

Abstract
Acorn squash (Cucurbita pepo) is an iconic fall vegetable in the United States, known for its unique fruit shape and also prized for its culinary properties. Little is known about the metabolism that underlies the development of fruit quality attributes such as color, sweetness, texture and nutritional qualities in acorn squash, or any other winter squash grown worldwide. To provide insight into winter squash fruit and seed development and add to the genomic resources in the Cucurbita genus, RNA sequencing was used to generate an acorn squash fruit and seed transcriptome from the cultivar Sweet REBA at critical points throughout fruit development. 141 838 600 high-quality paired-end Illumina reads were assembled into 55 949 unigenes. 85% of unigenes with predicted open reading frames had homology with previously identified genes and over 62% could be functionally annotated. Comparison with the watermelon and cucumber genomes provided confirmation that the unigenes are full-length and comprehensive, covering an average of 90% of the coding sequence of their homologs and 72% of the cucumber and watermelon exomes. Key candidate genes associated with carotenoid and carbohydrate metabolism were identified toward a resource for winter squash fruit quality trait dissection. This transcriptome represents a major advance in C. pepo genomics, providing significant new sequence information and revealing the repertoire of genes expressed throughout winter squash fruit and seed development. Future studies on the genetic basis of fruit quality and future breeding efforts will be enhanced by tools and insights developed from this resource.
Introduction
Winter squash (Cucurbita spp.) is an important vegetable crop known for its nutritional content and long storage life. It is eaten as a fresh market crop, processed to make frozen squash and canned pumpkin, and the seeds are consumed as a snack food and pressed for edible oil. Winter squash is an excellent source of nutrients, including carotenoids, ascorbic acid and vitamin C.1 It is most notable as a source of carotenoids, primarily β-carotene and lutein,2 which are beneficial as a pro-vitamin A compound and for eye health respectively.3 Additionally, squash seeds provide protein, lipids, tocopherols and phytosterols.4–6 Seeds were likely one of the oldest food uses of this crop, as many of the wild accessions have bitter, inedible pericarps.7 Winter squash is a unique source of these nutrients because it can be stored and consumed for many months past the growing season.
To increase the consumption of winter squash, culinary quality is a vital target of improvement through breeding. Fruit quality is determined by several characteristics including color, nutrient content, sweetness, flavor and texture. Color and nutritional value are dually controlled by carotenoid content,8,9 with a deeper yellow or orange color and a higher nutrient level seen as desirable. Sweetness is associated with sugar content, and higher sweetness is desirable for consumer acceptance and contributes to improved squash flavor.10 Winter squash fruit texture is highly correlated with starch content and dry matter, with higher starch content linked to a superior texture.10–12 These quality traits are quantitative and have a complex inheritance,13,14 yet are controlled by well-understood metabolic pathways. Understanding the genetic basis of these quality traits will allow for more insight into the breeding of squash for improved fruit quality.
Cucurbita species have a small but growing number of genomic resources that can be employed when studying these quality traits. Other members of the Cucurbitaceae family, namely, cucumber, watermelon and melon, have more extensive genomic resources, including sequenced genomes and many molecular markers.15–21 Cucurbita species are diploid (2n=2x=40) and have a genome size of approximately 500 Mb.22 Their small and numerous chromosomes are a remnant of a polyploidy event with persisting duplications23 that complicates their molecular breeding. Nevertheless, several genomic maps have been generated for squash24–26 including that of Esteras et al.,27 which was the first squash map to use single nucleotide polymorphism markers. Another major source of genomic information is a C. pepo transcriptome,28 comprised of 49 610 unigenes derived from flower, leaf and root tissue. More than 60% of the unigenes were annotated, aiding in the potential identification of genes of interest in the squash genome. Furthermore, the transcriptome was used to identify more than 10 000 potential simple sequence repeats and single nucleotide polymorphisms that could be used to generate molecular markers. In addition, a C. moschata transcriptome was sequenced from leaf, stem, and shoot tissue and assembled into 62 480 unigenes.29 Sixty-eight percent of the unigenes were annotated and almost 8000 potential simple sequence repeats were identified.
Transcriptome generation through RNA sequencing is a technology that can be used in the dissection of complex traits; fruit transcriptome analysis has been used to successfully identify transcripts involved in early fruit development of cucumber.30,31 Assembled transcriptomes also provide valuable sequence resources in species lacking a sequenced genome. However, one limitation of RNA-sequencing data is that it is specific to the plant line, tissue, developmental stage and physiological condition sequenced. For this reason, it is essential to use transcriptome data relevant to the experimental question of interest. The first C. pepo transcriptome,28 assembled from summer squash root, leaf and flower tissue, constituted a huge advance in C. pepo genomics. To study aspects of fruit quality, however, knowledge of fruit-specific genes is important. Fruit development and ripening is a process often associated with the expression of suites of genes at defined stages.32,33 A comprehensive squash fruit transcriptome must therefore sample these specific tissues and stages.
