

Abstract
Galectins, a family of β-galactoside-binding proteins, are expressed in many different phagocytic leukocytes (granulocytes, monocytes, and macrophages). A number of family members have been shown to play an important role in ingestion of particles (phagocytosis), thus contributing to clearance of damaged cells and host defense against pathogens. Here we describe procedures for analysis of the roles of galectins in phagocytosis by using galectin-3 as an example. We emphasize the function of endogenous galectin-3 as determined by comparison of phagocytosis by macrophages from galectin-3 knockout mice and wild-type mice. We focus on the role of galectin-3 in phagocytosis of pathogens and Fcγ receptor-mediated phagocytosis of opsonized cells and particles.
Keywords: Galectin, Phagocytosis, Opsonization, Macrophage
1 Introduction
Phagocytosis is defined by ingestion of particles (usually larger than 0.5 μm in diameter) by cells that results in invagination of the plasma membrane, followed by formation of phagosomes. Examples of particles ingested by phagocytes include dead cells (apoptotic bodies), pathogens, and inert beads (reviewed in [1]). Ingestion of dead cells results in digestion and clearance of self and unwanted cells/tissues which is critical in maintenance of tissue homeostasis. On the other hand, uptake of foreign pathogens and subsequent destruction/digestion of these microorganisms represents the protective role of phagocytosis. Phagocytosis assays can be used to identify the receptors involved in the recognition and ingestion of particles as well as elucidate the cellular mechanisms. The ingestion of particles by phagocytes can be through a nonspecific manner or via specific receptors. For example, Fcγ receptors on phagocytes can bind to IgG-coated particles and mediate the ingestion (opsonization).
Galectins are a β-galactoside-binding lectin family; some of the members are highly expressed by phagocytic leukocytes, such as macrophages and granulocytes. Galectin-3 was also reported to augment neutrophil phagocytosis of bacteria [2] and fungi such as Candida albicans [3]. Similarly, recombinant galectin-1 can enhance FcγRI expression on human monocytes and FcγRI-dependent phagocytosis [4]. In addition, galectin-1 can induce cell surface exposure of phosphatidylserine (PS) in neutrophils, thus facilitating phagocytosis of neutrophils by macrophages [5]. Of particular note is that the above-mentioned functions of galectins are mainly investigated with exogenously added galectins, which may not reveal the functions of endogenous galectins. Our work focuses on the roles of endogenous galectins in phagocytosis. We found that galectin-3 plays an important role in phagocytosis of opsonized red blood cells (RBC) by macrophages and it translocates to the cytosolic site of the phagosomes [6]. Galectin-9 was also found in the phagosomes as revealed by proteomic analysis [7]. Our preliminary data showed that galectin-9 is involved in phagocytosis by human monocytes (unpublished data).
This chapter describes assays to study the functions of galectin-3 in phagocytosis. Detailed procedures are provided for the Fcγ receptor-mediated phagocytosis of opsonized sheep red blood cells (SRBC) and inert latex beads (Subheading 3.1). The roles of galectin-3 in phagocytosis of Listeria monocytogenes by macrophages are also described (Subheading 3.2).
2 Materials
2.1 Fcγ Receptor-Mediated Phagocytosis
2.1.1 Phagocytosis of IgG-Opsonized Sheep Red Blood Cells (SRBC)
Wild-type bone marrow-derived macrophages (WT BMM) and galectin-3 knockout bone marrow-derived macrophages (Gal3KO BMM) (see ref. 6).
Culture medium: RPMI1640 medium with 10 % fetal bovine serum (FBS), 100 U penicillin, and 100 μg/mL streptomycin.
Sheep red blood cells.
Rabbit polyclonal IgG anti-SRBC antibody.
ACK lysing buffer: 0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM Na2 EDTA, pH 7.2.
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
2.5 % (v/v) glutaraldehyde in PBS.
1 % eosin solution.
96-well flat-bottom tissue culture plates.
15 mL conical plastic centrifuge tubes.
8-channel pipettor and disposable reservoirs.
Hemocytometer.
Mini-rotator.
Refrigerated centrifuge.
Inverted microscope.
Digital camera.
Incubator at 37 °C and 5 % CO2.
2.1.2 Phagocytosis of Inert Latex Beads
Opsonization and Fluorescence Labeling
Latex beads (see Note 1).
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
Total IgG.
Secondary antibody conjugated with fluorochrome.
DAPI or Hoechst 33342.
25G needle.
Syringe.
