

Abstract
Alzheimer’s disease is an increasingly prevalent neurodegenerative disorder whose pathogenesis has been associated with aggregation of the amyloid-β peptide (Aβ42). Recent studies have revealed that once Aβ42 fibrils are generated, their surfaces strongly catalyse the formation of neurotoxic oligomers. Here we show that a molecular chaperone, a Brichos domain, can specifically inhibit this catalytic cycle and limit Aβ42 toxicity. We demonstrate in vitro that Brichos achieves this inhibition by binding to the surfaces of fibrils, thereby redirecting the aggregation reaction to a pathway that involves minimal formation of toxic oligomeric intermediates. We verify that this mechanism occurs in living brain tissue by means of cytotoxicity and electrophysiology experiments. These results reveal that molecular chaperones can help maintain protein homeostasis by selectively suppressing critical microscopic steps within the complex reaction pathways responsible for the toxic effects of protein misfolding and aggregation.
Alzheimer’s disease (AD) is a progressive and fatal neurodegenerative disorder characterized by memory loss and personality changes1-5. This disease is one of over 40 amyloid related disorders6-8 that also include Parkinson’s and Huntington’s diseases. This class of disorders is associated with the misfolding of specific peptides or proteins and the subsequent formation of ordered amyloid fibrils having a common cross-β structure6,7,9-11. A central molecular species in AD is the 42 residue amyloid-β peptide, Aβ42, which is the dominant component of the plaques that are a defining histopathological characteristic of the brains of AD patients4. Studies over the past decade, however, have indicated that it is the pre-fibrillar oligomeric aggregates of amyloidogenic peptides and proteins such as Aβ42 which appear likely to be the major toxic agents causing neuronal cell death3,12-14.
Major advances have recently been made in understanding the molecular mechanisms that lead to the generation of such toxic oligomers. This problem is highly complex, as the process of peptide and protein aggregation involves multiple events occurring simultaneously in a multi-step nucleated polymerization reaction15-18 that results in the formation of high molecular weight fibrillar aggregates from the soluble monomeric peptide via non-fibrillar oligomeric species19-22. In this type of reaction, the homogenous primary nucleation21-24 of new oligomers from monomers is inherently a slow process, and is therefore unlikely to generally represent the major origin of toxicity. It has been shown, however, that the production of oligomers can be catalysed in a very effective manner by the surfaces of high molecular weight fibrillar aggregates19. This catalytic pathway takes the form of a secondary nucleation reaction25-27, involving both free monomers and fibrils, and can increase dramatically the overall rate of Aβ42 aggregation and oligomer formation19,28,29. As such, although not directly toxic themselves, Aβ42 fibrils provide a catalytic surface for the continuous generation of toxic oligomers, species that can also grow and convert into additional fibrils19,21,30, thus promoting further the formation of additional toxic species in a catalytic cycle. The fibrils, therefore, play a key role in the formation of oligomers by lowering the kinetic barriers that under normal circumstances hinder their de novo formation.
Because of the importance of the catalytic cycle in the production of Aβ42 oligomers, an attractive strategy to prevent the formation of these damaging assemblies would be the identification of inhibitors that can interfere with the catalytic activity of the fibril surfaces, although no agents with this specific effect have yet been identified. In this context, we describe the effect of the chaperone domain Brichos31 on the molecular mechanism underlying the aggregation of Aβ42. Molecular chaperones have been known for several decades to play a key role in aiding the folding in vivo of newly synthesised proteins into their native states, in their trafficking to specific locations in cells, and in the efficient assembly of molecular sub-units into functional multimeric structures32-34. Moreover, it is increasingly evident that the chaperone machinery plays an important role in maintaining protein homeostasis under a wide range of circumstances33,35. Several pathways, including chaperone-mediated disaggregation and stimulation of proteolyic degradation, have been identified as part of a complex network that regulates proteostasis33,34,36. In particular, a range of genetic and biochemical studies imply that chaperones play a critical protective role in relation to the aberrant protein aggregation processes associated with protein misfolding disorders6,7,33,37-41, although much remains to be established concerning the precise mechanisms of action of such processes.
Brichos, a protein domain of approximately 100 amino acids, was initially identified in the proteins Bri, related to familial British dementia, chondromodulin, associated with chondrosarcoma and lung surfactant C precursor protein (proSP-C)42, and has now been found in approximately ten distantly related protein families43-45. Brichos domains have been found experimentally to inhibit misfolding and aggregation both in vitro31,46 and in vivo47,48, and mutations in proteins containing Brichos domains are linked to a variety of forms of amyloid disease49,50. Here we reveal for Aβ42 the mechanism by which Brichos performs its anti-aggregation function and show that this molecular chaperone interferes highly selectively and dramatically with the key secondary nucleation reaction. Brichos is, therefore, an effective inhibitor of the catalytic mechanism underlying Aβ42 aggregation and can consequently suppress the toxicity associated with the aggregation reaction. Our results thus reveal that molecular chaperones possess capabilities that go well beyond their established ability to interact directly with aggregation prone species in solution2,32-34, and that they are able to suppress the toxicity associated with aggregation reactions by interacting with amyloid fibrils selectively to break the critical catalytic cycle through which it is generated.
RESULTS
Brichos inhibits surface-catalysed secondary nucleation of Aβ42
We first monitored the kinetics of the aggregation of Aβ42 under the quiescent conditions described previously19,20, and obtained data completely consistent with this earlier study. In order to establish well-defined initial conditions for the kinetic experiments, and hence to obtain reproducible data, we purified to very high levels the recombinant monomeric Aβ42 peptide by repeated applications of size-exclusion chromatography and controlled carefully the inertness of surfaces with which it made contact20. On addition of the Brichos domain from proSP-C50, at concentrations between 10% and 100% Aβ42 monomer equivalents, we observed a reduction in the overall rate of formation of fibrils and characteristic changes in the time course of the reaction (Fig. 1; see also Supplementary Fig. 1). To connect these observations with the underlying microscopic mechanisms that contribute to the macroscopic changes, we used an analytical approach to predict the effects of perturbing the different individual microscopic processes involved in Aβ42 aggregation on the global kinetics of aggregation51. Specifically, alterations in the rates of primary nucleation, fibril elongation, and secondary nucleation are each predicted to result in distinct and characteristic changes in the shape of the reaction profiles (Fig. 1).