To create a resource for the study of fruit ripening and development in winter squash, in this study, we sequenced and assembled the first Cucurbita pepo fruit and seed transcriptome from the acorn squash cultivar Sweet REBA. Acorn squash is a popular winter squash known for its unique ribbed shape. Of the diverse squash types in C. pepo, the best-characterized Cucurbita species, acorn squash is the longest-storing and has the highest fruit quality. ‘Sweet REBA’ in particular is an inbred line that is agriculturally successful as a cultivar, making it an ideal candidate for generating a transcriptome that can both build off of the existing C. pepo knowledge and be directly applicable to squash breeding. The transcriptome was generated from five different time points throughout fruit development and derived from both mesocarp and seed tissues. The raw sequence data were assembled into 55 949 unigenes, which were then functionally annotated and compared to the cucumber and watermelon genomes. In addition, putative fruit quality-related transcripts were identified.
Materials and methods
Plant material
‘Sweet REBA’, an acorn squash inbred cultivar (M. Jahn, Cornell University), was grown in the Guterman greenhouse facility at Cornell University, Ithaca, NY, USA using standard horticultural practices. Flowers were manually self-pollinated and three representative, randomly-selected fruits per time point were collected at 5, 10, 15, 20 and 40 days after pollination. Time points were selected to represent a range of squash fruit and seed developmental stages, including early fruit and seed development (5 and 10 days), seed coat formation (15 and 20 days) and fruit ripening, up until the typical harvest date (40 days).34 Fruit mesocarp and seed tissues were dissected, flash frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
RNA extraction and sequencing
RNA was extracted from individual fruit and seed samples using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). Equal amounts of total RNA were pooled together into three independent biological samples, each consisting of RNA from both fruit and seed tissue at all five time points. Library preparation and sequencing were performed by the Genomics Core Facility, Cornell University. The sequencing library was prepared using the TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, CA, USA) and paired-end reads were sequenced from the three independent samples on one lane of an Illumina HiSeq 2000.
Sequence processing and transcriptome assembly
Sequencing reads were filtered by removing those flagged as low quality by the Illumina software, then adapter sequences and low-quality bases were trimmed from the end of reads using the software Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic). Data quality was assessed using the program FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and all of the sequence data were combined for transcriptome assembly using the program Trinity.35 The resulting unigenes were further assembled using the program iAssembler36 and then screened using the program DeconSeq37 to remove any unigenes of bacterial, viral or mammalian origin. DeconSeq threshold values for removal were greater than or equal to 95% sequence identity and sequence coverage.
Transcriptome refinement and analysis
A BLAST search38 (July 2012) was performed to compare unigenes to the NCBI non-redundant protein database with a significance level of 1.0×10−6 using the blastx algorithm implemented by the program Blast2GO.39 Unigene expression estimates, measured as FPKM values, were calculated with the program RSEM,40 which aligned the raw reads to the assembled unigenes. Trinity35 was used to identify unigenes containing predicted open reading frames. These results were then applied to refine the set of unigenes. As described in the Trinity developers’ recommendations, the union of all transcripts with (i) significant homology to a known protein as determined through a blastx search; (ii) a predicted open reading frame; and/or (iii) an expression level, measured by the FPKM value, of greater than or equal to one were combined to remove likely assembly artifacts from the set of unigenes. To reduce the unigene set to representative transcripts, BLAST38 was used to query the 99 839 well-supported contigs against themselves. Perl scripts were written to merge self-alignments into groups likely originating from the same gene model.
Candidate coding regions within the transcripts were detected by ESTscan41 with the Arabidopsis thaliana scoring matrix. The resulting subset of transcripts was screened against the SWISS-PROT and TrEMBL42 protein databases using BLASTx with a cutoff of 1.0×10−20 to assign putative functions to unigenes. Predicted peptides were analyzed using InterProScan43 to identify functional protein domains and assign Gene Ontology (GO) terms. SWISS-PROT manual curation of GO terms was also utilized to assign terms.44 Transcripts were queried against the watermelon genome assembly v1 coding sequence (watermelon_v1.cds)21 and the cucumber genome assembly v2 coding sequence (cucumber_v2.cds)15 using BLASTn. Unigenes with a resulting e-value of less than 1.0×10−20 were considered to have a significant match. The 55 949 fruit and seed transcripts were compared to the Blanca et al.28 root, leaf and flower transcriptome by performing a reciprocal blast search of one against the other using BLASTn with a significance level of 1.0×10−25. Bioconductor, using the methods of Sanchez, Salicru and Ocana,45 was used to determine if there was a statistical difference in GO term representation between the unigenes unique to each transcriptome.46
Results and discussion
Transcriptome sequencing and assembly
RNA was extracted from fruit mesocarp and seed tissues of self-pollinated ‘Sweet REBA’ acorn squash fruit at 5, 10, 15, 20 and 40 days after pollination (Figure 1). All samples were pooled to create three biological replicates of a comprehensive fruit and seed sequencing library and paired-end reads were sequenced on one lane of an Illumina HiSeq 2000, resulting in a total of 156 540 465 pairs of 100 base pair (bp) reads (Table 1). After removing low-quality reads and trimming both adapter sequences and low-quality bases from the ends of reads, 141 838 600 paired reads remained, with an average length of 98.46 bp and an average quality score of 36.98, constituting a total of almost 28 Gbp of sequence (reads available in the NCBI Sequence Read Archive repository, run accession numbers SRR747836, SRR747920 and SRR747922). All sequence data were combined and assembled using the program Trinity35 to yield 161 780 contigs. These contigs were further assembled using the program iAssembler,36 which resulted in a total of 150 044 contigs. The contigs were then screened using the program DeconSeq37 to remove 2081 bacterial and mammalian sequence artifacts, leaving 147 963 unigenes with an average length of 1311 bp.
