

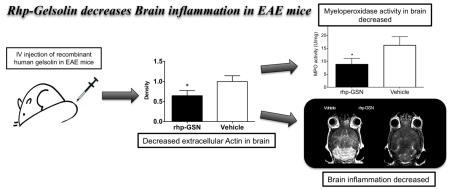
Abstract
Gelsolin is the fourth most abundant protein in the body and its depletion in the blood has been found in multiple sclerosis (MS) patients. How gelsolin affects the MS brain has not been studied. We found that while the secreted form of gelsolin (pGSN) decreased in the blood of experimental autoimmune encephalomyelitis (EAE) mice, pGSN concentration increased in the EAE brain. Recombinant human pGSN (rhp-GSN) decreased extracellular actin and myeloperoxidase activity in the brain, resulting in reduced disease activity and less severe clinical disease, suggesting that gelsolin could be a potential therapeutic target for MS.
Keywords: gelsolin, experimental autoimmune encephalomyelitis (EAE), actin, inflammation, myeloperoxidase, magnetic resonance imaging
Graphical Abstract
1. Introduction
Multiple sclerosis (MS) is the leading cause of non-traumatic neurological disability in young adults (Noseworthy et al., 2000). While the etiology of MS remains unknown, it is characterized by inflammatory demyelinating plaques in the CNS (Bruck, 2005, Frohman et al., 2006, Imitola et al., 2006, Noseworthy, Lucchinetti, 2000). A multitude of inflammatory factors has been investigated in MS, but relatively little is known about whether gelsolin (GSN), the fourth most abundant protein in the human body (Smith et al., 1987), has a role in MS.
Gelsolin is the first and most widely expressed member of a family of actin-severing proteins in humans (Yin and Stossel, 1979). Gelsolin is mainly produced from muscle tissue but is also expressed in the human central nervous system (Kwiatkowski et al., 1988, Tanaka et al., 1993). It is involved in actin homeostasis, cell exocytosis, cell motility, phagocytosis, apoptosis, platelet formation and activation (Lee and Galbraith, 1992, Osborn et al., 2007, Silacci et al., 2004). It has intracellular and secreted (termed pGSN) forms in the human body, which are structurally similar but not identical (Yin et al., 1984). Both forms are from the same gene on the 9th chromosome, but the secreted form differs from the intracellular form by a 25-amino acid signaling peptide and the presence of a disulfide bond between cysteine residues at positions 188 and 201 (Wen et al., 1996, Yin, Kwiatkowski, 1984).
While a decrease of total gelsolin level in the blood was found in multiple sclerosis patients (Kulakowska et al., 2010), it is yet unknown how multiple sclerosis affects secreted gelsolin in the brain (Yin, Kwiatkowski, 1984), and whether modulating gelsolin levels could have a therapeutic benefit for multiple sclerosis. Therefore, we aimed to evaluate the effects of neuroinflammation on the levels of pGSN in the brain in a mouse model of multiple sclerosis (experimental autoimmune encephalomyelitis [EAE]) and to determine whether gelsolin has a therapeutic role for EAE/MS.
2. Materials and Methods
2.1. EAE Induction
All experimental protocols were approved by the Institutional Animal Care and use Committee of Massachusetts General Hospital. 111 SJL female mice 6–10 weeks old (Jackson Laboratories, Bar Harbor, ME) were used for this study. EAE was induced with synthetic proteolipid protein (PLP139–151, Axxora, CA) according to (Greer et al., 1996). Briefly, 2mg of PLP and 8mg M. tuberculosis H37Ra (Difco, MI) in 1ml water and 1 ml of complete Freund’s adjuvant (Sigma-Aldrich, MO) were emulsified. Each mouse received 100μl of the emulsion (25μl each in the bilateral inguinal and axillary regions). On days 0 and 2, 0.1mg of Pertussis toxin was injected intravenously via the tail vein. The animals were monitored at least daily with the following clinical grading: 0=normal, 1= tail limpness, 2=hind limb weakness, 3=complete hind limb paralysis, 4=unresponsive and moribund, 5=found dead in cage. 15 mice died shortly after EAE induction and were removed from the study. Sham-induced mice (n=6) were induced with all of the above except PLP was not administered.
2.2. Blood and brain specimen preparation
Retro-orbital blood was centrifuged at 2,500rpm for 15min at 4°C. The supernatant was collected and mixed with 1% RIPA lysis buffer (Millipore, MA) in PBS v/v for 2 hours on ice and then centrifuged at 14,000rpm for 20 min. The supernatant was used for protein quantification with the bicinchoninic acid kit (Thermo Fisher Scientific, MA) and for Western blot of pGSN.