Figure 1. Kinetics of Aβ42 aggregation in the presence of Brichos.

(a-c) Reaction profiles from left (blue) to right (green) for aggregation in the absence of Brichos and in the presence of 10%, 15%, 35%, 50%, 75%, and 100% Aβ42 monomer equivalents of Brichos. The data show averages (points) and standard errors over five technical replicas. The effect of Brichos saturates at a stoichiometry of approximately one monomer equivalent of Aβ42 (see Supplementary Fig.1). The blue dashed line is the integrated rate law for Aβ42 aggregation in the absence of Brichos using the rate constants determined previously19. The green dashed lines show predictions for the resulting reaction profiles when each of (a) primary nucleation, (b) fibril elongation, and (c) secondary nucleation are inhibited by the chaperone (see Supplementary Fig. 2). The thin dotted lines in (c) are theoretical predictions for the reaction profiles at the intermediate Brichos concentrations using the association and dissociation rate constants determined for its binding by means of SPR (Fig. 3b). (d-f) Time evolution of the nucleation rate calculated from the kinetic analysis. The blue line corresponds to the situation in the absence of Brichos and the green dashed lines show predictions for the cases when each of (d) primary nucleation, (e) fibril elongation, and (f) secondary nucleation are inhibited by the chaperone. The insets show the relative number of oligomers generated during the aggregation reaction. The concentration of monomeric Aβ42 was 3μM.
Since all of the rate constants associated with the aggregation pathway of Aβ42 are known19, we can predict quantitatively the effects on the reaction profile accompanying any given perturbation, for example the inhibition of individual molecular steps (Fig. 1a-c, green dashed lines), without introducing any additional fitting parameters. In particular, the effects of the complete inhibition of secondary nucleation can be predicted (Fig. 1c, green dashed line) simply by setting the rate constant of this process to zero in the integrated rate law19 that describes Aβ42 aggregation (see Supplementary Fig. 2, and Supplementary Note for the rate law). Remarkably, in the presence of an excess of the Brichos domain, the data match precisely at all times the predictions for complete inhibition of secondary nucleation – but match none of the other mechanisms of inhibition that we tested by reducing the corresponding rate constants – revealing that this molecular chaperone acts specifically to inhibit completely this microscopic step in the aggregation reaction. In order to test whether or not this specificity of action – the selective inhibition of secondary nucleation – is a ubiquitous characteristic shared with other proteins, we performed experiments where the aggregation of the Aβ42 peptide was probed in the presence of a range of diverse control proteins, including calbindin D9k, calmodulin, the B1-domain of protein G and human serum albumin. These measurements (see Supplementary Fig. 3) revealed that none of these proteins interfered with Aβ42 aggregation in the same manner as Brichos.
A reduction in the rate of either primary nucleation, fibril elongation or secondary nucleation does not significantly affect the total quantity of mature fibrils formed over the duration of the Aβ42 aggregation reaction, since in all cases the soluble peptide is eventually converted almost entirely into fibrils19,20. Critically, however, kinetic analysis (Fig. 1d-f) reveals that the specific inhibition of each of the microscopic processes will not only change the time at which the plateau in the reaction profile of aggregation occurs, but will have very different effects on the generation of oligomers during the reaction. In particular, the reduction of the rate of primary nucleation delays the aggregation reaction (Fig. 1d) but does not change the total number of oligomers generated during the reaction, while the specific inhibition of the elongation process redirects the reactive flux involved in the conversion of the monomeric species to the fibrillar state towards secondary nucleation processes, therefore dramatically increasing the number of new oligomers that are generated (Fig. 1e). By contrast, suppressing the secondary nucleation process directs the reaction pathway towards elongation events, thereby preventing almost entirely the generation of oligomers (Fig. 1f). This analysis therefore predicts that the specific inhibition of secondary nucleation events, identified here for the Brichos domain, is an extremely effective strategy for preventing the formation of low molecular weight oligomeric species and hence for reducing dramatically the toxicity associated with the aggregation reaction. Conversely, simply inhibiting the rate of elongation of fibrils would increase the concentration of toxic species, and hence increase rather than decrease the toxicity associated with the aggregation process.
Brichos interacts specifically with fibrillar but not monomeric Aβ42
Inhibition of secondary nucleation requires perturbation of the interactions between soluble monomers and amyloid fibrils, and in principle either species could be targeted by Brichos. To identify the molecular species on which this molecular chaperone acts, therefore, we performed experiments in which newly formed Aβ42 fibrils, produced in the presence or absence of Brichos, were diluted and added to freshly prepared monomer solutions and then also incubated in the presence and absence of Brichos (Fig. 2). We observed that pristine fibrils that had not been exposed to the chaperone at any stage enhance the process of secondary nucleation and increase the rate of aggregation19 (Fig. 2a). By contrast, we have found in the present work that pre-formed fibrils that had been generated in the presence of Brichos (Fig. 2b) accelerate aggregation, even in the absence of Brichos in solution, to a significantly smaller extent than do fibrils formed in the absence of chaperone (Fig. 2a). Indeed, the kinetic analysis indicates a reduction of approximately 40% in the secondary nucleation rate in this latter case (Fig. 2b), showing that a significant fraction of the chaperones must have remained bound to the fibrils during the time course of the aggregation reaction. Furthermore, the surface catalytic activity of fibrils was observed to be entirely abolished when pre-formed aggregates generated in the absence of molecular chaperones were brought into contact with solutions containing both Brichos and monomeric Aβ42 at equal stoichiometry (Fig. 2c), demonstrating the ability of Brichos to arrest even ongoing reactions. Identical results were obtained when fibrils formed in the presence of Brichos were added to solutions containing both monomeric Aβ42 and Brichos (Fig. 2d). These results are consistent with observations that Brichos is able to arrest ongoing Aβ42 and Aβ40 aggregation reactions when added at various time-points.31
Figure 2. Inhibition by Brichos of Aβ42 surface-catalysed secondary nucleation of oligomers.