Figure 1.

‘Sweet REBA’ acorn squash fruit at five developmental time points. Self-pollinated fruit were harvested at 5, 10, 15, 20 and 40 days after pollinations. Photos are of the interior and exterior of representative fruit at each time point.
Table 1. Summary of transcriptome sequencing data.
| Reads | Total number of paired reads | Average read length (bp) | Total sequence length (bp) | Average quality score |
|---|---|---|---|---|
| Raw reads | 156 540 465 | 100 | 31 308 093 000 | 34.78 |
| High-quality reads | 141 838 600 | 98.5 | 27 929 628 643 | 36.98 |
The unigenes were filtered to remove any sequences likely resulting from possible sequencing or assembly errors by only retaining unigenes that had homology to a known protein, a predicted open reading frame and/or a FPKM expression estimate value of one or more. There were 83 221 unigenes with homology to a known protein, 57 598 with a predicted open reading frame, and 52 374 with an FPKM expression estimate value of greater than or equal to one. These overlapping subsets of unigenes were combined into a set of 99 839 unigenes, which were then clustered using self-BLAST to group alternatively-spliced transcripts originating from the same gene. An identity of 99.9% was determined to be the optimal value for merging transcripts likely transcribed from the same gene without merging putative paralogous genes. The longest unigene was chosen to represent each cluster in the final transcriptome, resulting in a total of 55 949 unigenes (Unigene sequences available in Supplementary File S1. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GBZI00000000. The version described in this paper is the first version, GBZI01000000.).
Transcriptome analysis, annotation and functional characterization
The final transcriptome consisted of 55 949 unigenes. The unigenes ranged in length from 201 bp to 17 024 bp, with an average length of 1315 bp, a median length of 876 bp and a total sequence length of 73 559 618 bp. The high average and median unigene lengths suggested that we were successful in the assembly of many full-length transcripts, although 18 561 unigenes measured 500 bp or less and were likely only partial transcripts. A BLAST search confirmed that the longest unigenes were homologous to known genes of similar length, indicating that they were likely correctly assembled. To verify that unigenes shorter than 500 bp were indeed mainly partial transcripts, the unigene coverage of matches was considered. A total of 5921 short unigenes had significant matches to sequences in the NCBI nr database. Of these matches, the average percent of coverage of the subject sequence was 24%. Only 7.6% of these unigenes matched 50% or more of the best nr subject, suggesting that these were truncated transcripts. With the future addition of more sequence data, it is likely that many of these short unigenes will be assembled together into full-length unigenes, reducing the total number of unigenes in the transcriptome.
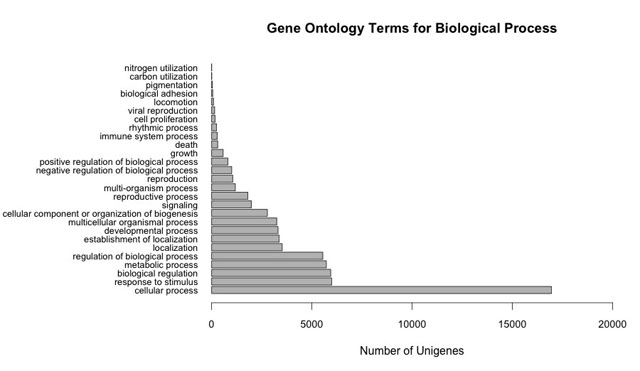
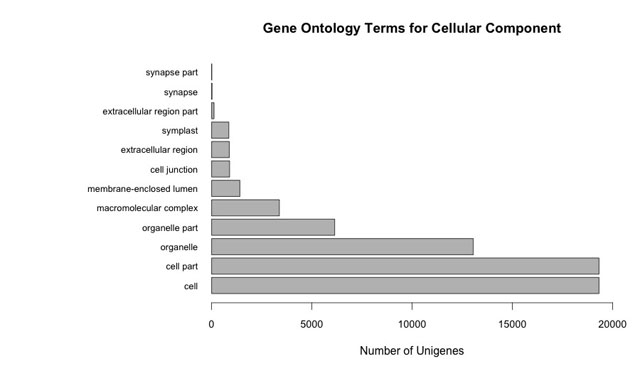
A total of 41 568 unigenes were predicted to have a single open reading frame and 18 unigenes were split into two open reading frames. This subset of unigenes was annotated based on homology to proteins found in SWISS-PROT and TrEMBL, with precedence given to SWISS-PROT annotations (for annotations, see Supplementary Table S1; for blast results, see Supplementary Table S2). The GenBank NR and TAIR10 databases were also queried, but results were not used in the final annotation. Of the four databases, the highest number of matches was found in GenBank NR (Table 2). Unigenes were translated into predicted proteins so that protein domains GO terms could be assigned to the predicted proteins, classifying them based on predicted involvement in biological processes, molecular functions, or cellular components.47 A total of 153 563 GO terms were assigned to 63% (26 049) of the predicted proteins (Supplementary Table S1). The number of GO terms assigned per unigene ranged from 1 to 68 for those proteins with GO annotations, with an average of six GO terms per unigene. These GO terms fell into three categories, with 40% assigned to biological process, 21% assigned to molecular function and 39% assigned to cellular component. A range of biological processes were represented, with large numbers of unigenes assigned to categories relevant to fruit and seed development, such as ‘seed development’, ‘embryo development’, ‘regulation of developmental process’, ‘macromolecule metabolic process’, ‘biosynthetic process’, ‘developmental growth’ and ‘multicellular organismal development’ (Supplementary Figures S1–S3).