Mouse brains were homogenized and proteins were extracted in 1% RIPA lysis buffer (Millipore, MA) in PBS v/v. The resultant suspension was sonicated for 15s and then incubated on ice for 2 hours. The suspensions were centrifuged at 14,000 rpm for 20 min, and used for protein quantification and Western blot of pGSN.
2.3. Western blot
25μg of protein from plasma or brain was loaded per well and samples were separated using Tris-HCL Gels (Bio Rad, CA) and then transferred onto PVDF membranes (IMMUNO-Blot PVDF Membrane, Bio Rad) using a wet tank blotting system (BioRad). Membranes were blocked in 5% milk at 4°C overnight and then incubated with a rabbit polyclonal antibody directed against plasma extension of mouse gelsolin at a dilution of 1:500 in 5% milk (Chou et al., 2011). This antibody does not recognize cytoplasmic gelsolin. To detect actin, membranes were incubated with 1:200 dilution of a rabbit polyclonal anti-actin antibody (Sigma-Aldrich, MO, #A2066). β-tubulin (rabbit polyclonal antibody at 1:500, Abcam, MA, #ab4074) and albumin (rabbit polyclonal antibody at 1:500, Abgent, CA, #ABIN390450) were used as loading controls in brain and serum samples, respectively. We used a chemiluminescence system and peroxidase-conjugated secondary antibody for detection of protein bands.
2.4. Rhp-GSN supplement therapy
Recombinant human plasma (rhp)-GSN was produced in Escherichia coli, refolded with oxidized glutathione, purified (Lee et al., 2007), and brought to a final stock protein concentration of 10 mg/ml in PBS (vehicle). For each mouse, 200mg/kg rhp-GSN split into two doses on either days 0 and 2 or days 8 and 10 of EAE induction was administered intravenously. Gelsolin has a blood half-life of about 2.3 days (Smith, Janmey, 1987) and was expected to remain elevated up to 7-9 days after administration.
2.5. Extracellular protein extraction
The protocol of extracellular protein extraction was adopted from (Hofstein et al., 1983, Pulli et al., 2013). After cardiac perfusion with 20ml cold saline in an anesthetized mouse, brains were harvested and washed in PBS three time. We then incubated organs in 4 times their weight HBSS with 0.32M sucrose, 1mM calcium acetate, 10U/ml heparin, and proteinase inhibitor on ice for 2 hours. After this procedure, we added four parts ice-cold acetone per part buffer to the samples and incubated for 1 hour at −20°C to precipitate proteins. Samples were then centrifuged at 4000g for 20min. The pellets were dried for 1-3 min and resuspended in 0.2mL of PBS and stored for further analyses.
2.6. Antibody-captured MPO activity assay
Antibody-captured MPO activity assay was performed according to (Pulli, Ali, 2013). 100μl of the 1:10 diluted samples were added per well into the MPO ELISA plates (Hycult Biotech, PA), incubated for 1 hour at 4°C. After washing five times with washing buffer, we added 50μl of 100μM Amplite ADHP (AAT Bio, prepared in DMSO), 49μl PBS and 1μl 0.03% H2O2 working solution into each well. We then acquired fluorescence using a plate reader for 5-10 minutes (10-50 cycles) with excitation=535nm, emission=590nm. The units of activity were computed according to the following formula: Activity=(ΔOD × Vt × 4)/(E × Δt × Vs), where ΔOD=change in absorbance; Vt=total volume; Vs=sample volume; E (extinction coefficient)=26.6mM−1; Δt=change in time.
2.7. MPO-Gd magnetic resonance imaging
To confirm the presence of myeloperoxidase (MPO) activity within inflammatory brain plaques in vivo and to evaluate the anti-inflammatory effect of rhp-GSN non-invasively in living animals, we performed MPO-Gd (bis-5-hydroxytryptamide-diethylenetriaminepentaacetate-gadolinium) molecular MRI on day 11 after induction, at the time of early acute disease onset, with and without rhp-GSN therapy (n=4 per group). MPO-Gd is an activatable MR imaging agent that reports extracellular MPO activity in vivo with high specificity and sensitivity (Breckwoldt et al., 2008, Chen et al., 2008). MR imaging was performed by using an animal 4.7-T MR scanner with a dedicated head coil (Bruker Instruments, Billerica, MA). Mouse axial T1-weighted (RARE sequence, TR=873,ms, TE=8.48 ms, fourteen averages, matrix size 192 × 192, field of view 2.5 × 2.5 cm2, slice thickness 0.7 mm, and 16 sections were acquired) images were obtained before and after intravenous administration of MPO-Gd (0.3mmol/kg). Post-contrast images were acquired for at least 60 minutes.
