

SUMMARY
Myelodysplastic syndrome (MDS) risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres but whether telomere erosion directly induces MDS is unknown. Here, we provide the genetic evidence that telomere dysfunction-induced DNA damage drives classical MDS phenotypes and alters common myeloid progenitor (CMP) differentiation by repressing the expression of mRNA splicing/processing genes, including srsf2. RNA-Seq analyses of telomere dysfunctional CMP identified aberrantly spliced transcripts linked to pathways relevant to MDS pathogenesis such as genome stability, DNA repair, chromatin remodeling and histone modification, which are also enriched in mouse CMP haploinsufficient for srsf2 and in CD34+ CMML patient cells harboring srsf2 mutation. Together, our studies establish an intimate link across telomere biology, aberrant RNA splicing and myeloid progenitor differentiation.
INTRODUCTION
Advancing age is associated with the accumulation of DNA damage and attendant cellular checkpoint responses of apoptosis and senescence, as well as declining mitochondrial function and oxidative defense (Jaskelioff et al., 2011). Model systems have established that these DNA damage corollaries contribute to tissue degenerative phenotypes (Sahin and DePinho, 2012). A source of age-associated DNA damage signaling can derive from progressive telomere erosion and damage which appears to provide a reservoir of persistent DNA damage signaling in the context of aging cells (Chin et al., 1999; di Fagagna et al., 2003; Karlseder et al., 2002; Rudolph et al., 1999). These processes are particularly evident in tissues with high cell turnover rate, including the hematopoietic system (Lee et al., 1998; Rudolph et al., 1999). Indeed, accumulating evidence supports the view that DNA damage checkpoints activated by telomere erosion can drive hematopoietic stem cell (HSC) decline, thereby compromising HSC self-renewal, repopulating capacity, and differentiation (Rossi et al., 2007; Wang et al., 2012). While acute DNA damage can trigger a p53-mediated apoptosis or senescence of hematopoietic progenitor cells (Insinga et al., 2013; Milyavsky et al., 2010), whether and how accumulating physiological or pathological DNA damage (including telomeres) might influence the differentiation decisions of hematopoietic progenitor cells has not been explored. Of relevance to this study, it is worth noting that the specific type of cellular response (e.g., apoptosis, cell cycle, etc.) in telomere dysfunctional mice can vary depending on cell type (Lee et al., 1998).
Myelodysplastic syndrome (MDS) is a very heterogeneous group of hematopoietic disorders characterized by ineffective myeloid differentiation, dysplasia and excessive DNA damage accumulation in stem/progenitor cells (Zhou et al., 2013). MDS incidence has risen dramatically in recent years (Rollison et al., 2008) and is associated with advanced age, shorter telomeres, cancer chemotherapy with alkylating agents, radiation and inherited syndromes related to abnormalities in DNA repair (Zhou et al., 2013). On the genomic level, MDS alterations include chromosomal abnormalities (loss of 5q, 7 or 7q, 20q, and/or Y, and trisomy 8), point mutations of NRAS or KRAS and/or TP53 as well as genes involved in DNA methylation (DNMT3A, TET2, IDH1, IDH2), chromatin remodeling (ASXL1, EZH2), splicing regulation (SF3B1, SRSF2, U2AF1, U2AF2, SF3A1, ZRSR2, SF1, PRPF40B) (Bejar et al., 2011; Larsson et al., 2013) and telomerase complex (TERT, RTEL1, TINF2). While the hematopoietic stem cells harboring these mutations are the cell-of-origin for MDS (Will et al., 2012) and are responsible for clonal dominance, derivative committed progenitors from these HSCs manifest skewed differentiation resulting in the morphological and clinical phenotype of MDS. Herein, we sought to understand if telomere dysfunction and persistent DNA damage signaling activation can be the primary instigator of MDS in the absence of MDS-associated gene mutations or genetic alterations, and how hematopoietic progenitor cells can contribute to ineffective differentiation that maintains MDS and eventually results in disease progression.
RESULTS
Telomere dysfunctional mice exhibit hallmarks of human MDS
Recognizing that the biological response to telomere dysfunction is highly cell type dependent (Lee et al., 1998), we sought to catalog the cell biological and molecular responses to telomere dysfunction in various hematopoietic lineages and assess their potential role in MDS pathogenesis. To model chronic physiological DNA damage in the hematopoietic system, we employed the inducible telomerase model, TERTER, engineered to encode a telomerase reverse transcriptase-estrogen receptor fusion protein that can be activated by 4-hydroxytamoxifen (OHT) treatment. Inter-generational crosses of TERTER/ER mice leads to progressive telomere erosion which by the fourth and fifth generations (G4/G5) elicits telomere dysfunction and associated DNA damage signaling and severe tissue degeneration (Jaskelioff et al., 2011). In the late generation G4/G5 TERTER/ER mice, systemic administration of OHT restores telomeres, quells DNA damage signaling and reverses tissue degeneration phenotypes (Jaskelioff et al., 2011).
We first audited in-depth the phenotypic impact of telomere dysfunction on the hematopoietic system in 3 month- and 7 month-old G4/G5 TERTER/ER mice. These analyses revealed significant cytopenias in the peripheral blood, a decline in lymphopoiesis, slight anemia, and moderate granulo-monocytosis in advancing age (Figure 1A). Bone marrow (BM) hyper-cellularity and increased myeloid-to-erythroid progenitor (M:E) ratio (Figure 1B), in the absence of increased apoptosis (Figure S1A), were consistent with a condition of myeloid-skewed differentiation and ineffective hematopoiesis. Further BM analysis revealed severe tri-lineage dysplasia (Figure 1C: hypersegmented neutrophils (29% ± 7.39%), erythroblasts (18.25% ± 7.13), and megakaryocytes (44.75% ± 20.27%), absence of ring sideroblasts (Figure S1B) and increase of immature, morphologically abnormal myeloid blasts (Figure 1D, top: % of blasts: 10–15%; myeloperoxidase positive blasts: 78% ± 10.81%) frequently with pronounced monocytic differentiation (Figure 1D, bottom; butyrate esterase positive blasts: 35.75% ± 3.30%), which recapitulated the hallmark features of refractory anemia with excess of blasts (RAEB) in 80% of cases or chronic myelo-monocytic leukemia (CMML) in 20% of cases, specific sub-groups of MDS that are characterized by a high propensity to develop acute myeloid leukemia (AML). Accordingly, approximately 5% of aged G4/G5 TERTER/ER mice progressed to AML, as demonstrated by a marked increase of BM myeloid blasts (more than 20% of BM cellularity) (Figure 1E), and infiltration of myeloid precursors into the splenic white-red pulp architecture, resulting in myeloid sarcoma with the complete effacement of lymphoid follicles (Figure 1F). Cytogenetic analysis of G4/G5 TERTER/ER BM cells showed chromosomal breaks and fusions (Figures S1C), which eventually fuel chromosomal translocations (Figure S1D).
Figure 1. The hematopoietic compartment of telomere dysfunctional mice recapitulates hallmark features of human myelodysplastic syndrome.