Phagocytosis
Primary neutrophils, macrophages, or macrophage cell lines, such as RAW264.7 mouse macrophage cell line and THP-1 human monocytic leukemia cell line (see Note 2).
Latex beads prepared in Subheading “Opsonization and Fluorescence Labeling.”
24-well plate.
Incubator at 37 °C and 5 % CO2.
Media: Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % fetal bovine serum and 4.5 g/L of glucose, L-glutamine, and antibiotics.
Internal/External Particles Discrimination
Media: Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % fetal bovine serum and 4.5 g/L of glucose, L-glutamine, and antibiotics.
Rhodamine labeled secondary antibody.
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
Cell Identification
4 % paraformaldehyde.
Hoechst 33342 or DAPI.
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
Determination of the Phagocytic Index
Imaging processing software.
Fluorescence microscope.
2.2 Phagocytosis of Bacteria
2.2.1 Preparation and Phagocytes and Phagocytosis Assay
Monocytes/macrophages: Primary monocytes/macrophages (e.g., bone marrow-derived macrophages or peritoneal macrophages) and monocytic cell lines (e.g., RAW264.7 or J774A.1 cells).
Mid-log phase (OD600=0.4–0.7) bacterial culture (e.g., Listeria monocytogenes 10403S).
Complete RPMI medium: RPMI1640 supplemented with 20 mM HEPES, 1× nonessential amino acids (NEAA), and 10 % FBS.
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
24-well tissue culture plates containing sterilized glass cover-slips (12 mm diameter).
2.2.2 Double-Cycle Immunofluorescence Staining
5 % casein blocking reagent (refer to Current Protocols in Immunology 18.13.23).
Antibody or antisera against the targeted bacteria (e.g., Listeria antisera, Denka Seiken).
Fluorochrome-conjugated secondary antibodies (e.g., Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 647-conjugated goat anti-rabbit IgG).
Washing buffer: PBS containing 1 % BSA.
Blocking/staining buffer: PBS containing 1 % BSA and 2.5 % casein.
Fixation buffer: 4 % paraformaldehyde in PBS.
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
Permeabilization buffer: PBS containing 1 % BSA, 2.5 % casein, and 0.05 % saponin.
Glass microscope slides.
Anti-fade mounting medium with DAPI (ProLong® Gold Antifade Reagent with DAPI, Molecular Probe).
Rhodamine-conjugated phalloidin.
Fluorescence microscope equipped with lasers and filter sets suitable for collecting fluorescence signals of 488, 555, and 647 nm.
2.3 Flow Cytometric Phagocytosis Assay
2.3.1 Label Bacteria with FITC
1 mg/mL FITC (fluorescein isothiocyanate) isomer 1 (Sigma) in PBS, pH 8.0.
1 % (v/v) glutaraldehyde in PBS.
1 M glycine in PBS.
2.3.2 Initiation of Phagocytosis and Analysis with Flow Cytometry
FITC-labeled bacteria prepared in Subheading 3.2.2, step 1.
Adherent phagocytes in 24-well culture plates prepared in Subheading “Opsonization and Fluorescence Labeling.”
Complete RPMI medium: RPMI1640 supplemented with 20 mM HEPES, 1× nonessential amino acids (NEAA), and 10 % FBS.
Phosphate-buffered saline (PBS): 140 mM NaCl, 5 mM KCl, 8 mM NaH2PO4, and 2 mM KH2PO4, adjusted to pH 7.4.
0.02 % EDTA in PBS.
1.25 mg/mL trypan blue in PBS.
0.5 % trypsin/EDTA or nonenzymatic cell dissociation solution such as Cell Stripper (Cellgro).
1 % FBS in PBS.
Polypropylene tube suitable for flow cytometry.
Flow cytometry.
3 Methods
3.1 Fcγ Receptor-Mediated Phagocytosis
3.1.1 Phagocytosis of IgG-Opsonized Sheep Red Blood Cells (SRBC)
Place wild-type bone marrow-derived macrophages (WT BMM) and galectin-3 knockout bone marrow-derived macrophages (Gal3KO BMM) in wells of 96-well plates (105 cells per well) and cultured at 37 °C in a CO2 incubator overnight.
Dilute 5 mL of sheep whole blood with 10 mL of PBS and centrifuge the cells at 800×g for 10 min at room temperature.
Remove the supernatant and buffy coat layer, and wash SRBC with 15 mL of PBS twice (see Notes 3 and 4).
Count SRBC in a hemocytometer and adjust the cell number to 108/mL in PBS.