MR images were independently evaluated by two independent authors blinded to the treatment groups using OsiriX. Contrast-to-noise ratios (CNR) were computed for each region of interest (ROI) according to the formula: CNR=(ROI lesion–ROI normal brain)/SDnoise, where ROI lesion is the ROI of an enhancing lesion, ROI normal brain indicates the ROI of an unaffected area of the brain, and SD noise is the standard deviation of noise from an ROI measuring empty. Comparisons of CNR, lesion number, lesion size and total lesion area were performed by visually counting the number of and area of enhanced lesions over the entire brain for each mouse, and the results were averaged.
2.8. Flow Cytometry
Mice were transcardially perfused with ice-cold PBS, brains were harvested and leukocytes were isolated by density centrifugation. Brains were mechanically dissociated in 7 ml of 30% Percoll using a douncer homogenizer and loose fitting pestle. The suspension was then filtered through a 40 μm cell strainer (BD Biosciences, San Jose, CA), underlayed with 3 ml of 70% Percoll and overlayed with 2 ml DPBS. The gradient was then centrifuged at 650 g, 18°C for 25 min. After centrifugation, the thick myelin layer at the 0/30 interface was discarded and the brain leucocytes at the 30/70 interface were collected. The cells were then stained for flow cytometry. All antibodies were purchased from BD Biosciences unless otherwise noted. Antibodies used were anti-CD90.2 (clone: 53-2.1); anti-NK1.1 (clone: PK136, eBioscience); anti-CD49b (clone: DX5); anti-Ly-6G, (clone: IA8); anti-TER119 (clone: TER119, eBioscience), anti-CD3 (clone: 17A2) and anti-CD11b (clone: M1/70,). After staining, cells were fixed with fixation buffer (BioLegend, San Diego, CA). CD90.2, NK1.1, CD49b, Ly-6G, and TER119 were used as lineage markers. T-Lymphocytes were identified as lineagepos, CD11bneg and CD3pos, neutrophils as lineagepos, CD11bpos, and macrophages/microglia as lineageneg, CD11bpos. Data was acquired on a flow cytometer (LSR II, BD Biosciences) and analyzed using FlowJo (FlowJo 8.7, Tree Star, Ashland, Ore).
2.9. Interaction of F98 cells with actin and rhp-GSN
We cultured astrocyte-like F98 glioma cells (ATCC®, #CRL-2397) in four different conditions: 1) control, only saline was added, 2) 1mg/ml rhp-GSN was added to the culture medium, 3) 50μg/ml actin protein was added, and 4) both 1mg/ml rhp GSN and 50μg/ml actin were added. Cells were incubated in RPMI-1640 with 10% FCS 37 °C, 95% air, 5% CO2 for 48 hours. After that, we stained cells with propidium iodide (1μg/ml) to check cell viability and counted the stained cell number in 4 areas at 4× high power fields from each well.
2.10. Interaction of rhp-GSN with human MPO
To determine whether rhp-GSN directly inhibited the MPO activity, we mixed 10μl of 10mg/ml or 1mg/ml rhp-GSN, or PBS with 10μl of 1.7μg/ml MPO (Lee Biosolutions, St. Louis, MO), than performed the MPO activity assay as above.
2.11. Statistical analysis
All measured data were evaluated with the non-parametric Mann-Whitney test. For multiple groups comparison, we applied Kruskal–Wallis one-way analysis of variance test and Dunn multiple comparison post-hoc test. All analyses were performed using Prism (GraphPad, La Jolla, CA, USA). P-values <0.05 were considered statistically significant.
3. Results
3.1. Serum pGSN level is decreased during acute EAE
Western blotting showed that serum pGSN level is decreased in EAE animals compared to that in sham mice (Fig. 1a). Quantification (Figure 1b) confirmed higher levels of pGSN in plasma from sham-induced mice compared to plasma from acute EAE mice (p=0.04).
Fig. 1.