(A) Complete blood count evaluation of age-matched young (3 month old) and old (7 month old) mice of indicated genotypes (top panel) (error bars denote s.e.m). Relative abundance of neutrophils and lymphocytes in total white blood cells of young and old mice of indicated genotypes (bottom panel) (error bars denote s.e.m).
(B) H&E stained sections of BM biopsies of representative G0 (left panel) and G5 (right panel) mice (scale bar, 50 μm).
(C) Pattern of multilineage differentiation in a representative G0 BM cytospin (left panel); Dysplastic neutrophil (N), erythroblast (E) and megakaryocyte (M) in a representative G5 BM cytospin (central and right panel) (scale bar, 15 μm).
(D) MPO (top, panel) and butyrate esterase (bottom panel) cytochemical staining of a representative G0 (on the left) or G5 (on the right) BM cytospin. Arrows indicate positive blasts (scale bar, 15 μm).
(E) Pattern of multilineage differentiation in a representative G0 BM cytospin (left panel); increased number of BM blasts (> 20% of BM cellularity) in the BM cytospin of a representative G5 mouse in transformation (right panel) (scale bar, 15 μm).
(F) H&E stained splenic section of a representative G0 mouse (top, left panel) or G5 mouse in transformation (top, right panel) (scale bar, 200 μm). Myeloid cells infiltrating the white-red pulp are positive for CD11b (bottom, left panel) and MPO (bottom, right panel) (scale bar, 15 μm).
See also Figure S1.
Telomerase reactivation rescues impaired progenitor cell differentiation in telomere dysfunctional mice
To study the cellular mechanisms underlying skewed myeloid differentiation induced by telomere attrition and given the important contribution of the hematopoietic progenitor compartment towards ineffective hematopoiesis and MDS phenotype, we examined the c-Kit+Sca−Lin− (KS−L) compartment that represents a key branch at which hematopoietic stem cell-derived progenitors commit to various myeloid lineages (Akashi et al., 2000) (Figure S2A). Compared to age- and gender-matched G0 TERTER/+ controls, the KS−L compartment of G4/G5 TERTER/ER mice showed a significant increase in the frequency (Figure 2A) and absolute number (Figure S2B) of granulocyte-macrophage progenitors (GMP; c-Kit+Sca−Lin−CD34+FcγRhi), with the concomitant loss of megakaryocyte-erythroid progenitors (MEP; c-Kit+Sca−Lin−CD34−FcγRlo) and slight reduction of common myeloid progenitors (CMP; c-Kit+Sca−Lin−CD34+FcγRlo), in the absence of increased apoptosis in these populations (data not shown). These observations gain added significance in light of recent findings that a significant expansion of the GMP compartment occurs in the RAEB stage of MDS patients with higher risk of leukemic transformation (Pang et al., 2013; Will et al., 2012). Notably, the GMP population further increased in the aged G4/G5 TERTER/ER mice (Figure S2C) or during leukemic transformation (data not shown). Further analysis of the hematopoietic subpopulations showed a preferential accumulation of γ-H2AX and 53BP1 DNA damage foci in the telomere dysfunctional CMP (Figure 2B and Figure S2D), but not in GMP or MEP (Figure S2E and S2F), suggesting that various subtypes of hematopoietic progenitors may respond differently to telomere erosion.
Figure 2. Skewed myeloid-erythroid differentiation of CMP is reversed by telomerase reactivation.

(A) KS−L frequency in the BM, as well as the CMP, GMP and MEP frequencies in the KS−L population of indicated genotypes and treatments (mean and s.e.m. of age-matched 3 month old mice from 6 independent experiments of telomerase reactivation in vivo; data are expressed as percentage of corresponding controls).
(B) Anti-γH2AX immunofluorescence in CMP sorted from mice of indicated genotypes and treatments: numbers of γH2AX foci per cell (upper panel; error bars denote s.d); representative images (bottom panel): α-γH2AX: green; DAPI: blue; scale bar, 20 μm.
(C) and (E) Frequency of the erythroid lineage in the BM (C, left panel) and spleen (E, upper panel) of indicated genotypes and treatments (mean and s.e.m of mice from 2 independent experiments of telomerase reactivation in vivo; data are expressed as percentage of corresponding controls); representative Ter119-stained sections of BM (C, right panel; scale bar, 100 μm) and spleen (E, bottom panel) from age-matched controls and experimental mice (scale bar, 200 μm).
(D) H&E stained BM section of a representative G5 mouse without (left panel) or with OHT treatment (right panel; scale bar, 15 μm).
See also Figure S2.
To determine whether elevated telomere associated DNA damage was the instigator for skewed myeloid-erythroid differentiation, we reactivated telomerase in 3 month-old telomere dysfunctional mice to restore telomere function and thus quell DNA damage signaling. Following 40 days of continuous OHT exposure, total BM cells showed increased telomere length as measured by Flow-FISH and a significant reduction of signal-free ends (Figure S2G and S2H). Telomere restoration was associated with a significant increase in MEP frequency (Figure 2A) and absolute number (Figure S2B) and a corresponding reduction of GMP. Furthermore, characterization of the progenitor compartment in the early generation telomerase deficient mice (G1) without telomere dysfunction showed no aberrant myeloid differentiation (Figure S2I), suggesting that skewed myeloid differentiation in G4/G5 mice is attributed to DNA damage activation, and not the absence of telomerase per se. Together, these data support the view that telomere attrition-induced DNA damage itself can serve as a driver of alter myeloid progenitor differentiation, resulting in a persistent accumulation of the myeloid lineage at the expense of the MEP population – a process fundamental to MDS pathogenesis.
Telomerase reactivation was also associated with significantly reduced γ-H2AX and 53BP1 foci of the CMP population (Figure 2B and S2D). The recovery of the MEP population upon sustained OHT treatment tracked with a significant restoration of BM Ter119+ erythroid progenitors (Figure 2C) in all developmental stages (data not shown), an improvement of M:E ratio and reduction of immature myeloid cells in the BM (Figure 2D), and decline of splenic extra-medullary erythropoiesis (Figure 2E). Thus, endogenous telomerase reactivation and extinction of DNA damage signaling restores the normal myeloid differentiation process, reinforcing the link between telomere dysfunction and aberrant myeloid differentiation. Nevertheless, the failure of telomerase reactivation to rescue the KS−L decline (Figure 2A) implicates the existence of additional upstream differentiation checkpoints involving the stem cell compartment as previously reported (Rossi et al., 2007; Wang et al., 2012). Consistent with the expansion of phenotypically primitive HSCs occurring in MDS (Will et al., 2012), the telomere dysfunctional HSC compartment (KSL) showed an increased expansion in the steady-state frequency and absolute number of long-term HSC (LT-HSC; c-Kit+Sca+Lin−CD34−flk2−) and short-term HSC (ST-HSC; c-Kit+Sca+Lin−CD34+flk2−), with the concomitant decrease of multipotent progenitor cells (MPP; c-Kit+Sca+Lin−CD34+flk2+) (Figure S2J, S2K and data not shown), and compromised repopulation capability upon BM competitive transplantation (Figure S2L).