Table 2. Results of squash fruit and seed transcriptome annotation based on homology.
| Database | Number of BLAST hits |
|---|---|
| SWISS-PROT | 22 934 (55%) |
| TrEMBL | 33 116 (80%) |
| TAIR10 | 30 590 (74%) |
| GenBank NR | 34 794 (84%) |
All unigenes with an open reading frame were blasted against four databases and the number of unigenes with a significant hit are reported. Blast hits were filtered by e-value less than 1.0×10−20.
Analysis of alternative splicing
Approximately 44% of unigenes were collapsed by self-blast while other overlap-based means of collapsing failed, suggesting that the variants may be alternative isoforms or mis-assemblies. To assess the structure of collapsed unigenes, the clusters were further analyzed; the average number of members in a cluster was 2.5±6.8, ranging from 1 member to 200 members (Supplementary Table S1). The clusters with the largest number of constituents were predicted to be DNA glycosylase and MYB genes. Both of these classes of genes are known to undergo alternative splicing,48,49 further reinforcing the notion that the isoforms identified were alternatively spliced transcripts and supporting our designation of these as such and their subsequent representation by the longest contig in each cluster.
Comparison with cucumber and watermelon genomes
A total of 31 307 unigenes had significant hits to 18 381 cucumber coding sequences, representing approximately 72% of the predicted cucumber coding sequences (Supplementary Table S1). The average coding sequence coverage by the homologous unigene was 90% for cucumber coding sequence with hits. A total of 4573 unigenes with matches to a cucumber coding sequence were shorter than 500 base pairs and 4351 of these covered less than 90% of their respective match, suggesting that these unigenes are likely partial transcripts. Similarly, 33 588 unigenes significantly matched 16 903 watermelon coding sequences, representing 72% of the watermelon predicted coding sequences (Supplementary Table S1). The average coverage of watermelon coding sequence by squash matches was 91%. A total of 4868 out of 5110 unigenes shorter than 500 bp had significant matches that covered less than 90% of their respective watermelon coding sequence match. Overall, these comparisons indicated that the majority of our unigenes were fully assembled, with the exception of those shorter than 500 bp, and that the transcriptome as a whole was comprehensive.
Comparison with root, leaf and flower C. pepo transcriptome
The C. pepo fruit transcriptome was compared with the previously published C. pepo transcriptome sequenced from root, leaf and flower tissue.28 The root, leaf and flower transcriptome was assembled from sequences derived from a scallop-type and a zucchini-type squash and consisted of 49 610 unigenes with an average length of 626 bp.28 The fruit and seed transcriptome had a similar number of unigenes as the previous transcriptome, though it had a longer average unigene length. The greater sequencing depth of the fruit and seed transcriptome allowed by the use of Illumina sequencing technology may have enabled the assembly of more full-length transcripts.
To more closely compare the two C. pepo transcriptomes, we conducted a reciprocal BLAST search38 in which each transcriptome was compared to the other transcriptome using the blastn algorithm with a significance threshold of 1.0×10−25 (Table 3). When the root, leaf and flower transcriptome was compared to the fruit and seed transcriptome, 96% (47 433) of the root, leaf, and flower unigenes were significantly similar to one or more of the unigenes in the fruit and seed transcriptome, with 4% not significantly similar. The matches corresponded to 22 749 fruit and seed unigenes, suggesting redundancy in the root, leaf and flower transcriptome. From the reciprocal BLAST search, we found that 61% (33 988) of the fruit and seed unigenes were significantly similar to one or more of the unigenes in the root, leaf and flower transcriptome, with 39% not significantly similar (unigenes not significantly similar are noted in Supplementary Table S1). These matches were to 22 812 of the root, leaf and flower transcripts. Additionally, 19 831 unigenes had the same reciprocal match in both blast results, suggesting that these could be orthologous unigenes between zucchini and acorn squash.
Table 3. Comparison of fruit and seed transcriptome with root, leaf and flower transcriptome.
| Transcriptome | Unigenes with no significant similarity to other transcriptome | Unigenes with significant similarity to other transcriptome |
|---|---|---|
| Fruit and seed | 21 961 (39%) | 33 988 (61%) |
| Root, leaf and flower | 2177 (4%) | 47 433 (96%) |
A reciprocal BLAST search was conducted to compare the two C. pepo transcriptomes. Each transcriptome was compared to the other transcriptome using the blastn algorithm with a significance threshold of 1.0×10−25.