Mix 108/mL SRBC with rabbit polyclonal anti-SRBC antibody (subagglutination concentration, see Note 5) and incubate at room temperature for 30 min with gentle rotation on a mini-rotator.
Wash the opsonized SRBC twice with 10 mL of PBS and resuspend the opsonized SRBC into 108/mL in culture medium.
Place the opsonized SRBC on ice for later usage (see Note 6).
Take out the 96-well plates containing WT BMM and Gal3KO BMM from the incubator and place the 96-well plates on ice for 30 min.
Remove the medium from the plate and add 100 μL of ice-cold opsonized SRBC (108/mL).
Centrifuge the plates at 300×g for 5 min at 4 °C to increase the contact between SRBC and macrophages.
Place the plates back into the 37 °C incubator and incubate the plates for different time periods (e.g., 0, 5, 10, 20, 40, and 60 min).
Remove the plates from the incubator and place on ice, after the indicated time periods.
Remove the cell medium and wash the cell monolayers with 200 μL of cold PBS followed by adding 100 μL of ACK lysis buffer and placing the plates on ice for 1 min to remove the SRBC not phagocytosed (see Note 7).
Remove the ACK lysis buffer and wash the cells with cold PBS again and fix cells by adding 100 μL of fresh cold 2.5 % glutaraldehyde for 15 min on ice.
After fixation, the fixed cells are washed twice with PBS and stain the cells with eosin for 5 min at room temperature.
Next, wash the cells with PBS three times and leave PBS in the wells.
Capture digital images under an inverted microscope.
Calculate the phagocytic index according to the following formula: Phagocytic index = (total number of engulfed cells/number of macrophages containing engulfed cells) × (number of macrophages containing engulfed cells/total number of counted macrophages) × 100.
3.1.2 Phagocytosis of Inert Latex Beads
Opsonization and Fluorescence Labeling (See Note 8)
Resuspend 40 μL of 10 % suspension of latex beads in 500 μL of PBS and pellet the beads by centrifugation for 30 s at 5,000×g in a microcentrifuge, discard the supernatant, and resuspend the beads in 500 μL of fresh PBS.
Repeat step 1 twice to wash the beads.
At the third wash, resuspend the beads in 190 μL of PBS and add 10 μL of IgG (stock concentration: 20 mg/mL) (see Note 9).
Mix gently and incubate the beads at room temperature for 60 min.
Wash the mixtures three times with PBS to remove unbounded antibody and resuspend the beads in 500 μL PBS.
Label the beads with fluorochrome for detection by fluorescence microscopy or flow cytometry by adding 5 μL of fluorochrome-conjugated (e.g., FITC) secondary antibody to 500 μL opsonized beads.
Mix gently and incubate at room temperature for 60 min.
Wash the mixtures four times with PBS to remove unbounded antibody and resuspend in 500 μL PBS (see Note 10).
Phagocytosis
Place cells in the wells of 24-well plates at 1×105 cells/well and incubate at 37 °C, 5 % CO2 overnight.
Add 15 μL of the labeled opsonized bead suspension to each well.
Shake the plate to ensure even distribution of the beads.
Centrifuge the plates at 300×g for 1 min to sink the beads onto the cells. Alternatively, place the plate on ice for 10 min to allow the beads to slowly settle on the cells without being internalized. Wash the cells once with media prewarmed to 37 °C and proceed to the following step.
Incubate the plates at 37 °C for 15 min to allow proper phagocytosis process. Different incubation times should be optimized for different experiments.
Internal/External Particles Discrimination (See Notes 11 and 12)
Place the plates on ice to stop phagocytosis and wash the cells with ice-cold DMEM three times to remove unbound beads.
Stain the external beads with a secondary antibody labeled with a fluorochrome different from the one used in Subheading “Opsonization and Fluorescence Labeling” (e.g., Rhodamine labeled). Dilute the secondary antibody 100-fold in cold PBS and add 200 μL of diluted antibody to each well and incubate the cells on ice for 10 min.
Wash the cells five times with ice-cold PBS.
Cell Identification (See Note 13)
Remove supernatant from cultured cells and then add 500 μL of 4 % paraformaldehyde in PBS into each well and incubate at room temperature for 20 min to fix the cell.
Stain the cell nuclei with Hoechst 33342 (8 μg/mL) in PBS for 10 min (see Note 8 (DAPI can also be used to stain nuclei).
Remove the excess dye by washing two times with PBS.
Maintain the cells in PBS until analysis by fluorescence microscopy.