Defective CMP differentiation is due to cell intrinsic DNA damage signaling activation
Impaired progenitor differentiation could occur as a result of a telomere dysfunctional systemic environment that limits HSC function and organ homeostasis (Ju et al., 2007) and/or cell intrinsic defects of telomere dysfunctional hematopoietic cells (Allsopp et al., 2003). To distinguish between the two possibilities, we transplanted LT-HSC isolated from 3 month-old G0 or G4/G5 mice into wild type congenic recipients and assayed recipient BM for donor-derived progenitor cell frequencies 2 months post-transplantation. Analysis of G4/G5 derived hematopoietic system revealed that the level of donor-derived skewed myeloid differentiation was comparable to that observed at steady state in the same young telomere dysfunctional mice before transplantation (Figure 3A). Furthermore, G4/G5 transplanted BM showed severe tri-lineage dysplasia and an increase of immature, morphologically abnormal myeloid blasts. Notably, one of the 6 mice transplanted with G5 HSC progressed to AML, as demonstrated by a marked increase of BM myeloid blasts, and infiltration of myeloid precursors into the splenic white-red pulp architecture (Figure 3B). Overall, these findings demonstrate that telomere dysfunction exerts a prominent cell-intrinsic effect on the differentiation commitment of progenitor cells, and are consistent with the existence of “diseased” stem cells that are capable of regenerating the MDS phenotype after transplantation in wild type mice.
Figure 3. Defective CMP differentiation is due to cell intrinsic DNA damage signaling activation.
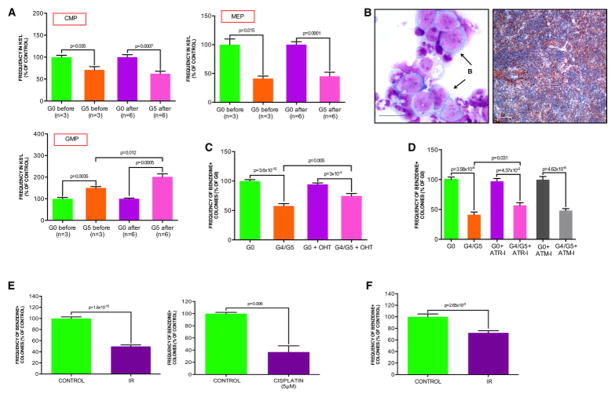
(A) CMP, GMP and MEP frequencies in the KS−L population of G0 or G5 mice before and after HSC transplantation in wild type recipient mice (error bars denote s.e.m.; data are expressed as percentage of corresponding controls).
(B) Representative BM cytospin of one recipient mouse transplanted with G5 HSCs (on the left). Arrows indicate blastic cells. Massive infiltration of CD11b positive myeloid precursors into the splenic white-red pulp architecture of a recipient mouse transplanted with G5 HSCs (on the right) (scale bar, 100 μm).
(C) Clonogenic myeloid colony formation in methylcellulose from sorted CMP of indicated genotypes cultured in the presence of vehicle or OHT. Erythroid cells were scored by benzidine staining and expressed as frequency of the total number of colonies (mean and s.e.m of replicates from 6 experiments of telomerase reactivation in vitro; each experiment includes equal number of CMP sorted from independent mice of indicated genotypes; data are expressed as percentage of G0 control).
(D) Clonogenic myeloid colony formation in methylcellulose from sorted CMP of indicated genotypes pre-treated with vehicle, ATR inhibitor (ATR-I, 1 μM) or ATM inhibitor (ATM-I, 2 μM) for 1 hr. Erythroid cells were scored by benzidine staining and expressed as frequency of the total number of colonies (mean and s.e.m of replicates from 3 independent experiments, each experiment includes equal number of CMP sorted from independent mice; data are expressed as percentage of G0 control).
(E) Clonogenic myeloid colony formation in methylcellulose from sorted wild type CMP isolated from control or irradiated (IR, 3 Gy) mice (left panel), or pre-treated with vehicle or cisplatin (5 μM) for 4 hr (right panel). Erythroid cells were scored by benzidine staining and expressed as frequency of the total number of colonies (mean and s.e.m of replicates from 3 and 2 independent experiments, respectively; each experiment includes CMP sorted from a pool of three wild type mice; data are expressed as percentage of corresponding controls).
(F) Clonogenic myeloid colony formation in methylcellulose from sorted CMP isolated from 9 month-old wild type mice 4 months after sub-lethal irradiation. Control mice indicate age-matched wild type mice without irradiation. Erythroid cells were scored by benzidine staining and expressed as frequency of the total number of colonies (mean and s.e.m of replicates from 4 or 5 mice for each condition; equal number of CMP was sorted from independent mice; data are expressed as percentage of control).
See also Figure S3.
Additionally, in the light of recent findings showing that megakaryocyte and megakaryocyte-erythroid progenitors can also directly differentiate from HSC (Yamamoto et al., 2013), we evaluated if the CMP is responsible for the skewed myeloid differentiation of telomere dysfunctional mice as suggested from the in vivo experiments. To this end, we sorted G0 and G4/G5 CMP and determined their differentiation potential in methylcellulose clonogenic assay. Consistent with the in vivo results (Figure 2A), there was a profound impairment of myeloid differentiation toward the erythroid lineage in favor of granulo-monocytic commitment in the telomere dysfunctional CMP which was partially rescued upon telomerase reactivation (Figure 3C; data not shown). Similar results were obtained in clonogenic assays of BM mononuclear cells (MNCs) (Figure S3A), as well as HSCs upon long-term culture (LTC-IC) (data not shown). On the basis of these in vivo and in vitro data, we conclude that telomere dysfunction affects myeloid differentiation. Next, we explored the nature of DNA damage signaling and its impact on CMP differentiation processes. We observed that a highly specific inhibitor of the ATR but not ATM kinase partially improved erythroid differentiation of telomere dysfunctional CMP (Figure 3D), a finding consistent with a known role for ATR in telomere dysfunction and aging-induced replicative stress signaling (Kastan and Bartek, 2004) (p=2.2×10−10, Figure S3B). Correspondingly, our clinical correlative studies showed that ATR phosphorylation (p-ATR) status in the CD34+ cells correlates with high risk MDS, which is characterized by an expanded GMP population at the expense of MEP (Pang et al., 2013; Will et al., 2012). We observed p-ATR signal in only 5 of 25 patients samples exhibiting low risk MDS versus 23 of 32 with high risk MDS (p=0.00014) (Figure S3C). Collectively, these data indicate the existence of a cell intrinsic telomere dysfunction-induced differentiation checkpoint, which occurs at the level of progenitor cells and contributes to ineffective hematopoiesis – a key feature of the MDS phenotype.