To characterize the unique sequences in the fruit and seed transcriptome, their matches in the SWISS-PROT, TrEMBL, nr and TAIR10 sequence databases were assessed (e-value 1.0×10−20). Of the unique fruit and seed unigenes, 53% (11 671) had a predicted open reading frame and 38% (8315) had significant similarity to previously identified sequences. Therefore, our transcriptome sequencing identified more than 8000 novel unigenes in the fruit and seed transcriptome that had not been previously sequenced in the species. To examine gene enrichment in the two datasets, GO terms for the root, leaf and flower transcriptome were obtained from Blanca et al.28 A total of 606 unique root, leaf and flower unigenes and 5437 unique fruit and seed unigenes had at least one associated GO term. Statistically significant differences between the GO terms represented in the two samples were found for the level 2 biological process, molecular function and cellular component groups (P<0.05). The unique fruit and seed unigenes were enriched for development-related GO terms, as was expected for the fruit and seed tissues sampled at five different developmental time points. These GO terms included ‘cellular developmental process’, ‘anatomical structure morphogenesis’, ‘developmental maturation’, ‘developmental process involved in reproduction’, ‘seed development’, ‘embryo development’ and ‘cellular process involved in reproduction’, confirming that unigenes unique to fruit and seed development were represented in our transcriptome.
Identification of fruit quality genes
Several metabolic processes that are crucial for winter squash fruit quality occur during fruit development, including the synthesis of carotenoids, sucrose and starch. To demonstrate the future utility of this transcriptome for the study of fruit quality, we identified genes involved in these processes in our transcriptome. A BLASTn search38 was conducted to query all biosynthetic genes known to be involved in carotenoid, sugar and starch metabolism against the fruit and seed transcriptome to identify squash homologs of the genes. Squash unigenes homologous to functionally characterized genes were identified for 18 genes involved in carotenoid synthesis, storage, and degradation (Table 4), 18 genes involved in sucrose metabolism (Table 5) and 14 genes involved in starch metabolism (Table 6), demonstrating that this transcriptome fully captures gene expression related to these metabolic processes of interest.
Table 4. Carotenoid metabolism gene homologs found in fruit transcriptome.
| Carotenoid metabolism gene | C. pepo unigene homolog(s) |
|---|---|
| β-carotene hydroxylase | CP112262 |
| Carotenoid cleavage dioxygenase | CP134539 |
| Carotenoid isomerase | CP040968 |
| DOXP reductoisomerase | CP135419 |
| DOXP synthase | CP135924, CP060201, CP060202 |
| ε-hydroxylase | CP029795 |
| Geranylgeranyl hydrogenase | CP056527, CP056528, CP072215, CP072216 |
| GGPP synthase | CP135982, CP031308 |
| IPP isomerase | CP096828, CP006070, CP135789 |
| Lycopene β-cyclase | CP073600 |
| Lycopene ε-cyclase | CP120024 |
| Orange gene | CP135849 |
| Phytoene desaturase | CP120142, CP120144 |
| Phytoene synthase | CP005213, CP097163 |
| Violaxanthin de-epoxidasea | CP068838 |
| ζ-carotene desaturase | CP071571 |
| ζ-carotene isomerase | CP003737 |
| Zeaxanthin epoxidase | CP041038 |
Homologs to functionally characterized carotenoid metabolism genes were identified in the fruit and seed transcriptome through a BLAST search. The best candidate(s) for each gene are listed, identified by unigene number.
Homology found to genes annotated with only putative function.
Table 5. Sugar metabolism gene homologs found in fruit transcriptome.
| Simple sugar metabolism gene | C. pepo unigene homolog(s) |
|---|---|
| Acid α-galactosidase | CP031132, CP041982 |
| Acid invertase | CP078022, CP039349 |
| Alkaline α-galactosidase | CP135438, CP082553, CP082552, CP097985 |
| Fructokinase | CP138498, CP138398 |
| Galactokinase | CP100962, CP100963 |
| Galactose-1-phosphate uridyltransferase | CP115252, CP115240 |
| Hexokinase | CP081918 |
| Hexose transporter | CP139122, CP032998, CP047946 |
| Neutral invertase | CP120038, CP120034, CP140685 |
| Phosphofructokinase | CP076642 |
| Phosphoglucomutase | CP124387, CP005960 |
| Sucrose synthase | CP039490, CP036200, CP033875 |
| Sucrose transporter | CP123643, CP123649, CP008293, CP009234 |
| Sucrose-phosphate phosphatase | CP033610, CP078010 |
| Sucrose-phosphate synthase | CP125836 |
| UDP-glucose epimerase | CP083217, CP083218, CP040136 |
| UDP-glucose/galactose pyrophosphorylase | CP134768 |
| UDP-glucose pyrophosphorylase | CP062357 |
Homologs to functionally characterized simple sugar metabolism genes were identified in the fruit and seed transcriptome through a BLAST search. The best candidate(s) for each gene are listed, identified by unigene number.