Western blotting results. (a) Western blots for pGSN of serum samples (upper lane) with albumin as internal control (lower lane). (b) Quantification of serum pGSN western blots (*: p<0.05). Sham (n=3) vs EAE (n=5) (c) Western blots for pGSN of brain samples (upper lane) with β-tubulin as internal control (lower lane). (d) Quantification of brain pGSN western blots (*: p<0.05). Sham (n=3) vs EAE (n=6). EAE = experimental autoimmune encephalomyelitis. pGSN=plasma gelsolin.
3.2. Brain pGSN level is increased during acute EAE
Next we asked if pGSN levels in the brain changed during acute inflammation in EAE. Unlike in the plasma, higher pGSN levels were found in the brains from EAE mice compared to sham mice (Fig. 1c). Quantification confirmed higher level of pGSN in brains from acute EAE mice brains compared to those of sham-induced mice (p=0.03, Fig. 1d).
3.3. Rhp-GSN administration decreased disease severity of EAE
To determine whether rhp-GSN has beneficial effects, mice were injected with either rhp-GSN or vehicle intravenously on day 0 and day 2 after induction, and monitored up to 30 days post induction. The overall survival rate in rhp-GSN treatment group was 100% compared to 62.5% in the vehicle-treated group (Figure 2c). Clinical severity (Figure 2a) was reduced in the rhp-GSN group (closed squares) compared to the vehicle-treated group (open circles). We also administered rhp-GSN on days 8 and 10, when there is developing neuroinflammation and early symptom onset(Pulli et al., 2015), and found that this delayed treatment also resulted in improvement in the clinical course, demonstrating that gelsolin’s beneficial effects were not from interfering with EAE induction. Interestingly, the delayed treatment resulted in a faster and more complete resolution of the clinical symptoms (Figure 2a, open squares), likely due to higher bioavailability compared to the earlier administration. The average daily scores also showed significant lower values in the rhp-GSN group than in the vehicle-treated group (P<.001, Fig. 2b).
Fig. 2.

Clinical Score and survival rates of recombinant human plasma gelsolin (rhp-GSN) early (day 0 and day 2) treated (■), late (day 8 and day 10) treated (□), and saline treated vehicle ( ) EAE mice over the 30-day study period. (a) Clinical scores and (b) average daily clinical score comparing day 0/2 rhp-GSN treated to saline-treated control EAE mice. (c) The survival curves in the groups. (***: p<0.001). Note that the survival curves do not include 15 mice that died within 2 days after induction that were deemed to be unrelated to EAE or treatments. Early treatment group (n=7), Late treatment group (n=8), and control group (n=14).
) EAE mice over the 30-day study period. (a) Clinical scores and (b) average daily clinical score comparing day 0/2 rhp-GSN treated to saline-treated control EAE mice. (c) The survival curves in the groups. (***: p<0.001). Note that the survival curves do not include 15 mice that died within 2 days after induction that were deemed to be unrelated to EAE or treatments. Early treatment group (n=7), Late treatment group (n=8), and control group (n=14).
3.4. Rhp-GSN administration decreased brain MPO activity
A key aspect of EAE and MS is neuroinflammation. Secreted extracellular MPO (intracellular MPO is stored in myeloid cell granules and thus not damaging to other cells) is a key marker of oxidative stress and inflammation (Forghani et al., 2012). This can be monitored noninvasively in living animals by MPO-Gd MRI, which reports on extracellular MPO activity (Chen, Breckwoldt, 2008, Forghani, Wojtkiewicz, 2012). We sought to determine if rhp-GSN administration ameliorated oxidative stress and neuroinflammation as a potential mechanism for its therapeutic effects. MPO-Gd MRI performed during peak acute inflammation showed larger lesions and stronger enhancement signal in the vehicle group compared to the treatment group (Fig. 3a and b). Total lesion load and mean individual lesion size were significant higher in the vehicle group than those in the rhp-GSN treated group (p=0.03 and 0.04 respectively; Fig. 3c and 3d). Because activated MPO-Gd is retained at the sites of elevated MPO activity, CNR at 60 min reveals the degree of MPO activity (Chen, Breckwoldt, 2008, Pulli, Ali, 2013). Accordingly, these delayed enhanced images showed significant higher values in control group than that in rhp-GSN treated group (p=0.03; Fig. 3e). These results indicated decreased MPO activity and inflammation in the brain after rhp-GSN treatment, suggesting that rhp-GSN can reduce brain oxidative stress and inflammation in EAE mice.
Fig. 3.