(a-d) Kinetics of aggregation when pre-formed Aβ42 fibrils, grown in the absence (a, c) or presence (b, d) of Brichos, were added to monomeric Aβ42 with (c, d) or without (a, b) Brichos in solution. Fibrils grown (b) in the presence of Brichos do not accelerate the reaction to the same extent as (a) fibrils that have never been exposed to Brichos, showing that Brichos binds to fibrils. When Brichos was added in to solutions in which aggregation was underway (c, d), it arrested the reaction and prevented the acceleration due to added fibrils even with fibrils grown in the absence of chaperone. The dashed lines show predictions19 for the reaction profiles with the secondary nucleation rate constant set to (a) the value measured previously in the absence of the chaperone19, (b) 60% of this value, and (c, d) zero. The solution concentrations of Aβ42 and Brichos were 3 μM; the concentration of pre-formed fibrils was 6 nM. All data show four technical replicas overlaid.
The kinetic data with and without pre-formed seed fibrils suggest a mechanism for inhibition of aggregation involving the non-covalent association of Brichos with Aβ42 fibrils. To probe this mechanism further, we used transmission electron microscopy (TEM) with nano-gold conjugated anti-Brichos antibodies. The resulting images (Fig. 3a) provide a striking visualization of the nature of the interactions and clearly reveal specific binding of Brichos along the fibril surface. The interactions between the chaperone and the fibrils were also studied using surface plasmon resonance (SPR) measurements. The data (Fig. 3b) reveal that Brichos has a high affinity for the fibrils, whereas no binding was observed to SPR sensor surfaces functionalised with monomeric Aβ42 or to reference surfaces lacking any Aβ42; the absence of detectable binding to monomeric Aβ42 (Fig. 3b) verifies that the mode of action of Brichos is through interactions with the fibrillar species rather than with soluble monomers. These measurements also provide estimates for the association (kon ≈ 5.1 × 103M−1s−1) and dissociation (koff ≈ 2.1 × 10−4 s−1) rate constants for the binding of the chaperone to Aβ42 fibrils (KD ≈ 40 nM). Remarkably, using these rate constants we are able to predict quantitatively (see Supplementary Note for a complete derivation of the rate equations) and accurately the experimental kinetic profiles at each of the intermediate concentrations of Brichos shown in Fig. 1c (the predictions are shows as thin dotted lines). Moreover, the rate constants predict that under the conditions used to study the effects of pre-formed seed fibrils in Fig. 2b, the maximum level of dissociation of Brichos from the seeds prepared in the presence of the chaperone will only be ca. 68% after two hours, a finding consistent with the observation (Fig. 2b) of a partially reduced catalytic effect of fibrils formed in the presence of Brichos even after several hours.
Figure 3. Brichos interacts with fibrillar but not monomeric Aβ42.
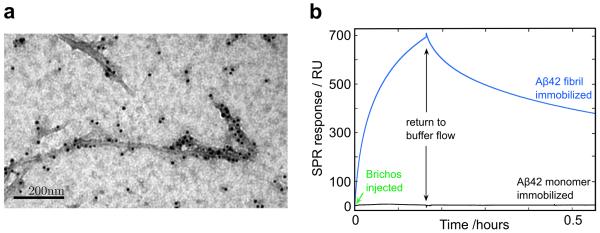
(a) Images using TEM with a nano-gold conjugated secondary antibody against anti-Brichos antibodies show that the chaperone binds to Aβ42 fibrils. (b) SPR analysis verifies the specific binding to fibrils and allows determination of the association (kon ≈ 5.1 × 103 M−1s−1) and dissociation (koff ≈ 2.1 × 10−4 s−1) rate constants, implying an apparent equilibrium dissociation constant KD ≈ 40 nM. No binding was observed to the monomer or to control in the absence of Aβ42.
Brichos blocks the catalytic cycle generating oligomers
The combination of kinetic studies and direct measurements of binding affinity discussed above demonstrates that Brichos is able to bind with high affinity to the surfaces of Aβ42 fibrils. These findings suggest that this molecular chaperone inhibits specifically secondary nucleation events occurring on the surfaces of fibrils. To probe explicitly the degree of inhibition of the production of low molecular weight oligomeric species that are created through this mechanism, we studied their concentrations during Aβ42 aggregation in the absence (Fig. 4a) and presence (Fig. 4b) of Brichos in solution. We monitored the generation of low molecular weight oligomeric species in each sample by removing aliquots at a series of time points and isolating in each case the fraction corresponding to low molecular weight species by means of size exclusion chromatography19. Eluted fractions were pooled in to three categories of samples: monomers, small oligomers (ca. 3-14 mers, equivalent to globular proteins of 14-65 kDA molecular weight) and large oligomers (ca. 15-20 mers, 66-90 kDa).
Figure 4. Brichos inhibits the formation of Aβ42 oligomers by suppressing secondary nucleation and redirecting the reaction pathway towards elongation events.

(a-b) Quantification of low molecular weight oligomers of Aβ42 in the absence (a) and presence (b) of Brichos using size-exclusion chromatography and the 6E10 antibody. The reduced intensities of the fractions corresponding to the oligomers are shown after incubation times of 15, 30, 45 and 60 min, verifying the reduction in the population of oligomers due to the chaperone. The soluble concentrations of Aβ42 and chaperone were 3 μM, and the concentration of pre-formed fibrils was 30 nM. (c-d) Corresponding predictions for the nucleation rate as a function of time with and without Brichos. (e-f) Cryo-TEM images of fibrils formed in the absence (e) and presence (f) of Brichos show that longer fibrils are formed in the presence of Brichos. Quantification of the fibril lengths over multiple images reveals that fibrils formed in the presence of Brichos are on average at least four times longer than those formed in the absence of Brichos. The samples contained 10 μM Aβ42 and 6 μM Aβ42 + 6 μM Brichos respectively, and were taken at the reaction end point (as measured by ThT) in each case.
We then probed the amount of Aβ42 present in these fractions using the Aβ42-sensitive 6E10 antibody and observed that the concentration of oligomers was indeed very substantially reduced in the presence of Brichos (Fig. 4a-b). Furthermore, by following the oligomer populations as a function of time, we confirmed that the population of oligomers that is reached during the aggregation process is reduced significantly in the presence of Brichos (Fig. 4b), consistent with our theoretical predictions (Fig. 4c-d). Thus, through removal of the secondary nucleation process that is normally responsible for the chain reaction associated with Aβ42 aggregation and which results in the rapid generation of oligomeric species19,29, the reaction pathway is fundamentally modified to generate fibrils from monomers via a reaction network that involves only primary nucleation and elongation, and thereby results in the generation of significantly fewer cytotoxic oligomers. As a consequence of the redirection of the reactive flux away from secondary nucleation and towards elongation events, we observed by cryo-TEM analysis (Fig. 4e-f) that Aβ42 fibrils formed in the presence of Brichos (Fig. 4f) are significantly longer than fibrils formed in the absence of chaperone (Fig. 4e).