Table 6. Starch metabolism gene homologs found in fruit transcriptome.
| Starch metabolism gene | C. pepo unigene homolog(s) |
|---|---|
| ADP-glucose pyrophosphorylase L | CP101035, CP101034 |
| ADP-glucose pyrophosphorylase S | CP126587 |
| α-amylase | CP135677 |
| α-glucosidase | CP144486, CP116493, CP116492, CP006281 |
| Amyloplastidial ATP/ADP translocator | CP098764, CP001903, CP098765, CP001904 |
| β-amylase | CP070598, CP070601, CP070602, CP074740 |
| Cell wall invertase | CP052558 |
| debranching enzyme | CP039826 |
| Glucose phosphate transporter | CP086414, CP081420, CP034210, CP034209, CP037833 |
| Phosphoglucose isomerase | CP109433 |
| Starch phosphorylase | CP033924, CP004861, CP136313 |
| Starch synthase—granule bound | CP118399, CP118397, CP118398, CP118396 |
| Starch synthase—soluble | CP113496, CP113492 |
| Starch-branching enzyme | CP033816, CP120408, CP120407 |
Homologs to functionally characterized starch metabolism genes were identified in the fruit and seed transcriptome through a BLAST search. The best candidate(s) for each gene are listed, identified by unigene number.
Structural genes known to control flux or act at pathway branch-points in carotenoid formation will be informative in future analyses of color and nutrient concentration in developing squash fruit. The carotenoid unigenes identified include the gene DOXP synthase, whose product is predicted to synthesize a regulatory step of carotenoid biosynthesis in tomato fruit.50 DOXP synthase is the first enzyme in the DOXP pathway that produces isopentenyl diphosphate, the precursor for carotenoids.51 The unigene for phytoene synthase, which performs the first committed step in carotenoid synthesis and has been shown to be rate-limiting in marigolds, canola and tomato,51 was also identified. An important branch-point in the squash carotenoid pathway involves lycopene ε-cyclase and lycopene β-cyclase, also identified in the transcriptome, which both encode enzymes that act on lycopene to direct flux towards either lutein or β-carotene,14 the two primary carotenoids in C. pepo.2 Natural variation in lycopene ε-cyclase in maize influences the partition of carotenoids between the two branches of the pathway,52 so these genes are also likely to impact the ratio of lutein to β-carotene in squash. The orange gene (Or) was also identified, which encodes an enzyme that is important for regulating the accumulation of carotenoids through the differentiation of chromoplasts and enables high levels of carotenoid accumulation in cauliflower and potato.53
The unigenes also included key genes involved in carbohydrate metabolism. Sucrose-phosphate synthase and sucrose-phosphate phosphatase were identified, which encode enzymes that act sequentially to synthesize sucrose from UDP-glucose and fructose 6-phosphate. Sucrose-phosphate synthase is a regulatory step for sucrose biosynthesis54 and its higher enzyme activity coincided with higher sucrose accumulation in pear55 and in muskmelon.56 Starch synthesis genes identified included ADP-glucose pyrophosphorylase and AATPT (amyloplastidial ATP/ADP translocator). ADP-glucose pyrophosphorylase synthesizes ADP-glucose from glucose-1-phosphate and is the first committed step in starch biosynthesis.57 AATPT transfers ATP and ADP between the cytosol and the amyloplast, providing the ATP needed for starch biosynthesis.57 Also identified was a unigene encoding α-amylase, which is the primary enzyme in squash that breaks down starch, yielding simple sugars that confer sweetness.58 Together, these genes form the core of hypotheses that will be tested in future comparative studies of winter squash quality.
Conclusion
In this study, we report the first Cucurbita pepo winter squash fruit and seed transcriptome, with more than 141 million high-quality paired-end sequencing reads compiled over five developmental time points and assembled into a final transcriptome of 55 949 unigenes. Approximately 85% of the unigenes with open reading frames shared homology with known proteins and 62% could be functionally annotated. This transcriptome was compared to the cucumber and watermelon genomes, as well as the previously sequenced C. pepo root, leaf and flower transcriptome, which confirmed that it was comprehensive, had a majority of full-length unigenes, and contained unigenes unique to fruit and seed development. This fruit and seed transcriptome represents a major contribution to C. pepo genomic resources, with more than 8000 C. pepo unigenes homologous to known genes that are new to the sequenced exome and will be useful in future genome annotation efforts. Further, the identification of likely candidates for carotenoid and carbohydrate metabolism genes suggests that this novel resource will enable further study of fruit quality and development to enhance future squash breeding efforts that seek to produce higher quality fruit with greater nutritional and culinary value.
Acknowledgments
We thank the Cornell University Biotechnology Resource Center and Giovanna Danies for bioinformatics support, Li Li and Zhangjun Fei for experimental design advice, Scott Anthony for providing plant care and Paige Roosa for comments on the manuscript. This research was supported through funds from Cornell University and the Vegetable Breeding Institute. Support for Lindsay Wyatt was provided by a Cornell University Presidential Life Sciences Fellowship, USDA National Needs Graduate Fellowship Competitive Grant No. 2008-38420-04755 from the National Institute of Food and Agriculture, and the Agriculture and Food Research Initiative Competitive Grant No. 2013-67011-21122 from the USDA National Institute of Food and Agriculture.
The authors declare no conflict of interest.