Determination of Phagocytic Index
After phagocytosis, external particle discrimination, and cell identification, the images of macrophages are acquired by fluorescence microscopy.
Quantitation can be performed by using image processing software or simply counting cells one by one.
The efficiency can be expressed as the phagocytic index, which is defined as the number of latex beads ingested by 100 macrophages:
3.2 Phagocytosis of Bacteria
3.2.1 Preparation of Phagocytes and Phagocytosis Assay
Microscopic quantification of phagocytosis of bacteria by double-cycle immunofluorescence staining (see Note 14).
The day before phagocytosis assay, seed macrophages at a density of 1.8–2×105 cells/mL in complete RPMI medium on sterile glass coverslips contained in 24-well cultures plates. It is best for the cells to reach ~75 % confluency at the time of experiment.
Measure optical density at 600 nm (OD600) of a mid-log phase bacteria culture with a spectrophotometer and estimate the bacteria number.
Transfer 109 bacteria to a new microcentrifuge tube. Sediment bacteria by centrifuging at maximum speed (~20,000×g) for 1 min with a table-top centrifuge.
Discard supernatant and resuspend with 1 mL sterile PBS.
Repeat the wash by sedimentation and resuspension in sterile PBS.
Dilute bacteria with prechilled complete RPMI medium to the desired density so that the multiplicity of infection (MOI) will be 5 or 10 when added to the cells.
Place the 24-well culture plates with macrophages on ice.
Aspirate culture medium and add the prepared bacteria suspension to the cells (MOI = 5 or 10 in 1 mL/well).
Centrifuge the culture plate at 600×g, 4 °C for 5 min to facilitate bacteria adherence to cells.
Wash the cells gently with ice-cold medium to remove unbound bacteria.
Remove the culture plate from ice. Initiate phagocytosis by replacing cold medium with 1 mL prewarmed (37 °C) medium per well. Place the culture plate in 37 °C incubator.
At 5, 10, 20, 30, 40, 60, 90, and 120 min postinfection (or any other desired time points), stop phagocytosis by placing the culture plate back on ice.
Wash the cells twice with ice-cold PBS and proceed to immunofluorescence staining.
3.2.2 Double-Cycle Immunofluorescence Staining
To discriminate between extracellular bacteria that adhere to the cell surface and intracellular bacteria that have been phagocytosed, two cycles of staining will be performed on the infected cells.
Keep cells on ice. Add blocking buffer to the cells and incubate for 10 min.
Remove blocking buffer and replace with primary antibody (e.g., rabbit-anti-Listeria) diluted in blocking buffer. Incubate for 45 min on ice, and then wash for three times with washing buffer.
Add appropriate fluorochrome-conjugated secondary antibody (e.g., Alexa Fluor 488-conjugated goat anti-rabbit IgG) diluted in blocking buffer. Incubate for 30 min on ice followed by three washes with washing buffer.
Fix cells with 4 % paraformaldehyde for 10 min on ice. Wash cells three times with washing buffer.
Incubate cells with permeabilization buffer for 10 min at room temperature (RT).
Remove blocking buffer and incubate cells with primary antibody (e.g., rabbit-anti-Listeria) diluted in blocking buffer for 45 min at RT. Wash three times with washing buffer.
Add secondary antibody conjugated with a fluorochrome different from that used in the first cycle of staining (e.g., Alexa Fluor 647-conjugated goat anti-rabbit IgG) diluted in blocking buffer. Incubate for 30 min at RT.
(Optional) Add rhodamine-conjugated phalloidin simultaneously with secondary antibody to assist identification of cell boundaries (see Note 15).
Wash cells for three times with washing buffer.
Mount samples on glass coverslides with antifade mounting medium.
View samples with a fluorescence microscope. Examine cells by using a 40× or greater oil-immersion objective. Count the number of single-labeled bacteria in at least 200 cells. Single-labeled bacteria represent intracellular bacteria, while double-labeled bacteria are extracellular bacteria.
Calculate phagocytic index according to this formula: (total number of engulfed bacteria/total number of counted macrophages) × (number of macrophages containing at least one bacterium/total number of counted macrophages) × 100.
3.3 Flow Cytometric Phagocytosis Assay (See Note 16)
3.3.1 Label Bacteria with FITC
Transfer 5×108 bacteria to a new microcentrifuge tube. Sediment bacteria by centrifuging at ~20,000×g for 1 min with a table-top centrifuge. Discard supernatant and resuspend with 1 mL sterile PBS.