Next, we sought additional evidence to cement the role of DNA damage in altering myeloid differentiation. Employing ionizing radiation and cisplatin treatment as distinct instigators of DNA damage signaling, sorted CMP from wild type mice subjected to irradiation (IR, 3 Gy) or cisplatin treatment (5 μM, 4 hr of cisplatin treatment) show impaired erythroid differentiation (Figure 3E). Notably, skewed myelo-erythroid differentiation occurred even 4 months after sub-lethal irradiation of wild type mice (Figure 3F), consistent with recent findings showing that brief exposure to a moderate level of DNA damage is sufficient to maintain chronic DNA damage signaling activation in hematopoietic cells (Insinga et al., 2013).
Telomere dysfunction induces aberrant RNA splicing by repressing splicing gene expression in CMP
Next, to determine the mechanistic bases of how telomere dysfunction-induced DNA damage may drive abnormal myeloid differentiation, we performed gene expression profiling analysis of sorted CMP cells from age- and gender-matched G0 or G4/G5 TERTER/ER mice. Gene set enrichment analysis (GSEA) of the differentially expressed genes of the G4/G5 TERTER/ER CMP revealed a significant enrichment of genes involved in mRNA splicing and processing (Figure 4A, Table S1), including the splicing factors U2AF2, SRSF2, SRSF10, SF3B2 and SF3A3. To exclude the possibility that the changes in splicing gene expression was due to cellular heterogeneity in the CMP, we performed single cell Fluidigm real-time PCR and observed a consistent repression of splicing gene expression in every G4/G5 CMP compared to the G0 control (Figure 4B, Table S2), indicating that telomere dysfunction leads to a homogeneous change in splicing gene expression during this particular stage of differentiation. That the splicing pathway was down-regulated in the G4/G5 CMP was further validated by western blot analysis (Figure S4A). In striking contrast, RNA splicing pathway was not altered in the G4/G5 TERTER/ER GMP (Figure S4B) or MEP (data not shown), a finding which aligns with a lack of a significant γ-H2AX and 53BP1 DNA damage foci increase in these subpopulations (Figure S2E and S2F) and underscores the context-specific impact of DNA damage signaling in the differentiating hematopoietic system. Interestingly, the CMP population also showed preferential and significant downregulation of genes belonging to the cohesin complex (RAD21, STAG1, SMC2 and SMC5) (Table S1) that is known to be involved in post-replicative DNA repair (Dorsett and Strom, 2012), and recurrently mutated or deleted in AML and MDS (Kon et al., 2013; Ley et al., 2013), suggesting that DNA damage accumulation in the CMP cells could be further potentiated by the impairment of pathways regulating the DNA damage repair.
Figure 4. Telomere dysfunction induces aberrant RNA splicing by repressing splicing gene expression in CMP.

(A) Significantly downregulated and upregulated pathways identified by GSEA in G4/G5 compared to G0 CMP (FDR =0.05).
(B) Fluidigm-based gene expression analysis of single cells (rows) for representative genes in the mRNA processing/ spliceosome pathways (columns) from GSEA, which are significantly altered in sorted CMP from the G0 and G5 mice with or without OHT treatment (n=3). Genes analyzed were (from left to right): ACTB, β2M, GAPDH (housekeeping genes; internal controls), U2AF2, SF3B2, SF3A3, SRSF2, SFPQ, SFRS10, SFRS2IP, CDC51, DDX46, WBP11, SMC1A, PAPOLA, SRRM1, FUS, RBM5 and NUP54. A full gene list is shown in Table S2. Color scale on the right shows correspondence between color code and Ct values.
(C) Interaction network of splicing genes significantly downregulated in G4/G5 CMP (blue color). Size of the nodes is proportional to the number of interactions of a given protein with other splicing components. * indicates splicing factors mutated in MDS.
(D) Fluidigm-based gene expression analysis of single cells (rows) for representative genes in the mRNA processing / spliceosome pathways (columns), which are significantly altered in sorted CMP from the G0 and G5 mice with or without ATR inhibitor treatment (n=2 or 3 mice for each condition). Genes analyzed were (from left to right): ACTB, β2M, GAPDH (housekeeping genes; internal controls), U2AF2, SF3B2, SF3A3, SRSF2, SFPQ, SFRS10, SFRS2IP, CDC51, DDX46, PAPOLA, SRRM1 and RBM5. Color scale on the right shows correspondence between color code and Ct values.
(E) Significantly enriched pathways relative to the 1,940 aberrantly spliced genes (p <0.05).
We observed an overrepresentation of splicing genes belonging to the same interaction network (Figure 4C), which is known to be involved in the 3′splice site recognition and to play a critical role for commitment complex E formation in U2-dependent splicing or for stabilization of splicing complex A. Remarkably, highly recurrent mutations in some of these same key components of the spliceosome machinery and their interacting partners have been recently reported in MDS (Yoshida et al., 2011), although little is known about how these mutations contribute to the pathogenesis of MDS or its transformation to AML.
To fortify the link between telomere dysfunction-induced DNA damage and suppressed expression of the mRNA splicing and processing components, we quantified the mRNA expression of these genes upon telomerase reactivation and observed that the downregulation of mRNA splicing and processing components, which was absent in G1 mice (data not shown), was rescued by telomerase reactivation in the G4/G5 mice (Figure 4B). The repression of mRNA splicing and processing components persisted in CMP after the transplantation of telomere dysfunctional HSC in wild-type recipient mice (Figure S4C), establishing that telomere dysfunction-induced DNA damage exerts a prominent cell-intrinsic effect on mRNA splicing regulation. Furthermore, splicing repression in the CMP was significantly rescued when telomere dysfunctional mice were treated with the specific ATR kinase inhibitor VE-821 (Figure 4D), a finding in accord with decreased p-ATR immunofluorescence staining of the CMP after VE-821 treatment (Figure S4D).
Together, these findings indicate that the cell intrinsic telomere dysfunction-induced DNA damage response can impact on the expression of genes involved in splicing regulation through the activation of ATR kinase. In correlative clinical studies, the link between splicing factor expression levels and ATR activation is further supported by a significant correlation between decreased srsf2 levels and the phosphorylation status of ATR in MDS CD34+ cells (n=50, p=0.04, data not shown).
Given that telomere dysfunction results in the down-regulation of splicing components, we performed RNA-Seq analysis on G0 and G4/G5 TERTER/ER CMP to assess if the RNA splicing process is indeed altered. Notably, this analysis detected a total of 2,489 aberrantly splicing events affecting 1,940 genes in the G4/G5 TERTER/ER CMP compared to G0 control (Table S3), among which 40.5% and 59.5% of aberrant splicing events would result in exon skipping and exon retention, respectively, consistent with the decreased expression of factors that define the exon-intron boundaries (Figure 4C). Furthermore, we found that 26.9% of aberrant splicing events are predicted to produce loss-of-function transcripts that arise mainly due to a premature stop codon (Table S4) or an in-frame deletion disrupting known functional domains (Table S5). There were no changes in intron retention (data not shown) which we speculate may result from increased expression of genes involved in the nonsense-mediated mRNA decay response present in G4/G5 TERTER/ER CMP (Zhang and Manley, 2013) (Figure 4A). Of note, the aberrantly spliced transcripts in the G5 CMP population are enriched in pathways that are highly relevant to the MDS phenotype, including the maintenance of genome stability, DNA damage response, chromatin remodeling and histone modifications (such as acetylation, methylation, sumoylation and ubiquitination) (Figure 4E).