{kind=link}
{kind=link}
{kind=link}
References
- Ferriol M, Picó B. Pumpkin and winter squash. In: Prohens J, Nuez F (ed.) Vegetables I. Vol. 1. New York: Springer, 2008: 317–349. [Google Scholar]
- Azevedo-Meleiro CH, Rodriguez-Amaya DB. Qualitative and quantitative differences in carotenoid composition among Cucurbita moschata, Cucurbita maxima, and Cucurbita pepo. J Agric Food Chem 2007; 55: 4027–4033. [DOI] [PubMed] [Google Scholar]
- Cazzonelli CI, Pogson BJ. Source to sink: regulation of carotenoid biosynthesis in plants. Trends Plant Sci 2010; 15: 266–274. [DOI] [PubMed] [Google Scholar]
- Idouraine A, Kohlhepp EA, Weber CW, Warid WA, MartinezTellez JJ. Nutrient constituents from eight lines of naked seed squash (Cucurbita pepo L). J Agric Food Chem 1996; 44: 721–724. [Google Scholar]
- Murkovic M, Mulleder U, Neunteufl H. Carotenoid content in different varieties of pumpkins. J Food Compos Anal 2002; 15: 633–638. [Google Scholar]
- Yoshida H, Shougaki Y, Hirakawa Y, Tomiyama Y, Mizushina Y. Lipid classes, fatty acid composition and triacylglycerol molecular species in the kernels of pumpkin (Cucurbita spp.) seeds. J Sci Food Agric 2004; 84: 158–163. [Google Scholar]
- Robinson RW, Decker-Walters DS. Cucurbits. New York: CAB International, 1997. [Google Scholar]
- Gajewski M, Radzanowska J, Danilcenko H, Jariene E, Cerniauskiene J. Quality of pumpkin cultivars in relation to sensory characteristics. Not Bot Hort Agrobot Cluj 2008; 36: 73–79. [Google Scholar]
- Itle RA, Kabelka EA. Correlation between L*a*b* color space values and carotenoid content in pumpkins and squash (Cucurbita spp.). HortScience 2009; 44: 633–637. [Google Scholar]
- Cumarasamy R, Corrigan V, Hurst P, Bendall M. Cultivar differences in New Zealand “Kabocha” (buttercup squash, Cucurbita maxima). NZ J Crop Hort Sci 2002; 30: 197–208. [Google Scholar]
- Corrigan VK, Hurst PL, Potter JF. Winter squash (Cucurbita maxima) texture: sensory, chemical, and physical measures. NZ J Crop Hort Sci 2001; 29: 111–124. [Google Scholar]
- Hurst PL, Corrigan VK, Koolaard J. Genetic analysis of sweetness and textural attributes in winter squash (Cucurbita maxima). NZ J Crop Hort Sci 2006; 34: 359–367. [Google Scholar]
- Irving DE, Hurst PL, Ragg JS. Changes in carbohydrates and carbohydrate metabolizing enzymes during the development, maturation, and ripening of buttercup squash (Cucurbita maxima D ‘Delica’). J Am Soc Hort Sci 1997; 122: 310–314. [Google Scholar]
- Lu S, Li L. Carotenoid metabolism: biosynthesis, regulation, and beyond. J Integr Plant Biol 2008; 50: 778–785. [DOI] [PubMed] [Google Scholar]
- Huang SW, Li RQ, Zhang ZH et al. The genome of the cucumber, Cucumis sativus L. Nat Genet 2009; 41: 1275–1281. [DOI] [PubMed] [Google Scholar]
- Clepet C, Joobeur T, Zheng Y et al. Analysis of expressed sequence tags generated from full-length enriched cDNA libraries of melon. BMC Genomics 2011; 12: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A, Fergany M, Formisano G et al. A consensus linkage map for molecular markers and Quantitative Trait Loci associated with economically important traits in melon (Cucumis melo L.). BMC Plant Biol 2011; 11: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mas J, Benjak A, Sanseverino W et al. The genome of melon (Cucumis melo L.). Proc Natl Acad Sci USA 2012; 109: 11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Zhao H, Kou Q et al. A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS One 2012; 7: e29453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WW, Pan JS, He HL et al. Construction of a high density integrated genetic map for cucumber (Cucumis sativus L.). Theor Appl Genet 2012; 124: 249–259. [DOI] [PubMed] [Google Scholar]
- Guo SG, Zhang JG, Sun HH et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 2013; 45: 51–58. [DOI] [PubMed] [Google Scholar]
- Arumuganathan K, Earle E D. Nuclear DNA content of some important plant species. Plant Mol Biol Rep 1991; 9: 208–218. [Google Scholar]
- Weeden F. Isozyme studies indicate that the genus Cucurbita is an ancient tetraploid. Cucurbit Genet Cooper Rep 1984; 7: 84–85. [Google Scholar]
- Zraidi A, Stift G, Pachner M, Shojaeiyan A, Gong L, Lelley T. A consensus map for Cucurbita pepo. Mol Breeding 2007; 20: 375–388. [Google Scholar]
- Gong L, Stift G, Kofler R, Pachner M, Lelley T. Microsatellites for the genus Cucurbita and an SSR-based genetic linkage map of Cucurbita pepo L. Theor Appl Genet 2008; 117: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Pachner M, Kalai K, Lelley T. SSR-based genetic linkage map of Cucurbita moschata and its synteny with Cucurbita pepo. Genome 2008; 51: 878–887. [DOI] [PubMed] [Google Scholar]