Representative MPO MRI: (a) Control, (b) rhp-GSN treated. Comparison of (c) total lesion area, (d) mean individual lesion size, (e) Contrast-to-Noise ratios showed significant lower values in the treatment group (*: p<0.05). rhp-GSN (n=4) vs vehicle (n=4). (f) Extracellular brain MPO activity. (*: p<0.05). rhp-GSN (n=6) vs vehicle (n=8).
To further verify the imaging results, we performed antibody-capture MPO activity assays on the extracellular fraction from EAE brain specimens (Pulli, Ali, 2013). Significant higher extracellular MPO activity in the control group than in the rhp-GSN treated group was observed (p=0.04, Fig. 3f). This result further confirmed that rhp-GSN treatment decreased MPO activity and corroborated the imaging findings.
3.5. Rhp-GSN does not directly inhibit MPO activity or affect inflammatory cell populations
To investigate whether the rhp-GSN can directly inhibit MPO activity, we performed MPO activity assay after incubating MPO with two different concentrations of rhp-GSN. The activity results showed no difference between rhp-GSN and PBS (Fig. S1), revealing that rhp-GSN does not directly inhibit MPO activity.
A change in inflammatory cell populations can also alter the brain MPO activity. We performed flow cytometry on brain tissues from EAE mice treated with either rhp-GSN or vehicle (Fig. 4, n=6 per group). However, we did not find a significant difference between the two groups for T-lymphocytes, neutrophils, or macrophages/microglia (p>0.05), in terms of both percentage of all leukocytes or absolute number of cells, though there was a trend toward lower numbers of T-lymphocytes and neutrophils.
Fig. 4.

Flow cytometry of EAE brain tissues. There was no significant difference between the treatment groups for T-lymphocytes, neutrophils, or macrophage/microglia (n=6 per group, p>0.05), although there was a trend toward lower number of T-lymphocytes and neutrophils. CD90.2, NK1.1, CD49b, Ly-6G, and TER119 were used as lineage markers. T-Lymphocytes were identified as lineagepos, CD11bneg and CD3pos, neutrophils as lineagepos, CD11bpos, and macrophages/microglia as lineageneg, CD11bpos. FSC=forward scatter.
3.6. Rhp-GSN administration decreased brain extracellular actin concentration
Based on the actin-scavenger functions of gelsolin in the plasma, we sought to determine if this mechanism played a role in the brain as a potential means to ameliorate inflammation and MPO activity. Indeed, we found lower extracellular actin level in the brains of rhp-GSN treated mice than in vehicle treated mice (p=0.03, Fig. 5).
Fig. 5.

Western blotting of extracellular actin in brains of rhp-GSN treated and vehicle treated EAE mice. (a) Representative western blots: upper lane=actin; lower lane=β-tubulin used as internal control. (b) Quantification. (*: p<0.05). rhp-GSN (n=8) vs vehicle (n=8).
3.7. Actin is cytotoxic to the F98 astrocyte-like cells and can be neutralized by rhp-GSN
To test if actin is toxic to brain cells and whether gelsolin can ameliorate actin-induced cytotoxicity, we cultured F98 astrocyte-like cells in four different conditions: control (saline), rhp-GSN only, actin only, and both actin and rhp-GSN (Fig. 6a-d). Actin robustly induced cell death as evidenced by an increased number of PI-positive cells (p=0.0005, Fig. 6e) compared to control. In contradistinction, the number of PI-positive cells was markedly reduced by the addition of rhp-GSN to the actin-supplemented cells (p=0.01 between actin and actin + rhp-GSN groups).
Fig. 6.

Fluorescence staining with propidium iodide (PI) for dead cell nuclei in (a) control group, (b) rhp-GSN supplement group, (c) actin supplement group and (d) actin plus rhp-GSN supplement group in 4× high power fields. (e) Significantly more PI-positive cells in actin supplement group was noted (***: p<0.001). The number of PI-positive cells was markedly reduced by adding rhp-GSN into actin-contained cell culture medium. (*: p= 0.01 between actin and actin + rhp-GSN groups).
4. Discussion
In this study, we found that while serum pGSN concentration decreased in EAE mice, there was an unexpected increase in pGSN concentration in the inflamed brain. This suggests either a shift of gelsolin from the periphery to the CNS or increased gelsolin expression in the CNS in response to inflammation. Cellular damage in inflammatory conditions is associated with release of actin into the extracellular space, and we found extracellular actin to be cytotoxic. By decreasing the amount of extracellular actin, rhp-GSN therapy indirectly reduced MPO activity, a key marker for the innate immune response and inflammation in the brain, and resulted in improved clinical scores and decreased imaging markers of active disease in EAE.