Epigenetic alterations and histone code changes drive aberrant differentiation of MDS cells, which are sensitive to drugs that modify the epigenome and influence DNA methylation (Issa, 2013). In this regard, it is notable that, among the aberrantly spliced epigenetic regulators in G5 CMP, the protein level of DNMT3A DNA methyltransferase decreased due to premature termination of protein translation (Figure S4A) as a result of a frameshift that occurs upon exon skipping (Table S4). In MDS patients, recurrent mutations in DNMT3A reduce the methyltransferase activity of the protein and are associated with rapid progression to AML (Walter et al., 2011). Supporting the view that the loss of DNMT3A may impact on DNA methylation, we found locus-specific changes in methylation patterns in the G5 CMP compared to G0 control, involving both gains and losses of methylation in CpG promoters, gene bodies, and intergenic regions (Figure S4E and S4F). These findings are consistent with previous studies showing that DNMT3A loss in the hematopoietic system results in both hypo and hyper-methylation at distinct loci, progressive impairment of differentiation (Challen et al., 2011) and hematological malignancies including MDS (Mayle et al., 2014).
SRSF2 haploinsufficiency impairs CMP differentiation in mice
Recognizing that an optimal stoichiometry of splicing components is critical for proper spliceosomal function (Caceres et al., 1994) and that either over-expressing or downregulating particular splicing factors alters splicing activity, we sought multi-level evidence of a causal link between decreased splicing factor activity/expression and abnormal myeloid progenitor differentiation. To that end, we evaluated whether pharmacological or genetic perturbation of the splicing machinery could induce defective myeloid differentiation similar to that observed in the telomere dysfunctional CMP. Using Pladienolide B (PLA-B, a natural macrolide that inhibits the spliceosome assembly through specific Sf3b complex binding (Kotake et al., 2007)), we observed that a transient (4 hr) pre-treatment of sorted wild type CMP cells prior to differentiation resulted in a profound impairment of differentiation towards the erythroid lineage (Figure S5A). Similar results were obtained with transient NSC663284 treatment that inhibits the catalytic activity of the splicing machinery (Berg et al., 2012) (Figure S5A).
To provide genetic evidence that decreased expression of splicing components impacts progenitor differentiation towards the erythroid lineage, we generated mice heterozygous for SRSF2 by crossing conditional SRSF2 knockout mice (SRSF2L/L) with hematopoietic cell-specific cre transgenic mice (Vav-cre). SRSF2 is a well-characterized splicing factor involved in both constitutive and regulated splicing (Lin and Fu, 2007), and is significantly down-regulated in telomere dysfunctional CMP and recurrently mutated in MDS patients (Yoshida et al., 2011). Although the Vav-cre/ SRSF2L/+ mice showed no obvious hematopoiesis defect in the peripheral blood (data not shown), analysis of the Vav-cre/ SRSF2L/+ BM revealed multilineage dysplasia (Figure 5A: hypersegmented neutrophils (20.5% ± 7.76%), erythroblasts (14.75% ± 2.06), and megakaryocytes (27.25% ± 10.93%), a slight increase in the number of morphological abnormal blasts (Figure 5A: 13% ± 7.57%) and myelo-monocytosis (Figure 5B: % of butyrate esterase positive cells is 7% ± 2% in the Vav-cre BM versus 16.33% ± 4.67% in the Vav-cre/SRSF2L/+ BM, p=0.003; % of CD11b positive cells is 41.66% ± 2.88% in the Vav-cre BM versus 67.5% ± 9.57% in the Vav-cre/SRSF2L/+ BM, p=0.008). Compared to the age- and gender-matched Vav-cre controls, the KS−L compartment of the Vav-cre/SRSF2L/+ mice showed an slight increase in the frequency of GMP with the concomitant loss of MEP (Figure 5C), which was further confirmed by in vitro methylcellulose clonogenic assay of sorted CMP (Figure 5D).
Figure 5. SRSF2 haploinsufficiency induces skewed myeloid differentiation of CMP.

(A) Dysplastic monolobated and hyperlobated (upper panel, on the left) or multinucleated (upper panel, on the right) megakaryocytes (M), dysplastic erythroblast (E, bottom panel on the left) and abnormal (bottom panel, in the middle) and mitotic (bottom panel, on the right) blasts (B) in a representative Vav-cre/ SRSF2L/+ BM cytospin (scale bar, 15 μm).
(B) Butyrate esterase cytochemical staining of a representative Vav-cre (upper panel, on the left) or Vav-cre/ SRSF2L/+ BM cytospin (upper panel, on the right). CD11b stained sections of BM biopsies of representative Vav-cre (bottom panel on the left) and Vav-cre/ SRSF2L/+ (bottom panel on the right) mice (scale bar, 15 μm).
(C) KS−L frequency in the BM, as well as the CMP, GMP and MEP frequencies in the KS−L compartment of 2 month old Vav-cre (n=5) or Vav-cre/ SRSF2L/+ (n=5) mice (error bars denote s.e.m.; data are expressed as percentage of the Vav-cre control).
(D) Clonogenic myeloid colony formation in methylcellulose from CMP sorted from Vav-cre or Vav-cre/ SRSF2L/+ mice. Erythroid cells were scored by benzidine staining and expressed as frequency of the total number of colonies (mean and s.e.m of replicates from 5 independent mice; data are expressed as percentage of Vav-cre control).
See also Figure S5.
Aberrant RNA splicing due to reduced SRSF2 expression induces telomere dysfunction
Next, RNA-Seq analysis of Vav-cre and Vav-cre/ SRSF2L/+ CMP confirmed altered RNA splicing associated with SRSF2 haploinsufficiency, detecting 1,682 aberrant splicing events for 1,357 genes (Table S6). Strikingly, these aberrantly spliced genes are involved in telomere maintenance, chromatin remodeling and DNA repair pathways and show overlap to those genes altered in the telomere dysfunctional CMP (Figure 6A). Consistent with the aberrant splicing and predicted loss-of-function of telomere maintenance genes including RTEL1 and TERF2IP (Figure S6A), SRSF2 deletion was associated with significantly increased number of telomere dysfunction-induced foci in the CMP population (Figure 6B), and decreased telomere length in BM cells (Figure 6C). Together, the concordance of RNA splicing profiles of SRSF2 haploinsufficient and telomerase deficient CMP strongly reinforce the intimate connection between RNA splicing, telomere biology, DNA repair and MDS phenotype.
Figure 6. Aberrant RNA splicing due to altered SRSF2 function induces telomere dysfunction.

(A) Significantly enriched pathways relative to the 1,357 aberrantly spliced genes (p <0.05) in the Vav-cre/ SRSF2L/+ mice.