- Esteras C, Gomez P, Monforte AJ et al. High-throughput SNP genotyping in Cucurbita pepo for map construction and quantitative trait loci mapping. BMC Genomics 2012; 13: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanca J, Canizares J, Roig C, Ziarsolo P, Nuez F, Pico B. Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics 2011; 12: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Luo S, Wang R et al. The first Illumina-based de novo transcriptome sequencing and analysis of pumpkin (Cucurbita moschata Duch.) and SSR marker development. Mol Breeding 2014; 34: 1437–1447. [Google Scholar]
- Ando K, Grumet R. Transcriptional profiling of rapidly growing cucumber fruit by 454-pyrosequencing analysis. J Am Soc Hort Sci 2010; 135: 291–302. [Google Scholar]
- Ando K, Carr KM, Grumet R. Transcriptome analyses of early cucumber fruit growth identifies distinct gene modules associated with phases of development. BMC Genomics 2012; 13: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenoni S, Ferrarini A, Giacomelli E et al. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol 2010; 152: 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmann J, Tohge T, Alba R et al. Combined transcription factor profiling, microarray analysis and metabolite profiling reveals the transcriptional control of metabolic shifts occurring during tomato fruit development. Plant J 2011; 68: 999–1013. [DOI] [PubMed] [Google Scholar]
- Loy JB. Morpho-physiological aspects of productivity and quality in squash and pumpkins (Cucurbita spp.). Crit Rev Plant Sci 2004; 23: 337–363. [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011; 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zhao LJ, Gao JP, Fei ZJ. iAssembler: a package for de novo assembly of Roche-454/Sanger transcriptome sequences. BMC Bioinformatics 2011; 12: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 2011; 6: e17288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011; 12: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseli C, Jongeneel C, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999; 1999: 138–148. [PubMed] [Google Scholar]
- Apweiler R, Martin MJ, O’Donovan C et al. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res 2013; 41: D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001; 17: 847–848. [DOI] [PubMed] [Google Scholar]
- Camon E, Magrane M, Barrell D et al. The gene ontology annotation (GOA) project: Implementation of GO in SWISS-PROT, TrEMBL, and InterPro. Genome Res 2003; 13: 662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, Salicru M, Ocana J. Statistical methods for the analysis of high-throughput data based on functional profiles derived from the Gene Ontology. J Stat Plann Infer 2007; 137: 3975–3989. [Google Scholar]
- Carlson M, Falcon S, Pages H, Li N. GO.db: a set of annotation maps describing the entire Gene Ontology. R package version 2.6.1 ed.
- Ashburner M, Ball CA, Blake JA et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TM, Gao M. Multiple forms of formamidopyrimidine-DNA glycosylase produced by alternative splicing in Arabidopsis thaliana. J Photochem Photobiol B-Biol 2001; 61: 87–93. [DOI] [PubMed] [Google Scholar]
- Li JG, Li XJ, Guo L et al. A subgroup of MYB transcription factor genes undergoes highly conserved alternative splicing in Arabidopsis and rice. J Exp Botany 2006; 57: 1263–1273. [DOI] [PubMed] [Google Scholar]
- Lois LM, Rodriguez-Concepcion M, Gallego F, Campos N, Boronat A. Carotenoid biosynthesis during tomato fruit development: regulatory role of 1-deoxy-d-xylulose 5-phosphate synthase. Plant J 2000; 22: 503–513. [DOI] [PubMed] [Google Scholar]
- Hirschberg J. Carotenoid biosynthesis in flowering plants. Curr Opin Plant Biol 2001; 4: 210–218. [DOI] [PubMed] [Google Scholar]
- Harjes CE, Rocheford TR, Bai L et al. Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science 2008; 319: 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, van Eck J, Zhou X et al. The cauliflower or gene encodes a DnaJ cysteine-rich domain-containing protein that mediates high levels of beta-carotene accumulation. Plant Cell 2006; 18: 3594–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber SC, Huber JL. Role and regulation of sucrose-phosphate synthase in higher plants. Annu Rev Plant Physiol Plant Mol Biol 1996; 47: 431–444. [DOI] [PubMed] [Google Scholar]
- Zhang HP, Wu JY, Qin GH et al. The role of sucrose-metabolizing enzymes in pear fruit that differ in sucrose accumulation. Acta Physiol Plant 2014; 36: 71–77. [Google Scholar]
- Hubbard NL, Huber SC, Pharr DM. Sucrose phosphate synthase and acid invertase as determinants of sucrose concentration in developing muskmelon (Cucumis melo L.) fruits. Plant Physiol 1989; 91: 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigenberger P, Stitt M, Fernie AR. Metabolic control analysis and regulation of the conversion of sucrose to starch in growing potato tubers. Plant Cell Environ 2004; 27: 655–673. [Google Scholar]
- Irving DE, Shingleton GJ, Hurst PL. Starch degradation in buttercup squash (I). J Am Soc Hort Sci 1999; 124: 587–590. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.