Breaking the catalytic cycle suppresses toxicity in brain tissue
We next analyzed whether or not the reduction in the population of oligomeric species resulting from the specific suppression of the secondary nucleation pathway by Brichos can be sufficient to reduce the high level of neurotoxicity associated with Aβ42 aggregation in brain tissue. To this effect, we used electrophysiology techniques on mouse brain slices to measure the degree of Aβ42-induced impairment of hippocampal gamma oscillations in the presence and absence of Brichos (Fig. 5). Gamma oscillations are important for higher brain functions such as cognition, learning and memory, and are found to be degraded in brain disorders that lead to cognitive decline in patients suffering from neurodegenerative disorders such as Alzheimer’s disease52.
Figure 5. Brichos reduces the toxicity associated with the aggregation of Aβ42 in brain slices.

(a) Solutions containing mature fibrils alone and chaperone alone do not show increased toxicity relative to the control (first gray bar, p = 0.3 and p = 0.5). The toxicity is, however, increased relative to the control for solutions (Δt = 15 min) undergoing aggregation from initially purely monomeric peptide (orange bar, p = 0.02, indicated *), and is furthermore dramatically increased in samples initially containing monomeric Aβ42 with pre-formed fibrils (red bar, p < 0.0001, indicated ***). In both of these aggregation reactions, the toxicity is strongly suppressed to a level comparable with the control by adding Brichos in solution (green bars, p = 0.3 and p = 0.2 relative to the control). The box-and-whisker plot is based on 8-16 repeats for each condition as indicated, with the boxes enclosing data from the first to the third quartile, the horizontal line being drawn at the median, the whiskers indicating the range, and the number of repeats given above or below. All p-values are based on two-tailed Mann-Whitney U-tests relative to the control. (b) Representative traces and power spectra of the kainate-induced gamma oscillation measurements for the control (gray), the monomeric peptide (orange) and the monomeric peptide supplemented with pre-formed fibrils (red).
The results reveal that incubation for 15 minutes of samples initially containing only soluble Aβ42 monomers generates limited but significant toxicity (Fig. 5a, orange bar, p = 0.02) relative to the control, but that the addition of small amounts of pre-formed fibrils to soluble monomers prior to incubation increases the toxicity substantially at the same time point (Fig. 5a, red bar, p < 0.0001; Fig. 5b shows representative power spectra), showing that the toxic species are overwhelmingly produced by fibril-catalysed processes. Remarkably, in the presence of Brichos (Fig. 5a, green bars), such toxicity does not develop, confirming that selective inhibition of the pathway generating oligomers reduces dramatically the toxicity associated with Aβ42 aggregation. This reduction in toxicity, to a level comparable with the controls, is observed both for reactions initiated from purely monomeric peptide (Fig. 5a, first green bar, p = 0.3) and for those initiated from solutions including pre-formed fibrils (Fig. 5a, second green bar, p = 0.2), demonstrating that the inhibition of secondary nucleation by Brichos reduces toxicity even when Aβ42 fibrils have already been formed. We also performed a range of cell viability and cytotoxicity measurements by MTT and by apoptosis based assays, respectively, and found consistent trends; critically, in all cases Brichos was observed to suppress the toxicity associated with Aβ42 aggregation leading to readouts close to those of the controls across this range of assays (Supplementary Fig. 4).
DISCUSSION
The results that have emerged from this study show that the chaperone Brichos targets a highly specific molecular process in the aggregation pathway of Aβ42, that of fibril-induced secondary nucleation, which is predominantly responsible for the generation of toxic oligomers. Indeed, by inhibiting secondary nucleation, the reaction network underlying the Aβ42 aggregation process (Fig. 6a) is fundamentally altered such that the modified pathway leading from monomeric peptide to fibrils proceeds through primary nucleation and elongation-related processes alone (Fig. 6b), and results in significantly lower concentrations of toxic oligomers (Fig. 4b). The reduction in the population of these species originates from the fact that out of the two processes generating oligomers, primary and secondary nucleation, the chaperone has specifically removed the source that is dominant in Aβ42 aggregation19, namely secondary nucleation (Fig. 6b). It is this ability to turn off the otherwise continuing and increasing generation of oligomers through secondary nucleation that explains the efficacy of this process. It is interesting to note, however, that this reduction in oligomer population occurs even though the eventual quantities of mature fibrils, which are now generated through primary nucleation and elongation alone, remain unaffected by the presence of Brichos19,20.
Figure 6. Brichos inhibits the catalytic cycle that generates toxic Aβ42 oligomers.
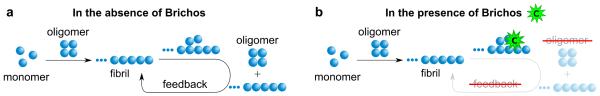
Schematic diagram showing (a) the molecular pathways – primary and secondary nucleation – involved in oligomer formation in Aβ42 aggregation and (b) the mechanism by which Brichos suppresses the formation of toxic oligomers, in which the secondary nucleation pathway is specifically inhibited to suppress the remove source of oligomers.
The inhibition of secondary pathways identified here represents a mechanism through which a specific molecular chaperone can act to control a single step in the complex process of protein aggregation, in this case the step that is very largely responsible for the formation of toxic oligomers. This mechanism is effective both in retarding de novo aggregation and in delaying aggregation even in the presence of pre-formed aggregates, and is therefore an effective means for reducing the toxicity associated with Aβ42 self-assembly at any stage in the process. Most importantly, however, by inhibiting secondary nucleation Brichos is able to reduce dramatically the production of toxic species that otherwise accelerates as the aggregation reaction proceeds. Moreover, since this approach targets the catalytic sites on the fibrils at which secondary nucleation occurs, and does not rely on interactions with the soluble species, it is possible to reach high efficacy even for highly sub-stoichiometric ratios of chaperone to Aβ42 molecules, particularly if the chaperone is able to bind specifically to the active catalytic sites in preference to other locations on the fibril surface. Indeed, the identification of Brichos as a specific fibril-binding partner presents a potential approach to establish the precise surface features that permit secondary nucleation.