(B) Telomere-FISH and anti-γH2AX immunofluorescence in CMP sorted from 5 month-old mice of indicated genotypes (telomere: red; anti-γH2AX: green; co-localization: yellow; n=5 Vav-cre and n=7 Vav-cre/ SRSF2L/+); numbers of telomere dysfunction-induced foci per cell (left panel) (error bars denote s.e.m.); representative images (right panel) (scale bar, 10 μm).
(C) Mean value of telomere length in primary BM cells of 5 month old mice of indicated genotypes, as determined by flow-FISH analysis (error bars denote s.e.m.; data are expressed as percentage of the Vav-cre control).
(D) Significantly enriched pathways relative to the 1,355 aberrantly spliced genes (p <0.01) in CMML patients with SRSF2 (P95) mutation.
Finally, we asked if this functional link was indeed relevant to the human counterpart of MDS, which harbors somatic mutations of spliceosomal genes in over half of all patients (Yoshida et al., 2011). Since CMML patients are also characterized by skewed myeloid differentiation towards the myelo-monocytic lineage and present high rate of SRSF2 mutation (hence potentially affecting RNA splicing) (Itzykson et al., 2013), we evaluated SRSF2 mutation (P95)-specific exon usage patterns by RNA-seq analysis of SRSF2 mutant (n=6) and wild type (n=9) CD34+ cells isolated from CMML patients, to understand the functional consequences of SRSF2 mutation on RNA processing/splicing. This analysis detected a total of 1,536 aberrant splicing events affecting 1,355 genes (Table S7), mainly involved in DNA repair and telomere maintenance pathways (Figure 6D), which overlapped with the telomere dysfunctional (Figure 4E) and SRSF2 haploinsufficient CMP RNA-Seq datasets (Figure 6A). Specifically, we found that transcripts encoding ACD, which is a component of the shelterin telomeric complex, and TNKS, which interacts with TRF1, to be aberrantly spliced in CD34+ cells with SRSF2 mutation, with predicted loss of function of these telomere maintenance genes. Together, these findings strongly support the view that perturbation of RNA splicing either through loss of SRSF2 expression or SRSF2 mutation results in the aberrant splicing of a specific subset of genes, including those that influence telomere dynamics, which is predicted to induce telomere dysfunction and exacerbate aberrant RNA splicing.
DISCUSSION
Our study provides multi-level evidence that telomere dysfunction can be a critical factor driving MDS, and that diminished splicing factor expression induced by telomere dysfunction drives myeloid differentiation processes in a manner that contributes to the high risk MDS phenotype. On the mechanistic level, genetic, pharmacological and correlative clinical data establishes that telomere dysfunction represses the expression of genes governing RNA splicing in part through ATR. Notably, on the clinical level, increased p-ATR staining is strongly correlated with high risk MDS in patients, a finding of therapeutic clinical utility in the prevention and treatment of aging-associated or therapy-induced MDS.
An unexpected finding from our study is the highly specific impact of telomere dysfunction and DNA damage on progenitor differentiation and RNA splicing. We propose that accumulating levels of physiological DNA damage perturb RNA splicing and thus impair the differentiation of specific progenitor subpopulations, possibly via de-regulation of epigenetic and DNA repair factors. This is consistent with previous studies which have demonstrated that genetic alterations in DNA repair genes can result in bone marrow failure syndromes and familial MDS by altering differentiation of hematopoietic cells (Geiselhart et al., 2012; Owen et al., 2008). Furthermore, it is tempting to speculate that defective DNA repair in these progenitors may also fuel secondary events that predispose to AML transformation. This may be of considerable relevance for the disease progression and subsequent AML transformation that is frequently seen in MDS patients, and is consistent with the view that leukemic stem cells often display a progenitor rather than a stem cell phenotype in AML (Goardon et al., 2011).
Through pharmacologic and genetic perturbation of RNA splicing, we provided evidence that aberrant RNA splicing alone impairs CMP differentiation, supporting the view that aberrant RNA splicing upon telomere dysfunction is one of the mechanisms underlying the skewed myeloid differentiation (and MDS phenotypes) seen in vivo. Indeed, reduced SRSF2 expression in the Vav-cre/ SRSF2L/+ mouse model partially recapitulates the MDS features observed in the telomere dysfunctional mice. An unanticipated finding is that, both SRSF2 haploinsufficiency and SRSF2 mutation resulted in the aberrant splicing of genes that are involved in telomere maintenance, DNA repair, and chromatin remodeling - pathways extremely relevant to MDS pathogenesis - which are also altered in the telomere dysfunctional CMP as a consequence of DNA damage-induced downregulation of splicing factor expression. Furthermore, our observation that splicing deregulation induces telomere dysfunction underscores a connection between RNA splicing and telomere biology, which warrants further investigation.
Despite significant convergence of pathways with mutant SRSF2 and SRSF2 haploinsufficiency, we also observed differences in the aberrantly spliced transcripts, suggesting that the process of RNA splicing is executed differentially with SRSF2 mutation compared to its loss of function (e.g. different splice site recognition) which may explain the weaker MDS phenotype observed in Vav-cre/ SRSF2L/+ mice. This observation concurs with what has been shown for the splicing gene PRPF8 (Kurtovic-Kozaric et al., 2014), and is consistent with many other examples of cancer relevant genes, such as the TP53 tumor suppressor gene, whereby TP53 deletion and TP53 mutation share many commonalities in cancer pathogenesis but are not identical in function (Lang et al., 2004).
In conclusion, our studies reveal an intimate relationship between telomeres and mRNA splicing in the control of cellular differentiation of specific hematopoietic cell subpopulations but the molecular details are still to be clarified. We propose that telomere dysfunction causes aberrant RNA splicing (by down-regulating splicing factors), which exacerbates telomere erosion (by aberrantly splicing of telomere maintenance genes), impairing progenitor cell differentiation, culminating in the eventual progression to AML. Our results are consistent with the long-standing clinical observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage accumulation in myeloid progenitor cells and that therapy-related MDS can result from cancer treatments utilizing alkylating agents or ionizing radiations. The improved understanding of DNA-damage induced perturbations in the splicing of specific transcripts and linked pathways reported here should provide highly specific risk biomakers and therapeutic targets for the prevention or treatment of this incurable disease.
EXPERIMENTAL PROCEDURES
Generation and analysis of mice
Mice were maintained in specific pathogen-free (SPF) conditions at MD Anderson Cancer Center. All manipulations were performed with IACUC approval. The heterozygous (G0 TERTER/+) and late generation homozygous (G4/G5 TERTER/ER) mice were generated based on standard breeding protocol of successive generations of telomerase-deficient mice (Jaskelioff et al., 2011). All studies were performed on adult (12–16 week old) G0 TERTER/+ and telomere dysfunctional G4/G5 TERTER/ER mice, unless otherwise noted. OHT time-release pellets (2.5 mg; Innovative Research of America) were inserted subcutaneously to reach steady state blood levels of 1 ng ml−1 OHT. The conditional deletion of SRSF2 in the hematopoietic compartment was accomplished by crossing Vav-cre mice (Jackson laboratories) with the srsf2L/L mice (Jackson laboratories) to generate heterozygous Vav-cre/ SRSF2L/+ mice. Animals were autopsied, and the BM and spleen tissues were examined regardless of their pathological status. Details are described in Supplemental Experimental Procedures.
