete paralysis in a few days and had to be killed (Fig. 7b). Consistent with observations in primary triple-transgenic mice, untreated transplant recipients had massive spleen enlargement owing to lymphoma cell infiltration (Fig. 7c), with abundant p53.1224 siRNA expression in whole spleen and lymph nodes (Fig. 7d). Similarly, after just 2 d of doxycycline treatment, the paralysis resolved, and we observed a marked change in spleen histopathology including substantial lymphoma cell clearance and partial restoration of normal tissue architecture (Fig. 7c). Doxycycline treatment abolished p53.1224 siRNA expression in the spleen (Fig. 7e) and produced a marked increase in cells expressing high levels of Trp53 or undergoing apoptosis (Fig. 7c). These results are consistent with observations in cultured primary Eμ-Myc/Eμ-tTA/TRE-p53.1224 lymphoma cells, which lose p53.1224 siRNA expression, show restored Trp53 expression and undergo massive apoptosis within 2 d of doxycycline treatment (Supplementary Fig. 7 online). Notably, the lymphoma regression and apoptosis we observed were due to Trp53 reactivation and were not a result of doxycycline toxicity, as mice bearing triple-transgenic lymphomas transduced with a constitutive Trp53 shRNA showed only a minimal increase in survival and retained Trp53 siRNA production after doxycycline treatment (Fig. 7b,e). Together, these data suggest that ongoing Trp53 knockdown in established lymphomas is required for their survival and progression.
Although all tumors derived from triple-transgenic animals underwent massive involution in response to doxycycline treatment, they invariably relapsed within several weeks, often with enlarged lymph nodes reminiscent of control Eμ-Myc transgenic mice (Fig. 7b and data not shown). In most relapsed lymphomas derived from triple-transgenic animals (1/2 primary and 6/7 transplanted), the p53.1224 siRNA was barely detectable or absent, implying that these lymphomas eventually escape their dependence on Trp53 knockdown through additional genetic alterations, consistent with a previous report33. However, one relapsed lymphoma arising in a primary triple-transgenic animal showed high amounts of the p53.1224 siRNA (data not shown), perhaps reflecting changes that allow constitutive activation of the TRE-p53.1224 transgene, as has been reported in some tet-regulated oncogene models upon tumor relapse34. Nevertheless, the potent biological effects we observe in tumorigenesis and upon relapse highlight the power of conditional RNA interference technology for studying biological phenomena in transgenic mice and point to its utility for probing molecules and pathways that are potential candidates for drug development.
Discussion
By adapting a tet-responsive system previously used for conditional cDNA overexpression and synthetic microRNAs capable of silencing a gene of interest, we demonstrate a rapid and simple method for generating transgenic mice with tissue-specific and reversible RNAi-mediated inhibition of endogenous gene expression. We have shown that standard transgenic techniques can introduce a tet-regulatable shRNA expression cassette into the mouse genome, where it remains latent until mice are crossed with existing transgenic tet transactivator mouse lines. These double-transgenic mice produce shRNA and endogenous gene knockdown in a tissue-specific manner that can be regulated by doxycycline in the drinking water. Using this system to knock down Trp53, we were able to regulate endogenous Trp53 expression in a tissue-specific, conditional, reversible manner, producing profound biological phenotypes in an entirely transgenic setting.
In developing this approach, we were concerned that the levels of tet transactivator produced in existing mouse strains would be insufficient to drive functional shRNA expression or, alternatively, that overexpression of the exogenous microRNA would saturate the RNAi machinery leading to off-target effects as has recently been described in an experimental gene therapy setting35. However, we observed abundant p53.1224 siRNA production and Trp53 protein knockdown in multiple tissue types of TRE-p53.1224 mice when we crossed them with the general tet-on CMV-rtTA mouse strain and in the liver and pre-B cell Eμ-Myc lymphomas when we crossed them with the tissue-specific tet-off strains LAP-tTA and Eμ-tTA, respectively. In some cases, p53.1224 siRNA was produced in equivalent amounts as observed in tumors resulting from retroviral p53.1224 expression, in which selection for maximal Trp53 knockdown has probably occurred20. Moreover, we showed that Trp53 shRNA production by either Eμ-tTA or LAP-tTA in vivo does not interfere with endogenous microRNA maturation. Although not all tet-transactivator mouse lines may be useful for gene knockdown, it seems that tet-regulated shRNA production can effectively harness but not saturate the endogenous RNAi machinery in vivo, potentially affording it a similar experimental versatility as tet-regulated cDNA overexpression but allowing regulation of endogenous gene expression.