It is well established that other molecular chaperones are important in assisting in the efficient folding of newly synthesised proteins, and in binding to soluble misfolded monomeric or multimeric species, targeting them for degradation and inhibiting primary nucleation32-35, the essential first step in the production of aggregates. Our results, however, show that specific chaperones exist that are able to suppress subsequent steps in the process of protein aggregation by inhibiting further proliferation of aggregates, thereby generating a series of protective mechanisms to minimize the risk to an organism of the consequences of misfolding events. In the case of Brichos and Aβ42 aggregation, this step is secondary nucleation, which is responsible for the production of those species that are most toxic to living cells. The fact that a range of other chaperones have been observed to bind to fibril surfaces53 suggests that this potent mechanism may be quite general.
This study demonstrates the power of targeting the specific molecular-level processes that are responsible for the toxicity associated with protein aggregation, in contrast to untargeted approaches that focus on the non-specific suppression of the overall aggregation reaction or processes that do not generate significant levels of toxic oligomers. In particular, a reduction in the final fibril load may not be associated with a reduced population of toxic species, indicating that molecular chaperones have emerged during biological evolution to have distinctive roles in suppressing specific steps in the process of aggregation. The present results also suggest that strategies designed to identify potential drug targets by screening for suppression of fibril formation may, at least in some cases, lead to compounds that are not able to suppress, or can even increase, the levels of toxic species. Indeed the results show that the populations of cytotoxic oligomers can be successfully reduced without affecting the final fibril load, as shown in Fig. 1, and it is interesting to note that this decoupling between the total aggregate load and the observed toxicity could provide a molecular rationalisation for the well-established lack of direct correlation between aggregate formation and disease progression3,6,54-57.
Finally, our findings also reveal that elucidating the specific molecular processes by which toxicity occurs is a vital step in understanding the nature of biological defence mechanisms, and hence potentially for the rational design of effective therapeutic strategies to combat Alzheimer’s disease; moreover, given the existence of common features of the mechanisms of protein aggregation2,7, such approaches are likely to be important for addressing misfolding diseases more generally. Indeed, our results suggest that nature has evolved this type of targeted strategy for suppressing toxicity through molecular chaperones that are not only capable of promoting the folding and suppressing the misfolding of proteins in solution, but which also have the ability to interfere selectively in the different steps in the complex reaction pathways associated with peptide and protein aggregation, and in the case of Brichos, studied here, to block the catalytic production of toxic species.
Online Methods
Materials
We expressed the Aβ(M1-42) peptide (MDAEFRHDSGYEVHHQKLVFFAEDVGSNKGA-IIGLMVGGVVIA) in Escherichia coli from a synthetic gene and purified the peptide essentially as described previously58. In short, the purification procedure involved sonication of E. coli cells, dissolution of inclusion bodies in 8 M urea, ion exchange in batch mode on DEAE cellulose resin, lyophylization, and gel filtration on a 3.4 × 200 cm gel filtration column at 4°C. The purified peptide was frozen as identical 3 mL aliquots and lyophylized. pro-SPC Brichos was expressed in E. coli and purified as described previously31. Purification of the control proteins is described elsewhere for human calmodulin59, fatty-acid free human serum albumin60, bovine calbindin D9k61, and the B1 domain of streptococcal protein G62.
Kinetic assays
For kinetic experiments, aliquots of purified Aβ42 were dissolved in 6 M GuHCl, and the monomer was isolated by two rounds of gel filtration on a Superdex 75 column in 20 mM sodium phosphate buffer, pH 8, with 200 μM EDTA and 0.02% NaN3. The centre of the monomer peak was collected on ice and lyophilized. The sample was again dissolved in 6 M GuHCl, and the monomer isolated by gel filtration on a Superdex 75 column in 20 mM sodium phosphate buffer, pH 8, with 200μM EDTA and 0.02% NaN3 was typically found to have a concentration (determined by quantitative amino acid analysis purchased from BMC Uppsala) of 5-12 μM. The gel filtration step removes traces of pre-existing aggregates and exchanges the buffer to the one used in the fibril formation experiments. The monomer generated in this way was diluted with buffer, or with buffer and Brichos, and supplemented with 6 μM ThioflavinT (ThT) from a 1.2 mM stock to prepare a series of samples with the same Aβ42 concentration but different Brichos concentrations. All samples were prepared in low-bind Eppendorf tubes (Axygen, California, USA) on ice using careful pipetting to avoid introduction of air bubbles. Each sample was then pipetted into multiple wells of a 96 well half-area plate of black polystyrene with a clear bottom and PEG coating (Corning 3881, Massachusetts, USA), 100 μL per well. Assays were initiated by placing the 96-well plate at 37°C in a plate reader (Fluostar Omega, Fluostar Optima or Fluostar Galaxy, BMGLabtech, Offenburg, Germany). A series of control experiments19,20 demonstrated that under the conditions used, the fluorescence from ThT is linearly related to the Aβ42 aggregate mass concentration.
Kinetic assays with pre-formed fibrils
Kinetic experiments were set up as above for multiple samples of Aβ42 in 20 mM sodium phosphate buffer, pH 8, with 200 μM EDTA, 6 μM ThT and 0.02% NaN3. The fluorescence of added ThT was monitored for 1.5 h to verify the formation of fibrils. The samples were then collected from the wells into low-bind Eppendorf tubes (Axygen, California, USA) and sonicated for 2 min in a sonicator bath at room temperature to disrupt any fibril clusters. Fresh monomer was isolated by gel filtration as above and diluted to 3 μM, with or without 3 μM Brichos in 20 mM sodium phosphate, pH 8, containing 200 μM EDTA, 6 μM ThT, 0.02% NaN3. Samples were prepared containing the sonicated pre-formed fibrillar seeds at concentrations corresponding to 0, 0.04, 0.2, and 1% of the highest monomer concentration in the dilution series, and therefore the concentration of Brichos remaining in solution with the pre-formed fibrils was below 0.03 μM. The ThT fluorescence was monitored in the plate reader every 60 s under quiescent conditions at 37°C. The previously established rate constants19 for elongation and secondary nucleation in Aβ42 aggregation show that at the concentrations of pre-formed fibrils applied here, the accelerating effect on the reaction is due primarily to secondary nucleation events catalyzed by the surfaces of the added fibrils, rather than due to elongation processes induced by the added reactive fibril ends, a conclusion verified in Fig. 2c-d where secondary nucleation is inhibited.