Human primary samples
Human MDS or CMML bone marrow specimens were obtained from patients referred to the Department of Leukemia at MD Anderson Cancer Center following protocol LAB01–473, which was approved by MD Anderson’s Institutional Review Board. Written informed consent was obtained from donors. MDS bone marrow cells were collected from 57 MDS and 15 CMML patients. Diagnosis was confirmed by a dedicated hematopathologist and patients were classified as lower or higher risk MDS according to the International Prognostic Scoring System (IPSS). Isolation of CD34+ cells was performed using MicroBead Kit (Miltenyi), following manufacturer’s instructions. An aliquot of purified CD34+ cells was used to prepare cytospin slides; the remainder was subjected to centrifugation at 300xg for 10 min and resuspended in Trizol (Invitrogen) for RNA extraction.
Flow cytometry analysis
Single-cell suspensions were prepared from spleen and bone marrow (from femoral and tibial bones). For FACS sorting and analysis we used described staining protocols and published stem and progenitor cell definitions (Amrani et al., 2011; Flach et al., 2014). Details are described in Supplemental Experimental Procedures.
Indirect immunofluorescence microscopy and Tif assay
Sorted progenitor cells were resuspended in PBS, spotted on immunofluorescence slides, fixed and immuno-stained with anti phospho-γH2AX (1:200; clone number JBW301, Millipore), 53BP1 (1:200, catalog number IHC00001, Bethyl Laborathories) or phospho-ATR (1:50; catalog number 2853, Cell Signaling). For Tif assay, cells were co-stained with phospho-H2AX and the telomere specific PNA probe using the Telomere PNA FISH Kit/Cy3, according to the manufacturer’s instructions. Details are described in Supplemental Experimental Procedures.
Treatments and colony forming assay
Mononuclear cells (MNCs) (20 × 104/replicate) or sorted CMP (500 cells/replicate) were seeded into cytokine supplemented methylcellulose medium, or pre-treated with a specific inhibitor of ATR, ATM, cisplatin, Pladienolide B or NSC663284 prior to seeding. Colonies were counted after 7–10 days. Erythroid cells were scored by benzidine staining. Details are described in Supplemental Experimental Procedures.
Microarray and pathway analysis
Bone marrow CMP and GMP cells were sorted from 2 paired pools of G0 TERTER/+ or G4/G5 TERTER/ER mice (5,000–20,000 cells per sample) and RNA was extracted using Trizol. Gene expression profiling was performed with GeneChip® Mouse Genome 430 2.0 Array (Affymetrix) and enriched pathways were identified using Gene Set Enrichment Analysis (GSEA; http://www.broadinstitute.org/gsea/msigdb/annotate.jsp). Details are described in Supplemental Experimental Procedures.
Single-cell gene expression profiling
Single CMP cells were sorted directly into 96-well plates before Fluidigm-based real-time PCR analysis. Details are described in Supplemental Experimental Procedures.
RNA-Seq sequencing and analysis
Total RNA from sorted CMP isolated from 3 independent G0 TERTER/+ and 4 G4/G5 TERTER/ER, 3 Vav-cre and 6 Vav-cre /Srsf2L/+ mice, as well as CMML patient-derived CD34+ cells (n=15) was isolated by Trizol, before RNA amplification and RNA-Seq library construction. Transcriptomic sequencing (RNA-Seq) was performed on the Illumina HiSeq platform using the standard paired-end protocol. Mapping of RNA-seq reads was performed with Tophat2; and the NCBI RefSeq gene model and HTSeq software were used to quantify the gene-level expression, exon-specific expression and intron retention levels. The differential analyses for gene/isoform expression and intron retention were analyzed with DESeq2 (Anders and Huber, 2010), while exon usage was analyzed with DEXSeq (Anders et al., 2012). Pathway enrichment analysis was performed with Pathway Studio. Details are described in Supplemental Experimental Procedures.
Western blotting
Western blotting in small amount of cells was performed as previously described (Nakada et al., 2010). Antibodies were anti-sfrs2 (clone number 1SC-4F11, Millipore), anti-sf3b2 (clone number 5D2, Sigma), anti-dnmt3a (clone number H-295, Santa Cruz), anti terf2ip (clone number D9H4, Cell Signaling) and anti-vinculin (clone number hVIN-1, Sigma). Details are described in Supplemental Experimental Procedures.
Bone Marrow Transplantations
Experiments of BM transplantation were performed as previously described (Jaskelioff et al., 2011). Donor derived peripheral blood reconstitution (i.e. chimaerism) was assessed after 2 or 4 months following transplantation by FACS analysis of nucleated peripheral blood cells stained with anti-CD45.1 and anti-CD45.2-specific antibodies. Blood chimaerism for each recipient was calculated as the percentage of all CD45+ cells that were CD45.2+. Details are described in Supplemental Experimental Procedures.
Statistical analysis
All the data were analyzed by a two-tailed Student’s t-test (p<0.05 is considered to be statistically significant). For all experiments with error bars, standard error mean was calculated to indicate the variation within each experiment and data, and values represent mean ± s.e.m or mean ± s.d., as indicated in the figure legends.
Supplementary Material
SIGNIFICANCE.
Myelodysplastic syndrome (MDS) is a heterogeneous group of hematopoietic neoplastic disorders. While MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, whether DNA damage signaling can directly provoke MDS is unknown. Employing an inducible telomerase reverse transcriptase-estrogen receptor (TERTER) model, we demonstrate that persistent physiological DNA damage (from eroded telomeres) drives classical MDS features and biases common myeloid progenitor (CMP) differentiation towards the myeloid lineage. This defective CMP differentiation was attributed to repression of expression of mRNA splicing/processing genes, resulting in aberrant RNA splicing. Our studies establish an intimate link across telomere biology, aberrant RNA splicing and cellular differentiation, and suggest that strategies that mitigate DNA damage signaling may be useful for prevention/treatment of MDS.
HIGHLIGHTS.
Mice with dysfunctional telomeres exhibit hallmark features of human MDS.
DNA damage directly induces skewed myeloid progenitor differentiation.
Telomere dysfunction represses RNA splicing gene expression in CMP.
Aberrant RNA splicing due to reduced srsf2 expression induces telomere dysfunction.
Acknowledgments
We thank all members of the DePinho, Chin and Draetta laboratories for fruitful suggestions and discussions. This work is supported by the NCI (RO1 CA084628 to R.A.D, and MD Anderson TCGA Genome Data Analysis Center, grant number CA143883). D.O. is an Odyssey Fellow at MD Anderson Cancer Center, and supported by the Laura and John Arnold Foundation. Flow Cytometry was done with the assistance of the South Campus Flow Cytometry and Cell Sorting Core Laboratory, which is supported in part by NIH P30 CA16672. Dr. Cooper has patents with Sangamo BioSciences with artificial nucleases. He consults with Targazyme, Inc. (formerly American Stem cells, Inc.) and receives compensation. He also consults with GE Healthcare, and Ferring Pharmaceuticals. He receives honoraria from Miltenyi Biotec. He has licensed technology to Intrexon and Ziopharm and receives compensation.