Centrifuge again, discard supernatant, and resuspend in 1 mL of 1 mg/mL FITC in PBS, pH 8.0. Incubate at RT on an end-over- end rotator for 40 min.
Wash labeled-bacteria for three times with PBS as in step 1. After last wash, fix bacteria in 1 mL of 1 % glutaraldehyde. Incubate for 20 min at room temperature.
Quench free glutaraldehyde by adding 100 μL of 1 M glycine. Incubate for 10 min at RT.
Wash with PBS as in step 1 and resuspend in 500 μL sterile PBS. Keep on ice.
3.3.2 Initiation of Phagocytosis and Analysis with Flow Cytometry (See Note 17)
Dilute FITC-labeled bacteria with ice-cold complete medium and add to adherent phagocytes grown in wells of a 24-well culture plate with a MOI of 10.
Centrifuge the culture plate at 600×g, 4 °C for 5 min to accelerate bacteria deposition and adherence to cells.
Wash cells gently with ice-cold medium to remove unbound bacteria.
Remove the culture plate from ice. Initiate phagocytosis by replacing cold medium with 1 mL prewarmed (37 °C) medium per well. Place the culture plate in 37 °C incubator. As a negative control, keep one sample at 4 °C the whole time so phagocytosis is not initiated.
At desired time points (e.g., 30 and 60 min postinfection), stop phagocytosis by adding 200 μL per well of ice-cold 0.02 % EDTA/PBS and move the culture plate back on ice. Wash the cells twice with ice-cold PBS.
Detach cells from the culture plate with 0.5 % trypsin/EDTA or nonenzymatic cell dissociation buffer and collect cells in a new microcentrifuge tube. Spin down cells and resuspend in 240 μL of PBS containing 1 % FBS. Pipet gently repeatedly to ensure cells are dispersed into a single-cell suspension.
Keep cells on ice and add 60 μL (1/5 total volume) of 1.25 mg/mL trypan blue to quench extracellular FITC.
Transfer cells to a polypropylene tube suitable for use with flow cytometry and analyze immediately.
Fig. 1.
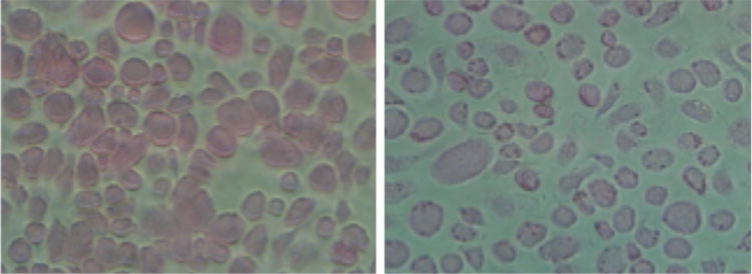
Non-opsonized SRBC (left panel) and opsonized SRBC (right panel) incubated with macrophages for 10 min at 37 °C
Footnotes
The particle size used for phagocytosis ranges from 0.5 to 3 μm, which needs to be optimized for different experiments.
The assays described in the section of phagocytosis of inert beads are performed with any of these phagocytic cells.
After centrifugation, SRBC will only form loose pellets. Be careful when removing the supernatant.
It is better to use SRBC less than 1 week old. Aged SRBC are sticky. It is better to include a set of non-opsonized SRBC as a negative control (Fig. 1). In addition to nuclear staining, cell identification can also be accomplished by using cytosol/cytoplasm labeling. There are several different methods to label the cytosol, for example, the Qtracker kits from Life Technologies can be used to label the cell and the labeling is stable through several generations. The cells can be labeled before or after phagocytosis experiments.
Usually the agglutination titer of the anti-SRBC antibody is provided from the commercial source. Use the concentration just below the agglutination titer.
Opsonized SRBC can be stored at 4 °C and should be used within several hours.
Macrophages are also destroyed by ACK lysis buffer, if the cells are exposed to the buffer for too long.
The opsonization of inert latex beads is important for the phagocytes to recognize the inert beads as foreign particles. By the use of fluorochrome-conjugated antibodies that recognize the antibodies coated on the surface of the beads, we can determine the phagocytic index.
In this protocol, we mainly focus on using IgG to coat the latex beads for FcγR-mediated phagocytosis. One can also use complement component C3bi to opsonize the beads to study complement-mediated phagocytosis.
The opsonized beads can be stored at 4 °C for 5 days. Before performing phagocytosis, the particles should be vortexed vigorously for 5 s and pass through a 25-gauge needle to disrupt any text aggregates.