We chose to target the Trp53 tumor suppressor gene as a proof of principle, as the gene is well characterized in vitro and in vivo. In addition, by addressing whether Trp53 loss was required for the continued progression of tumors in vivo, we could assess the utility of regulated RNAi knockdown to validate a pathway that is a potential target for new therapies. Using our ability to conditionally regulate endogenous Trp53 expression in vivo, we show that Eμ-Myc lymphomas invariably respond to Trp53 reactivation through apoptosis and tumor involution, although these tumors eventually relapse. Extension of survival by 64 d in one mouse suggests complete tumor clearance upon Trp53 restoration, with relapse potentially due to de novo Eμ-Myc lymphomagenesis. Our results are generally consistent with recent studies from our group and other groups33,36,37 using different approaches and tumor types, highlighting the potential of therapies that reactivate Trp53 but also pointing toward the potential for resistance. Although other strategies have proved effective for conditionally regulating Trp53, they do not allow reversible regulation of Trp53 in a tissue-specific fashion. More notably, the system described herein should be readily portable to any other gene.
As many tet-transactivator transgenic mouse strains are available, the conditional RNAi approach described here holds promise as a general method for controlling endogenous gene expression in vivo. The pronuclear injection transgenesis strategy we used is rapid and simple, although variations on this strategy such as targeted knock-ins or BAC transgenics may increase the frequency of founder animals capable of effective gene knockdown. Furthermore, microRNA-based shRNAs efficiently silence gene expression when expressed in the 3′ UTR of protein-coding cDNAs21, which should allow coregulation of a protein-coding cDNA and shRNA of interest from the same transcript or incorporation of a green fluorescent protein (GFP) or bioluminescence reporter to monitor shRNA production in vivo. Although tet-regulated RNAi does not produce the null genotype achievable with Cre/loxP-based strategies, its reversibility and ability to produce hypomorphic phenotypes opens up new experimental possibilities. For example, as illustrated here, it should be possible to study the action of any endogenous gene in tumor maintenance. These include testing potential drug targets in mouse models of cancer, as RNAi probably mimics drug action more faithfully than complete gene disruption, and, like drug administration, its effects can be rapidly reversed. Other potential uses involve assessing the spatial or temporal requirement for endogenous genes during development. Therefore, in principle, the strategy described here may allow more effective use of mice as vehicles for understanding gene function and as preclinical models of human disease.
Methods
Transgenic mice
A 948-bp transgene fragment was excised from the TMP-p53.1224 vector20 using BglII and AgeI. Transgenic mice were generated by standard pronuclear injection procedures. Founder line A (mainly used in this study) was generated on a C57BL/6;SJL F1 background and subsequently backcrossed onto C57BL/6. Founder line D was generated on an FVB/N background. Mice were genotyped by PCR using the primers miR30fwd and TREmiR30genoR (Supplementary Table 1 online), yielding a 300-bp product. Other mouse lines used were Eμ-Myc27, LAP-tTA9, Eμ-tTA31, Rosa26-M2rtTA11 and CMV-rtTA23. Doxycycline (Clontech) was administered to mice at 2 mg ml−1 (4 mg ml−1 for tet-on studies) in a 1% sucrose solution in drinking water. MRI was performed at the Memorial Sloan-Kettering Cancer Center Animal Imaging Facility. The Cold Spring Harbor Animal Care and Use Committee approved all procedures described in this work.