Kinetic rate laws
Following our previous analysis19,26, the generation of fibril mass, M, when both primary and secondary nucleation events occur is described by the integrated rate law:
| (1) |
where two particular combinations of the rate constants for primary nucleation (kn), elongation (k+), and fibril-catalysed secondary nucleation (k2) define much of the macroscopic behaviour; these parameters are related to the rate of formation of new aggregates through primary pathways and through secondary pathways . Indeed, Eq. 1 depends on the rate constants through these two parameters, λ and κ, alone since , C± = ±λ2/(2κ2), and . The initial concentration of soluble monomers is denoted m(0) and the exponents describing the dependencies of the primary and secondary pathways on the monomer concentration are given as nc and n2 respectively.
The effect of an inhibitor can in the first instance be semi-empirically captured by altering the microscopic rate constants for primary nucleation (kn), elongation (k+) and secondary nucleation (k2) in the integrated rate law Eq. 1. Perturbing the different microscopic events results in characteristic changes in the shape of the macroscopic reaction profile that can be used to identify the mechanism of action of an inhibitor (Fig. 1a-c). This approach is exact for complete inhibition of a particular process, as shown for secondary nucleation in Fig. 1c (green dashed line). Using this approach (Fig. 1a-c and Supplementary Fig. 2), we were able to identify a Brichos domain which affects specifically the secondary nucleation rate.
In order to further quantify the inhibitory effect of the Brichos domain at chaperone concentrations where inhibition of secondary nucleation is not complete and predict the kinetic profiles at intermediate chaperone concentrations (thin dotted lines, Fig. 1c), we then used a first-principles approach by introducing into the reaction scheme the reversible binding of the chaperone along the fibril surface. Such binding decreases the surface of the fibrils available to catalyse the oligomer formation, therefore reducing the secondary nucleation rate. A complete derivation of the rate equations including this Langmuir-type adsorption mechanism is provided in Supplementary Note.
Transmission electron microscopy
Samples from the kinetic experiments were taken at the end-point of the experiment (ThT maximum) for reactions involving Aβ42 alone and reactions involving Aβ42 together with 0.7 equivalents of Brichos. Aliquots of 2 μL volume were loaded on nickel-coated grids, and any excess sample was removed. The grids were placed on a drop of 1% BSA in TBS, incubated for 30 min at room temperature, and then washed three times for 10 min with TBS. The grids were then placed on drops of rabbit anti-C-terminal proSP-C (kindly provided by Prof. T. Weaver, Cincinnati, USA) that was diluted 1:200 in TBS, and incubated over night at +4°C. After washing five times for 10 min, the grids were placed on goat anti-rabbit IgG coupled to 10-nm gold particles (kindly provided by Prof. G. Westermark, Uppsala University, Sweden) that was diluted 1:40 in TBS, and incubated for 2 h at room temperature. The grids were then washed five times as before, followed by negatively staining with 2% uranyl acetate in 50% ethanol, and the immuno-labeled fibrils were examined using a Hitachi H7100 TEM operated at 75 kV. The lengths of approximately 150 fibrils across multiple images were measured using Digital Micrograph software. For the case of Aβ42 in the presence of Brichos, many fibrils were so long as to have one end outside the field of view of our images, and were excluded from the analysis. By contrast, several of the fibrils formed in the absence of Brichos were too short and tangled with one another to be measured accurately and were excluded. The reported ratio of the average length of fibrils formed in the presence of Brichos to that in the absence of Brichos is hence to be taken as a lower bound for its exact value.
Cryogenic transmission electron microscopy
To ensure a stable temperature and to avoid the loss of solution during sample preparation a controlled environment vitrification system was used. Samples were prepared as thin liquid films (<300 nm thick) on glow-discharge treated lacey carbon film coated copper grids and plunged into liquid ethane at -180°C. In this way the original microstructures are preserved as component segmentation and rearrangement is avoided in addition to water crystallisation as the samples are vitrified. Samples were stored under liquid N2 until measured and then transferred using an Oxford CT3500 cryoholder and its workstation into the electron microscope (Philips CM120 BioTWIN Cryo) equipped with a post-column energy filter (Gatan GIF100). An acceleration voltage of 120kV was used and images were recorded digitally with a CCD camera under low electron dose conditions.
Surface plasmon resonance assays
Aβ42 monomers or fibrils were immobilized on C1 sensorchips (GE Healthcare) using amine coupling. A fresh mixture of 0.05 M NHS and 0.2 M EDC was added to the sensorchip surface for activation, followed by washing and incubation with 1 μM Aβ42 monomers or fibrils outside of the instrument at room temperature for 0.5 and 2.0h, respectively, and finally blocking by ethanolamine. Blank channels for negative controls were prepared by omitting protein in the coupling step. Binding of Brichos was measured using a BIACORE 3000 instrument by injecting 100μL of a 1 μM solution of proSP-C Brichos, followed by buffer flow to monitor dissociation. The flow rate was 10μL/min throughout the experiment.
Monomer and oligomer fraction analysis using monoclonal antibodies
Aggregation was monitored by ThT fluorescence for samples of 5 μM Aβ42 with 50 nM of pre-formed seeds with and without 5 μM pro-SPC Brichos in 20 mM sodium phosphate buffer, pH8, with 200 μM EDTA and 0.02% NaN3 with 6 μM ThT. Samples were taken from reactions without Brichos after 15 min, and from reactions with Brichos after 15, 30, 45 and 60 min (Fig. 4). The samples were collected from the wells and immediately injected into a 1 × 30 cm Superdex 75 column. Eluted fractions (1 mL per fraction) were pooled in three samples: monomers (elution volume between 11.5 and 15 ml), small oligomers (elution volume between 8 and 11.5 ml) and large oligomers (elution volume between 5 and 8 ml). According to the calibration curve of the SEC column performed with globular proteins, small and large oligomers correspond to species with molecular weights in the range 14-65 kDa (3-14 mers) and 66-90 kDa (15-20 mers) equivalent of globular proteins, respectively, with dimers eluting in the tail of the monomer peak. Pooled samples were pulled through a PVDF membrane (200 nm) using a vacuum device for semi-quantitative analysis using a 6E10 primary mouse antibody (Signet SIG-39300) and goat-anti-mouse secondary antibody (Dako) conjugated with an IR probe and fluorescence detection after excitation at 800 nm in a LI-COR Odyssey CLx instrument.