The fluorescence can also be quenched by using trypan blue at a concentration of 250 μg/mL in PBS. 0.5 mL of trypan blue is added to each well and the mixtures are kept for 2 min at room temperature.
When phagocytosis is complete, it is important to determine whether those particles are internalized or just adhere to cell membrane. This can be achieved by using differential labeling the external beads with different fluorochrome-labeled secondary antibody. Since the internal beads cannot be detected by the secondary antibody, the internal beads will show only the color of the first fluorochrome, while the external beads will show two different fluorescence signals.
To quantitate phagocytosis, it is important to determine the exact number of phagocytes as well as the internal and external particles. There are several different ways to identify the cells. Here in this protocol, we use nuclear staining to identify individual cells.
This protocol describes a method to measure the ability of adhered phagocytes to ingest bacteria. Bacterial suspensions are added to cells grown on glass coverslips and centrifuged to enhance contact between the bacteria and cells. Extracellular bacteria are removed by washing (these steps are performed at 4 °C). Phagocytosis is initiated by adding a fresh medium pre-warmed at 37 °C and continued at 37 °C, and it is terminated at different time points by placing the cells back on ice. Bacteria are then immunostained and visualized by using a fluorescence microscope. Despite the washing step, some extracellular bacteria may remain adhered to the cell surface, and it may be difficult for the experimenter to distinguish these extracellular bacteria from those truly internalized. A double-cycle immunofluorescence staining method solves this problem by staining extracellular bacteria first, followed by cell permeabilization and a second round of staining of bacteria. Using secondary antibodies conjugated with different fluorochromes that emit different colors, one can easily discriminate intracellular single-stained bacteria from extracellular double-stained bacteria.
Phalloidin selectively binds to F-actin with high affinity. Combining phalloidin staining with bacteria staining allows visualization of the cell boundaries and quantitation of the number of engulfed bacteria per cell. Some intracellular bacteria (e.g., Listeria monocytogenes and Shigella flexneri) are capable of escaping from the vacuole and polymerizing host actin to facilitate their movement inside the cells. Phalloidin also stains the actin tail of these bacteria.
An alternative approach to analyzing bacterial phagocytosis is to allow the cells to phagocytose fluorescence-labeled bacteria and measure the fluorescence intensity of the cells by flow cytometry. Most gram-positive bacteria can be easily stained by fluorescein isothiocyanate (FITC). Following internalization of FITC-labeled bacteria by cells, extracellular FITC on the bacteria bound to the cell surface but not internalized can be quenched by trypan blue. Therefore, the fluorescence detected is from the phagocytosed FITC-labeled bacteria only.
Compared to the control samples kept at 4 °C throughout the experiment, cells that have phagocytosed FITC-labeled bacteria show a shift in fluorescence intensity. Although this method does not reveal the absolute number of bacteria phagocytosed by each cell, the relative phagocytosis ability can be compared according to the fluorescence intensity shift.
References
- 1.Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 2.Farnworth SL, Henderson NC, Mackinnon AC, Atkinson KM, Wilkinson T, Dhaliwal K, Hayashi K, Simpson AJ, Rossi AG, Haslett C, Sethi T. Galectin-3 reduces the severity of pneumococcal pneumonia by augmenting neutrophil function. Am J Pathol. 2008;172:395–405. doi: 10.2353/ajpath.2008.070870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linnartz B, Bodea LG, Neumann H. Microglial carbohydrate-binding receptors for neural repair. Cell Tissue Res. 2012;349:215–227. doi: 10.1007/s00441-012-1342-7. [DOI] [PubMed] [Google Scholar]
- 4.Barrionuevo P, Beigier-Bompadre M, Ilarregui JM, Toscano MA, Bianco GA, Isturiz MA, Rabinovich GA. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J Immunol. 2007;178:436–445. doi: 10.4049/jimmunol.178.1.436. [DOI] [PubMed] [Google Scholar]
- 5.Stowell SR, Karmakar S, Arthur CM, Ju T, Rodrigues LC, Riul TB, Dias-Baruffi M, Miner J, McEver RP, Cummings RD. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol Biol Cell. 2009;20:1408–1418. doi: 10.1091/mbc.E08-07-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I, Izui S, Liu FT. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112:389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buschow SI, Lasonder E, Szklarczyk R, Oud MM, de Vries IJ, Figdor CG. Unraveling the human dendritic cell phagosome proteome by organellar enrichment ranking. J Proteomics. 2012;75:1547–1562. doi: 10.1016/j.jprot.2011.11.024. [DOI] [PubMed] [Google Scholar]