Cell culture and expression analysis
Cell culture, small RNA blotting, and protein blotting were performed as described previously20. To test founder lines for Trp53 knockdown, we infected MEFs with pRev-TetOFF-IN (Clontech) and selected in G418 before harvesting. For all Trp53 protein blots in Figure 1 and Figure 2, MEFs were treated with 0.5 μg ml−1 adriamycin for 4-8 h to induce Trp53. The p53.1224 siRNA blot probe sequence is listed in Supplementary Table 1. miR-122 and miR-143 probe sequences were as described previously25. Lymphoma immunotypes were assessed by flow cytometry after incubation of cultured lymphoma cells with fluorescently conjugated antibodies recognizing hematopoietic lineage markers. Thymocyte apoptosis assays were performed as described previously38. For these experiments, doxycycline was administered in the drinking water of the mouse before harvesting, but not during thymocyte culture.
Adenovirus preparation and in vivo infection
To generate high titer viral stocks, 2 × 108 HEK293 packaging cells were infected at 5 to 10 plaque-forming units (pfu) per cell (Adeno-Cre, Vector Biolabs). Adenovirus propagation and preparation was performed as described previously39. Before infection, virus was dialyzed twice against a solution containing 10 mM Tris (pH 8), 1 mM MgCl2 and 140 mM NaCl at 4 °C Infection of the mice was carried out by administration of 0.25 ml virus solution into the tail vein at a total virus load of 5 × 109 viral particles per gram body weight. Ten hours after injection, livers were fixed in OCT compound and stored at −80 °C before sectioning and assessment of Trp53 immunofluorescence.
Supplementary Material
Acknowledgments
We thank H. Bujard (Heidelberg University) and D. Felsher (Stanford University) for LAP-tTA mice; D. Felsher for Eμ-tTA mice; R. Jaenisch (Whitehead Institute) for Rosa26-M2rtTA mice; H. Varmus, F. Cong and R. Sotillo (Memorial Sloan-Kettering Cancer Center) for CMV-rtTA mice and J. Adams (Walter and Eliza Hall Institute) for Eμ-Myc mice. Many thanks to L. Bianco, J. Coblentz and the Cold Spring Harbor Laboratory animal house staff; M. Lupu and C. Le at the Memorial Sloan-Kettering Cancer Center for MRI; M.S. Jiao, K. Manova and E. de Stanchina at Memorial Sloan-Kettering Cancer Center for histology and immunohistochemistry; members of the Hannon laboratory for advice on small RNA blotting and members of the Lowe laboratory for advice and discussions. This study was supported by a Mouse Models of Human Cancer Consortium grant, a program project grant from the National Cancer Institute and the Don Monti Memorial Research Foundation. K.M is a Robert and Theresa Lindsay Fellow and the Leeds scholar of the Watson School of Biological Sciences. D.J.B is an Engelhorn Scholar of the Watson School of Biological Sciences. V.K. is a Leukemia and Lymphoma Society Fellow.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Author Contributions: R.A.D. and S.W.L. designed the study; R.A.D., K.M., E.H., P.K.P, V.K. and L.Z. performed experiments; S.Y.K. performed pronuclear injections; D.J.B. provided reagents; C.C.-C. supervised and interpreted histopathology; G.J.H. and S.W.L. supervised experiments and data interpretation and R.A.D. and S.W.L. wrote the paper.
Competing Interests Statement: The authors declare no competing financial interests.