MPO is a key modulator of inflammation (Heinecke, 1999, Klebanoff, 1970) and is closely related to active demyelination (Chen, Breckwoldt, 2008, Forghani, Wojtkiewicz, 2012). Furthermore, in vivo MPO activity reported by MPO-Gd imaging reflects the underlying changes in the innate immune response (Pulli, Bure, 2015). We found that MPO activity was markedly decreased after rhp-GSN administration both in vivo (MPO-Gd imaging, Fig. 3) and ex vivo (antibody-captured MPO activity assay, Fig. 4), demonstrating that additional exogenous gelsolin improved clinical outcome through decreasing inflammation. However, gelsolin did not directly modulate MPO activity and thus must ameliorate inflammation in the brain via a different pathway.
When we examined for changes in the inflammatory cell populations, we did not find a significant difference between treated and untreated groups for lymphocytes, macrophages/microglia, and neutrophils. This suggests that the beneficial effects we observed from gelsolin treatment was not due to a change in inflammatory cells in the brain.
Actin is a potential target for gelsolin. It is the most abundant protein in mammalian cells, and is important to maintaining cell size, shape and motility. However, when released from cells as a consequence of cell damage and death, it polymerizes and can cause adverse consequences (Erukhimov et al., 2000, Lee and Galbraith, 1992, Teunissen et al., 2005). Extracellular actin filaments have been seen in both extracellular fluid (Wen, Corina, 1996) and circulation (Accinni et al., 1983) in several diseases, including multiple sclerosis (Lee, Waxman, 2007, Semra et al., 2002, Teunissen, Dijkstra, 2005). Gelsolin can act directly on actin filaments to shorten them (Lamb et al., 1993, Yin et al., 1980) and enhance its clearance (Accinni, Natali, 1983, Haddad et al., 1990, Lamb, Allen, 1993, Lee and Galbraith, 1992, Lind et al., 1986, 1988, Yin, Zaner, 1980). Interestingly, previous studies found that rhp-GSN administration did not diminish total actin concentration (Lee, Waxman, 2007). Since only extracellular actin is potentially deleterious, we focused on extracellular actin levels in our study. In this study, we found that gelsolin decreases extracellular actin in the inflamed brain to improve clinical outcome. Our observation in astrocyte-like F98 cell culture further confirmed that extracellular actin is toxic to CNS cells, and that this toxicity can be alleviated by rhp-GSN. Interestingly, astrocytes have been found to play an important role in neuroinflammation and EAE such that inhibition of astrocytosis led to a worsening of EAE (Toft-Hansen et al., 2011, Voskuhl et al., 2009). Thus, the beneficial effects of gelsolin in EAE also may be in part due to decreasing the toxicity of actin on astrocytes.
Our study showed that the predominant site of action for gelsolin in EAE is the brain. However, we also found a trend toward decreased number of T-lymphocytes and neutrophils with gelsolin treatment. Because the percentages of these cells did not change, we believe that this slightly decreased recruitment of the peripheral immune cells is a consequence of decreased extracellular actin toxicity from gelsolin treatment. Nonetheless, we cannot exclude that gelsolin may also have a direct effect on the peripheral response, though it would be unlikely that a decrease in neutrophils and T-lymphocytes could cause a decrease in extracellular actin level in the brain.
In conclusion, our results indicated that rhp-GSN acted to decrease extracellular actin, thereby decreasing its toxicity and inflammatory affects to lessen disease burden in the brain and improve outcome. Our findings point to the importance of the secreted form of gelsolin in neuroinflammation. In a separate study using 125I-labeld rhp-GSN in naïve mice, we found that there was increased radiotracer uptake over time (data not shown), demonstrating that gelsolin can cross intact blood-brain barrier. Therefore, plasma gelsolin represents an interesting area for future studies as well as a potential therapeutic target for multiple sclerosis and other neuroinflammatory diseases.
Supplementary Material
Highlights.
In EAE, secreted (plasma) gelsolin levels are decreased in the serum but increased in the brain
The inflammatory enzyme myeloperoxidase’s activity is decreased by recombinant human gelsolin (rhp-GSN) administration
Geloslin decreased the amount of toxic extracellular actin, thus indirectly reduced myeloperoxidase activity and inflammation.
Rhp-GSN administration improved disease course and imaging metrics of disease activity in EAE.
Gelsolin may be a potential therapeutic target for multiple sclerosis and other neuroinflammatory diseases.