Footnotes
ACCESSION NUMBERS
All data sets generated in this study using Microrray and RBBS are now accessible at GEO under GSE62393, while RNA-Seq is accessible at ftp://ftp.ncbi.nih.gov/pub/TraceDB/misc/tmp/SRP048846_SRP048858.
Supplemental information includes Supplemental Experimental Procedures, six figures and six tables and can be found online.
AUTHOR CONTRIBUTION
S.C., D.O. G.G-M. and R.A.D. designed and guided the research; S.C., D.O., Y.O., A.V., P.S., Y.W., H.Y., D.C.W., N.G., S.H., Y.W.H., B.H., G.G., P.P., S.J., A.C., L.N., M.D.A., and I.G-G. performed research. M.M. performed and analyzed immunofluorescence experiments. N.A.M., M.C.R., L.Z. and H.L. analyzed RNA-sequencing data. S.A.A performed and analyzed Fluidigm experiments. K.C-D. and K.R. analyzed flow cytometry data. C.A.B. analyzed microarray data. A.S.M. performed cytogenetic analysis. M.E., S.G. and T.G. analyzed methylation data. C.B-R. performed cytochemical and histological analyses. J.W.H., T.P.H., P.J., H.K., L.J.N.C., Y.A.W. and L.C. provided critical intellectual contributions throughout the project. S.C., D.O., and R.A.D. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Allsopp RC, Morin GB, DePinho RA, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood. 2003;102:517–520. doi: 10.1182/blood-2002-07-2334. [DOI] [PubMed] [Google Scholar]
- Amrani YM, Gill J, Matevossian A, Alonzo ES, Yang CW, Shieh JH, Moore MA, Park CY, Sant’Angelo DB, Denzin LK. The Paf oncogene is essential for hematopoietic stem cell function and development. Journal of Experimental Medicine. 2011;208:1757–1765. doi: 10.1084/jem.20102170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11 doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Research. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. New England Journal of Medicine. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Wan LL, Younis I, Diem MD, Soo M, Wang CL, Dreyfuss G. A Quantitative High-Throughput In Vitro Splicing Assay Identifies Inhibitors of Spliceosome Catalysis. Molecular and Cellular Biology. 2012;32:1271–1283. doi: 10.1128/MCB.05788-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres JF, Stamm S, Helfman DM, Krainer AR. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science. 1994;265:1706–1709. doi: 10.1126/science.8085156. [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nature Genetics. 2011;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- di Fagagna FD, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Dorsett D, Strom L. The Ancient and Evolving Roles of Cohesin in Gene Expression and DNA Repair. Current Biology. 2012;22:R240–R250. doi: 10.1016/j.cub.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte YF, Forsberg EC, et al. Replication stress is a potent driver of functional decline in ageing hematopoietic stem cells. Nature. 2014;512:198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiselhart A, Lier A, Walter D, Milsom MD. Disrupted Signaling through the Fanconi Anemia Pathway Leads to Dysfunctional Hematopoietic Stem Cell Biology: Underlying Mechanisms and Potential Therapeutic Strategies. Anemia. 2012 doi: 10.1155/2012/265790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, Woll P, Mead A, Alford KA, Rout R, et al. Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell. 2011;19:138–152. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Insinga A, Cicalese A, Faretta M, Gallo B, Albano L, Ronzoni S, Furia L, Viale A, Pelicci PG. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP. The myelodysplastic syndrome as a prototypical epigenetic disease. Blood. 2013;121:3811–3817. doi: 10.1182/blood-2013-02-451757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, Ades L, Fenaux P, Platzbecker U, Gagey O, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121:2186–2198. doi: 10.1182/blood-2012-06-440347. [DOI] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102–U1700. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Z, Jiang H, Jaworski KM, Rathinam C, Gompf A, Klein C, Trumpp A, Rudolph KL. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nature Medicine. 2007;13:742–747. doi: 10.1038/nm1578. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nature Genetics. 2013;45:1232–U1187. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nature Chemical Biology. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, Nakashima M, Hsi ED, Yoshida K, Shiraishi Y, et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia. 2014 doi: 10.1038/leu.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Larsson CA, Cote G, Quintas-Cardama A. The Changing Mutational Landscape of Acute Myeloid Leukemia and Myelodysplastic Syndrome. Molecular Cancer Research. 2013;11:815–827. doi: 10.1158/1541-7786.MCR-12-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG, Hoadley K, Triche TJ, Laird PW, Baty JD, et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. New England Journal of Medicine. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SR, Fu XD. SR proteins and related factors in alternative splicing. Alternative Splicing in the Postgenomic Era. 2007;623:107–122. doi: 10.1007/978-0-387-77374-2_7. [DOI] [PubMed] [Google Scholar]
- Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2014 doi: 10.1182/blood-2014-08-594648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, Lechman E, Hermans KG, Eppert K, Konovalova Z, et al. A Distinctive DNA Damage Response in Human Hematopoietic Stem Cells Reveals an Apoptosis-Independent Role for p53 in Self-Renewal. Cell Stem Cell. 2010;7:186–197. doi: 10.1016/j.stem.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, SJM Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia - a review. British Journal of Haematology. 2008;140:123–132. doi: 10.1111/j.1365-2141.2007.06909.x. [DOI] [PubMed] [Google Scholar]
- Pang WW, Pluvinage JV, Price EA, Sridhar K, Arber DA, Greenberg PL, Schrier SL, Park CY, Weissman IL. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3011–3016. doi: 10.1073/pnas.1222861110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollison DE, Howlader N, Smith MT, Strom SS, Merritt WD, Ries LA, Edwards BK, List AF. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008;112:45–52. doi: 10.1182/blood-2008-01-134858. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nature Reviews Molecular Cell Biology. 2012;13:397–404. doi: 10.1038/nrm3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schimdt H, Kalicki-Veizer J, O’Laughlin M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–1158. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D, et al. A Differentiation Checkpoint Limits Hematopoietic Stem Cell Self-Renewal in Response to DNA Damage. Cell. 2012;148:1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- Will B, Zhou L, Vogler TO, Ben-Neriah S, Schinke C, Tamari R, Yu YT, Bhagat TD, Bhattacharyya S, Barreyro L, et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120:2076–2086. doi: 10.1182/blood-2011-12-399683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, Nakauchi H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Zhang J, Manley JL. Misregulation of Pre-mRNA Alternative Splicing in Cancer. Cancer Discovery. 2013;3:1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Hasty P, Walter CA, Bishop AJR, Scott LM, Rebel VI. Myelodysplastic syndrome: An inability to appropriately respond to damaged DNA? Experimental Hematology. 2013;41:665–674. doi: 10.1016/j.exphem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.