Electrophysiology
Experiments were carried out in accordance with an ethical permit granted to A.F. by Norra Stockholms Djurförsöksetiska Nämnd (N45/13). C57BL/6 mice of either sex (postnatal days 14-23, supplied from Charles River, Germany) were used in all experiments. Immediately after slicing sections were transferred to a submerged incubation chamber containing standard ACSF (artificial cerebrospinal fluid): 124 mM NaCl, 30 mM NaHCO3, 10 mM Glucose, 1.25 mM NaH2PO4, 3.5 mM KCl, 1.5 MgCl2, 1.5 mM CaCl2. The chamber was held at 34°C for at least 20 minutes after dissection. It was subsequently allowed to cool to ambient room temperature (19-22°C) for a minimum of 40 minutes. Aβ42 monomer was at 5-10 μM isolated by gel filtration in 20 mM sodium phosphate buffer pH 8.0 followed by filtration through a 200 nm sterile filter and stored on ice until use. Fibrils were prepared by incubation of a 5 μM Aβ42 solution in a non-binding plate at 37°C for 1 h under quiescent condition. Peptides (final concentration 50 nM monomer or 50 nM monomer plus 0.3 nM fibrils) were added to the incubation solution 15 minutes before transferring slices to the interface-style recording chamber. While incubating slices were continuously supplied with carbogen gas (5% CO2, 95% O2) bubbled into the ACSF. Recordings were carried out in hippocampal area CA3 with borosilicate glass microelectrodes pulled to a resistance of 3-7MΩ. Local field potentials (LFP) were recorded at 34°C using microelectrodes filled with artificial cerebrospinal fluid placed in stratum pyramidale. LFP oscillations were elicited by applying kainic acid (100 nM) to the extracellular bath. The signals were sampled at 10 kHz, conditioned using a Hum Bug 50 Hz noise eliminator (Quest Scientific, North Vancouver, BC, Canada), software low-pass filtered at 1 kHz, digitized and stored using a Digidata 1322A and Clampex 9.6 software (Molecular Devices, CA, USA). Power spectral density plots (from 60 s long LFP recordings) were calculated in averaged Fourier segments of 8192 points using Axograph X (Kagi, Berkeley, CA, USA). The oscillation power was calculated by integrating the power spectral density between 20 and 80 Hz. For statistical analysis the Mann-Whitney U-test was used. Full experimental protocols are provided in the Supplementary Note.
Cell viability and cytotoxicity
Cell viability was measured using a MTS reagent, and cytotoxicity was measured using caspase-3/7 activity. These assays are commonly used, although the analysis is less straightforward compared to the electrophysiology experiments. Both assays suffer from the limitation that the single colorimetric readout represents the global effect of many possible cellular processes. For example, an increase of caspase signal is commonly associated with an increase of apoptosis activity of the cells, although caspase activation has been observed in the absence of apoptosis63. Assays were performed on SHSY-5Y human neuroblastoma cells cultured under standard conditions at 37°C in a humidified incubator with 5% CO2. Cells were seeded at a density of 25,000 per well in a white walled, clear bottomed 96 well plate and cultured for 24 hours in DMEM/10% FBS. The culture media was then replaced with pre-warmed phenol red free DMEM without serum into which the peptide samples or NaH2PO4 buffer were diluted 1:4. Peptide monomer was isolated by gel filtration in 20 mM sodium phosphate buffer at pH 8.0 followed by filtration through a 200 nm sterile filter and stored on ice until added to the cells. Seeds were prepared by placing a monomer solution in a 96-well non-binding plate (Corning 3881) at 37°C for 1 h. Completion of fibril formation was confirmed by ThT fluorescence on a withdrawn aliquot. Seeds were then diluted 1:100 into fresh monomer. The experiments for monomer and monomer plus seeds were repeated in the presence of Brichos. Buffer and Brichos were sterile filtrated (200 nm). The cells were cultured in the presence of the peptides or buffer (five-fold diluted in medium) for a further 24 hours before the cytotoxicity and viability assays were performed. Caspase-3/7 activity was measured using the Apo-ONE Homogeneous Caspase-3/7 assay (Promega, Southampton, UK). The fluorogenic caspase-3/7 substrate was diluted 1:100 in the lysis buffer provided and added to the cell medium at a 1:1 ratio. The reagent/cell mix was then incubated for 1 hour before measuring the fluorescence at ex. 480 nm/em. 520 nm in an Optima FluoStar plate reader. Cell viability was measured using the Cell Titer 96 Aqueous One MTS reagent from Promega. The MTS reagent was added to the cell culture medium and incubated with the cells at 37°C in a humidified incubator with 5% CO2 before the absorbance at 495 nm was measured in an Optima Fluostar plate reader. All values given for both assays are normalized relative to the buffer treated cells. Additional experimental details are provided in the Supplementary Note.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge financial support from the Schiff Foundation (SIAC), the Swedish Research Council (SL, JJ, JP) and its Linneaus Centre Organizing Molecular Matter (SL), the Crafoord Foundation (SL), Alzheimerfonden (SL), the Frances and Augustus Newman Foundation (PA, TPJK), the European Research Council (TPJK, SL), the BBSRC (TPJK), nmc@LU (SL), the KID PhD studentship grant (FRK, LD), the Swedish Medical Association (AF), the Brain Fund (AF), the Strategic Program in Neurosciences at the Karolinska Institute (AF), the Swiss National Science Foundation (PA) and the Wellcome Trust (CMD, TPJK).
Footnotes
Supplementary Information is available in the online version of this paper.