Acknowledgments
This work is supported by grants from the National Institute of Health (R01NS070835 and R01NS072167).
Abbreviations
- EAE
experimental autoimmune encephalomyelitis
- MPO
myeloperoxidase
- GSN
gelsolin
- pGSN
plasma gelsolin
- rhp
recombinant human plasma
- rhp-GSN
recombinant human plasma gelsolin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accinni L, Natali PG, Silvestrini M, De Martino C. Actin in the extracellular matrix of smooth muscle cells. An immunoelectron microscopic study. Connect Tissue Res. 1983;11:69–78. doi: 10.3109/03008208309015012. [DOI] [PubMed] [Google Scholar]
- Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105:18584–9. doi: 10.1073/pnas.0803945105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol. 2005;252(Suppl 5):v3–9. doi: 10.1007/s00415-005-5002-7. [DOI] [PubMed] [Google Scholar]
- Chen JW, Breckwoldt MO, Aikawa E, Chiang G, Weissleder R. Myeloperoxidase-targeted imaging of active inflammatory lesions in murine experimental autoimmune encephalomyelitis. Brain. 2008;131:1123–33. doi: 10.1093/brain/awn004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SH, Lee PS, Konigsberg RG, Gallacci D, Chiou T, Arai K, et al. Plasma-type gelsolin is decreased in human blood and cerebrospinal fluid after subarachnoid hemorrhage. Stroke. 2011;42:3624–7. doi: 10.1161/STROKEAHA.111.631135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erukhimov JA, Tang ZL, Johnson BA, Donahoe MP, Razzack JA, Gibson KF, et al. Actin-containing sera from patients with adult respiratory distress syndrome are toxic to sheep pulmonary endothelial cells. Am J Respir Crit Care Med. 2000;162:288–94. doi: 10.1164/ajrccm.162.1.9806088. [DOI] [PubMed] [Google Scholar]
- Forghani R, Wojtkiewicz GR, Zhang Y, Seeburg D, Bautz BR, Pulli B, et al. Demyelinating diseases: myeloperoxidase as an imaging biomarker and therapeutic target. Radiology. 2012;263:451–60. doi: 10.1148/radiol.12111593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942–55. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Greer JM, Sobel RA, Sette A, Southwood S, Lees MB, Kuchroo VK. Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J Immunol. 1996;156:371–9. [PubMed] [Google Scholar]
- Haddad JG, Harper KD, Guoth M, Pietra GG, Sanger JW. Angiopathic consequences of saturating the plasma scavenger system for actin. Proc Natl Acad Sci U S A. 1990;87:1381–5. doi: 10.1073/pnas.87.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke JW. Mechanisms of oxidative damage by myeloperoxidase in atherosclerosis and other inflammatory disorders. J Lab Clin Med. 1999;133:321–5. doi: 10.1016/s0022-2143(99)90061-6. [DOI] [PubMed] [Google Scholar]
- Hofstein R, Hesse G, Shashoua VE. Proteins of the extracellular fluid of mouse brain: extraction and partial characterization. J Neurochem. 1983;40:1448–55. doi: 10.1111/j.1471-4159.1983.tb13589.x. [DOI] [PubMed] [Google Scholar]
- Imitola J, Chitnis T, Khoury SJ. Insights into the molecular pathogenesis of progression in multiple sclerosis: potential implications for future therapies. Arch Neurol. 2006;63:25–33. doi: 10.1001/archneur.63.1.25. [DOI] [PubMed] [Google Scholar]
- Klebanoff SJ. Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science. 1970;169:1095–7. doi: 10.1126/science.169.3950.1095. [DOI] [PubMed] [Google Scholar]
- Kulakowska A, Ciccarelli NJ, Wen Q, Mroczko B, Drozdowski W, Szmitkowski M, et al. Hypogelsolinemia, a disorder of the extracellular actin scavenger system, in patients with multiple sclerosis. BMC Neurol. 2010;10:107. doi: 10.1186/1471-2377-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Mehl R, Izumo S, Nadal-Ginard B, Yin HL. Muscle is the major source of plasma gelsolin. J Biol Chem. 1988;263:8239–43. [PubMed] [Google Scholar]
- Lamb JA, Allen PG, Tuan BY, Janmey PA. Modulation of gelsolin function. Activation at low pH overrides Ca2+ requirement. J Biol Chem. 1993;268:8999–9004. [PubMed] [Google Scholar]