References
- 1.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507–512. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- 2.Glaser S, Anastassiadis K, Stewart AF. Current issues in mouse genome engineering. Nat Genet. 2005;37:1187–1193. doi: 10.1038/ng1668. [DOI] [PubMed] [Google Scholar]
- 3.Lewandoski M. Conditional control of gene expression in the mouse. Nat Rev Genet. 2001;2:743–755. doi: 10.1038/35093537. [DOI] [PubMed] [Google Scholar]
- 4.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 5.Loonstra A, et al. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci USA. 2001;98:9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson JD, et al. Validating cancer drug targets. Nature. 2006;441:451–456. doi: 10.1038/nature04873. [DOI] [PubMed] [Google Scholar]
- 7.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furth PA, et al. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc Natl Acad Sci USA. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kistner A, et al. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci USA. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin MK, Levorse JM, Ingram RS, Tilghman SM. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature. 1999;402:496–501. doi: 10.1038/990040. [DOI] [PubMed] [Google Scholar]
- 11.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Felsher DW. Reversibility of oncogene-induced cancer. Curr Opin Genet Dev. 2004;14:37–42. doi: 10.1016/j.gde.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Wiznerowicz M, Szulc J, Trono D. Tuning silence: conditional systems for RNA interference. Nat Methods. 2006;3:682–688. doi: 10.1038/nmeth914. [DOI] [PubMed] [Google Scholar]
- 14.Xia XG, Zhou H, Samper E, Melov S, Xu Z. Pol II-expressed shRNA knocks down Sod2 gene expression and causes phenotypes of the gene knockout in mice. PLoS Genet. 2006;2:e10. doi: 10.1371/journal.pgen.0020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao MK, et al. Tissue-specific RNAi reveals that WT1 expression in nurse cells controls germ cell survival and spermatogenesis. Genes Dev. 2006;20:147–152. doi: 10.1101/gad1367806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ventura A, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci USA. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fritsch L, et al. Conditional gene knock-down by CRE-dependent short interfering RNAs. EMBO Rep. 2004;5:178–182. doi: 10.1038/sj.embor.7400064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coumoul X, Shukla V, Li C, Wang RH, Deng CX. Conditional knockdown of Fgfr2 in mice using Cre-LoxP induced RNA interference. Nucleic Acids Res. 2005;33:e102. doi: 10.1093/nar/gni100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J, McMahon AP. Reproducible and inducible knockdown of gene expression in mice. Genesis. 2006;44:252–261. doi: 10.1002/dvg.20213. [DOI] [PubMed] [Google Scholar]
- 20.Dickins RA, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- 21.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci USA. 1980;77:7380–7384. doi: 10.1073/pnas.77.12.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sotillo R, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Midgley CA, et al. Coupling between gamma irradiation, p53 induction and the apoptotic response depends upon cell type in vivo. J Cell Sci. 1995;108:1843–1848. doi: 10.1242/jcs.108.5.1843. [DOI] [PubMed] [Google Scholar]
- 25.Lagos-Quintana M, et al. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 26.Kubicka S, et al. p53 represses CAAT enhancer-binding protein (C/EBP)-dependent transcription of the albumin gene. A molecular mechanism involved in viral liver infection with implications for hepatocarcinogenesis. J Biol Chem. 1999;274:32137–32144. doi: 10.1074/jbc.274.45.32137. [DOI] [PubMed] [Google Scholar]
- 27.Adams JM, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 28.Hsu B, et al. Evidence that c-myc mediated apoptosis does not require wild-type p53 during lymphomagenesis. Oncogene. 1995;11:175–179. [PubMed] [Google Scholar]
- 29.Schmitt CA, McCurrach ME, deStanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999;13:2670–2677. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemann MT, et al. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet. 2003;33:396–400. doi: 10.1038/ng1091. [DOI] [PubMed] [Google Scholar]
- 31.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 32.Ceccarelli AV, Rozengurt N. Outbreak of hind limb paralysis in young CFW Swiss Webster mice. Comp Med. 2002;52:171–175. [PubMed] [Google Scholar]
- 33.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 34.Kim M, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 36.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 38.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 39.Kuhnel F, et al. NFkappaB mediates apoptosis through transcriptional activation of Fas (CD95) in adenoviral hepatitis. J Biol Chem. 2000;275:6421–6427. doi: 10.1074/jbc.275.9.6421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.