REFERENCES
- 1.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat. Rev. Drug. Discov. 2010;9(3):237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426(6968):900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 5.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 7.Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15(6):384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 8.Sipe JD, et al. Amyloid fibril protein nomenclature: 2012 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2012;19(4):167–170. doi: 10.3109/13506129.2012.734345. [DOI] [PubMed] [Google Scholar]
- 9.Goldschmidt L, Tenga PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U S A. 2010;107(8):3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18(10):1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Koo EH, Lansbury PT, Jr., Kelly JW. Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. U S A. 1999;96(18):9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416(6880):507–11. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 13.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 14.Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 15.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2(10):e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarrett JT, Lansbury PT., Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73(6):1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 17.Knowles TPJ, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 18.Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149(5):1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen SIA, et al. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U S A. 2013;110(24):9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hellstrand E, Boland B, Walsh DM, Linse S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chemical Neuroscience. 2010;1:13–18. doi: 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011;7(9):602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serio TR, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289(5483):1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 23.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 2011;18(3):328–336. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oosawa F, Asakura S. Thermodynamics of the Polymerization of Protein. Academic Press; 1975. [Google Scholar]
- 25.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J. Mol. Biol. 1985;183(4):611–631. doi: 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- 26.Cohen SIA, Vendruscolo M, Dobson CM, Knowles TPJ. Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J. Chem. Phys. 2011;135(6):065106. doi: 10.1063/1.3608917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruschak AM, Miranker AD. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. U S A. 2007;104(30):12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arosio P, Cukalevski R, Frohm B, Knowles TPJ, Linse S. Quantification of the concentration of abeta42 propagons during the lag phase by an amyloid chain reaction assay. J. Am. Chem. Soc. 2014;136(1):219–225. doi: 10.1021/ja408765u. [DOI] [PubMed] [Google Scholar]
- 29.Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, Dietler G. Novel mechanistic insight into the molecular basis of amyloid polymorphism and secondary nucleation during amyloid formation. J. Mol. Biol. 2013;425(10):1765–1781. doi: 10.1016/j.jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell. 2014;7(1):261–268. doi: 10.1016/j.celrep.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 31.Willander H, et al. Brichos domains efficiently delay fibrillation of amyloid beta-peptide. J. Biol. Chem. 2012;287(37):31608–31617. doi: 10.1074/jbc.M112.393157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381(6583):571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 33.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 34.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 35.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 36.Doyle SM, Genest O, Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol. 2013;14(10):617–629. doi: 10.1038/nrm3660. [DOI] [PubMed] [Google Scholar]
- 37.Auluck PK, Chan HYE, Trojanowski JQ, Lee VMY, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a drosophila model for Parkinson’s disease. Science. 2002;295(5556):865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 38.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292(5521):1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 39.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc. Natl. Acad. Sci. U S A. 2002;99(Suppl 4):16412–16418. doi: 10.1073/pnas.182426899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaffar G, et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol. Cell. 2004;15(1):95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 42.Sánchez-Pulido L, Devosi D, Valencia A. Brichos: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem. Sci. 2002;27(7):329–332. doi: 10.1016/s0968-0004(02)02134-5. [DOI] [PubMed] [Google Scholar]
- 43.Hedlund J, Johansson J, Persson B. Brichos - a superfamily of multidomain proteins with diverse functions. BMC Res. Notes. 2009;2:180. doi: 10.1186/1756-0500-2-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knight SD, Presto J, Linse S, Johansson J. The brichos domain, amyloid fibril formation, and their relationship. Biochemistry. 2013;52(43):7523–7531. doi: 10.1021/bi400908x. [DOI] [PubMed] [Google Scholar]
- 45.Willander H, Hermansson E, Johansson J, Presto J. Brichos domain associated with lung fibrosis, dementia and cancer-a chaperone that prevents amyloid fibril formation? FEBS J. 2011;278(20):3893–3904. doi: 10.1111/j.1742-4658.2011.08209.x. [DOI] [PubMed] [Google Scholar]
- 46.Nerelius C, Gustafsson M, Nordling K, Larsson A, Johansson J. Anti-amyloid activity of the c-terminal domain of prosp-c against amyloid beta-peptide and medin. Biochemistry. 2009;48(17):3778–3786. doi: 10.1021/bi900135c. [DOI] [PubMed] [Google Scholar]
- 47.Coomaraswamy J, et al. Modeling familial danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A. 2010;107(17):7969–7974. doi: 10.1073/pnas.1001056107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J, et al. Bri2 (itm2b) inhibits abeta deposition in vivo. J.Neurosci. 2008;28(23):6030–6036. doi: 10.1523/JNEUROSCI.0891-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vidal R, et al. A stop-codon mutation in the bri gene associated with familial British dementia. Nature. 1999;399(6738):776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 50.Willander H, et al. High-resolution structure of a brichos domain and its implications for anti-amyloid chaperone activity on lung surfactant protein c. Proc. Natl. Acad. Sci. U S A. 2012;109(7):2325–2329. doi: 10.1073/pnas.1114740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen SIA, et al. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J. Chem. Phys. 2011;135(6):065105. doi: 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schnitzler A, Gross J. Normal and pathological oscillatory communication in the brain. Nat. Rev. Neurosci. 2005;6(4):285–296. doi: 10.1038/nrn1650. [DOI] [PubMed] [Google Scholar]
- 53.Knowles TPJ, et al. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318(5858):1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 54.Lue LF, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J.Pathol. 1999;155(3):853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLean CA, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999;46(6):860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 56.Näslund J, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283(12):1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 1999;158(2):328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- 58.Walsh DM, et al. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 2009;276(5):1266–1281. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waltersson Y, Linse S, Brodin P, Grundström T. Mutational effects on the cooperativity of Ca2 binding in calmodulin. Biochemistry. 1993;32(31):7866–7871. doi: 10.1021/bi00082a005. [DOI] [PubMed] [Google Scholar]
- 60.Cedervall T, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. U S A. 2007;104(7):2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johansson C, et al. Biophysical studies of engineered mutant proteins based on calbindin D9k modified in the pseudo EF-hand. Eur. J. Biochem. 1990;187(2):455–460. doi: 10.1111/j.1432-1033.1990.tb15325.x. [DOI] [PubMed] [Google Scholar]
- 62.Lindman S, et al. Salting the charged surface: pH and salt dependence of protein G B1 stability. Biophys. J. 2006;90(8):2911–2921. doi: 10.1529/biophysj.105.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burguillos MA, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472(7343):319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.