- Lee PS, Waxman AB, Cotich KL, Chung SW, Perrella MA, Stossel TP. Plasma gelsolin is a marker and therapeutic agent in animal sepsis. Crit Care Med. 2007;35:849–55. doi: 10.1097/01.CCM.0000253815.26311.24. [DOI] [PubMed] [Google Scholar]
- Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med. 1992;326:1335–41. doi: 10.1056/NEJM199205143262006. [DOI] [PubMed] [Google Scholar]
- Lind SE, Smith DB, Janmey PA, Stossel TP. Role of plasma gelsolin and the vitamin D-binding protein in clearing actin from the circulation. J Clin Invest. 1986;78:736–42. doi: 10.1172/JCI112634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind SE, Smith DB, Janmey PA, Stossel TP. Depression of gelsolin levels and detection of gelsolin-actin complexes in plasma of patients with acute lung injury. Am Rev Respir Dis. 1988;138:429–34. doi: 10.1164/ajrccm/138.2.429. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Osborn TM, Dahlgren C, Hartwig JH, Stossel TP. Modifications of cellular responses to lysophosphatidic acid and platelet-activating factor by plasma gelsolin. Am J Physiol Cell Physiol. 2007;292:C1323–30. doi: 10.1152/ajpcell.00510.2006. [DOI] [PubMed] [Google Scholar]
- Pulli B, Ali M, Forghani R, Schob S, Hsieh KL, Wojtkiewicz G, et al. Measuring myeloperoxidase activity in biological samples. PLoS One. 2013;8:e67976. doi: 10.1371/journal.pone.0067976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulli B, Bure L, Wojtkiewicz GR, Iwamoto Y, Ali M, Li D, et al. Multiple sclerosis: myeloperoxidase immunoradiology improves detection of acute and chronic disease in experimental model. Radiology. 2015;275:480–9. doi: 10.1148/radiol.14141495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semra YK, Seidi OA, Sharief MK. Heightened intrathecal release of axonal cytoskeletal proteins in multiple sclerosis is associated with progressive disease and clinical disability. J Neuroimmunol. 2002;122:132–9. doi: 10.1016/s0165-5728(01)00455-6. [DOI] [PubMed] [Google Scholar]
- Silacci P, Mazzolai L, Gauci C, Stergiopulos N, Yin HL, Hayoz D. Gelsolin superfamily proteins: key regulators of cellular functions. Cell Mol Life Sci. 2004;61:2614–23. doi: 10.1007/s00018-004-4225-6. [DOI] [PubMed] [Google Scholar]
- Smith DB, Janmey PA, Herbert TJ, Lind SE. Quantitative measurement of plasma gelsolin and its incorporation into fibrin clots. J Lab Clin Med. 1987;110:189–95. [PubMed] [Google Scholar]
- Tanaka J, Kira M, Sobue K. Gelsolin is localized in neuronal growth cones. Brain Res Dev Brain Res. 1993;76:268–71. doi: 10.1016/0165-3806(93)90217-x. [DOI] [PubMed] [Google Scholar]
- Teunissen CE, Dijkstra C, Polman C. Biological markers in CSF and blood for axonal degeneration in multiple sclerosis. Lancet Neurol. 2005;4:32–41. doi: 10.1016/S1474-4422(04)00964-0. [DOI] [PubMed] [Google Scholar]
- Toft-Hansen H, Fuchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia. 2011;59:166–76. doi: 10.1002/glia.21088. [DOI] [PubMed] [Google Scholar]
- Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:11511–22. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D, Corina K, Chow EP, Miller S, Janmey PA, Pepinsky RB. The plasma and cytoplasmic forms of human gelsolin differ in disulfide structure. Biochemistry. 1996;35:9700–9. doi: 10.1021/bi960920n. [DOI] [PubMed] [Google Scholar]
- Yin HL, Kwiatkowski DJ, Mole JE, Cole FS. Structure and biosynthesis of cytoplasmic and secreted variants of gelsolin. J Biol Chem. 1984;259:5271–6. [PubMed] [Google Scholar]
- Yin HL, Stossel TP. Control of cytoplasmic actin gel-sol transformation by gelsolin, a calcium-dependent regulatory protein. Nature. 1979;281:583–6. doi: 10.1038/281583a0. [DOI] [PubMed] [Google Scholar]
- Yin HL, Zaner KS, Stossel TP. Ca2+ control of actin gelation. Interaction of gelsolin with actin filaments and regulation of actin gelation. J Biol Chem. 1980;255:9494–500. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.