

Abstract
DNA methylation plays a major role in the epigenetic regulation of gene expression. Although a few DNA methylation profiling studies of porcine genome which is one of the important biomedical models for human diseases have been reported, the available data are still limited. We tried to study methylation patterns of diverse pig tissues as a study of the International Swine Methylome Consortium to generate the swine reference methylome map to extensively evaluate the methylation profile of the pig genome at a single base resolution. We generated and analysed the DNA methylome profiles of five different tissues and a cell line originated from pig. On average, 39.85 and 62.1% of cytosine and guanine dinucleotides (CpGs) of CpG islands and 2 kb upstream of transcription start sites were covered, respectively. We detected a low rate (an average of 1.67%) of non-CpG methylation in the six samples except for the neocortex (2.3%). The observed global CpG methylation patterns of pigs indicated high similarity to other mammals including humans. The percentage of CpG methylation associated with gene features was similar among the tissues but not for a 3D4/2 cell line. Our results provide essential information for future studies of the porcine epigenome.
Keywords: pig, DNA methylation, methylome, epigenetics, RRBS
1. Introduction
DNA methylation is a mechanism behind epigenetic changes in gene expression in diverse species including mammals.1,2 5-Methylcytosine plays an important role in gene silencing, genomic imprinting,3,4 X-chromosome inactivation,5 cancer progression,6,7 embryonic development and tissue differentiation.8–11 Recent studies reported the tissue- or cell line-specific difference of DNA methylation which contributes to regulating tissue- or cell-specific gene expression.12–16 Cell immortalization can lead to the accumulation of DNA methylation at promoter regions, resulting in corresponding changes in gene expression.17
In mammals, DNA methylation occurs preferentially at cytosine and guanine dinucleotides (CpGs), which tend to cluster in highly enriched short regions (0.2–2 kb) known as CpG islands (CGIs); ∼50–60% of promoter regions are associated with CGIs.18,19 The levels of CpG methylation at CGI and promoter regions are lower than those at other CpG sites19,20 and play an important role in regulating gene expression.21
Several methods including bisulfite sequencing, reduced representation bisulfite sequencing (RRBS),19,22 methylated DNA immunoprecipitation (MeDIP-seq),23 methyl-binding domain sequencing (MBD-seq, MethylCap-seq)24–26 and BeadChip array (Infinium27) have been developed to analyse patterns of DNA methylation across diverse genomes.2,28,29 Aberrant methylation patterns on chromosomes or a part of genome can cause developmental abnormalities or diseases.30 Several large-scale projects including the International Human Epigenome Consortium (http://www.ihec-epigenomes.org/), human ENCODE project (https://www.encodeproject.org/) and Blueprint Epigenome (http://www.blueprint-epigenome.eu/) are in progress for a deeper understanding of the functional importance of epigenetic changes including DNA methylation. Recently, the International Swine Methylome Consortium (ISMC) was organized to generate the swine reference methylome map to illuminate the importance of epigenetic changes to phenotypic variations in production traits31–34 and disease progression.30,35–39 Although genome-wide methylation studies of farm animals have recently been published for sheep,40 cattle,41,42 horses43 and pigs,44 the depth of data and tissue coverage are still limited compared with that of humans and mice.
The importance of domestic pigs (Sus scrofa domesticus), as large animal biomedical models for human diseases, has recently been recognized for its similarity to humans in organ size and structure, general physiology and disease phenotypes.45 Advantages in animal husbandry, large litter size and short generation interval of pigs make them more suitable as animal models over primate models, although the phylogenetic relationship of pigs to humans is more distant than that of primates to humans.
The completion of the draft reference genome of the pig46 has allowed production of previously unachievable results using sequence-based genome-wide studies on variations or evolutionary changes.47–49 Although the analysis of global genome methylation using MeDIP-seq50 and RRBS44 was reported in pigs, the results from more diverse tissues at single base-pair resolution are necessary to have better understanding of the methylation profiles of the pig genome related to chromosomal variations, gene features, CGI association and non-biparental expression.
In this study, we analysed pig methylome of five different tissues including the neocortex, spleen, muscle, liver and olfactory epithelium, and a pulmonary alveolar macrophage (PAM) cell line (3D4/2) by using the RRBS method. We compared the methylation profiles across different tissues and described the similarity and differences of the pig methylome from human and mice methylomes in previously reported RRBS analysis. Further, we included the results of methylome analysis from several previously unreported tissues and demonstrated the successful use of RRBS on pig genome.
2. Materials and methods
2.1. Ethics statement
The experimental procedure was approved and supervised by the Institute of Animal Care and Use Committee of Konkuk University, Seoul, Korea. Three individuals of 9-week-old Landrace × Yorkshire crossbreed pigs in good health were purchased from local farm, deeply anaesthetized and sacrificed.
2.2. Sample preparation and genomic DNA isolation
Target tissues including the neocortex, spleen, liver, femoral muscle and olfactory epithelium were dissected out, snap frozen in liquid nitrogen and kept at −80°C until use. Pig pulmonary alveolar macrophages cell line 3D4/2 (ATCC® CRL-2845™) was cultured in T75 cell culture flasks (SPL Life Sciences, Pocheon, Korea) using an RPMI 1640 culture medium (ATCC modification) (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and 1% Penicillin–Streptomycin (Life Technologies) and 0.1 mM 2-mercaptoethanol. Genomic DNA was extracted from 0.5 g tissue or from 3 × 107 cells. Briefly, tissues were incubated in 400 µl of tissue lysis buffer (0.1 M Tris–HCl, 200 mM NaCl, 5 mM EDTA and 0.2% SDS) with 250 µg/ml proteinase K at 55°C for 6 h, and cell pellets were incubated in 400 µl lysis buffer (10 mM Tris–HCl, 100 mM EDTA and 0.5% SDS) at 37°C for 6 h, followed by phenol extraction. Isolated DNA was incubated with 20 µg/ml DNase-free RNase and purified using a PowerClean® DNA Clean-Up Kit (MO BIO, Carlsbad, CA, USA) according to the manufacturer's protocol.
2.3. Sequencing and reads alignment
The construction of RRBS libraries and paired-end sequencing using a HiSeq2000 analyzer (Illumina, San Diego, CA, USA) was performed at BGI-Shenzhen (Shenzhen, China) according to the manufacturer's instructions. Briefly, 100 ng of pooled genomic DNA with an equal amount from three individuals was used for the construction of each RRBS library. Genomic DNA was digested with MspI enzyme (recognition site C^CGG, NEB, Ipswich, MA, USA) at 37°C for 16 h. After purification, single A nucleotides were added to the digested blunt-end products, followed by ligation to methylated adapter with T overhang. Ligated products corresponding to DNA fragments 40–220 bp long were purified by 2% agarose gel electrophoresis. Bisulfite conversion was conducted using a ZYMO EZ DNA Methylation-Gold™ Kit (ZYMO, Irvine, CA, USA). The final libraries were generated by PCR amplification. RRBS libraries were analysed by an Agilent 2100 Bioanalyzer (Agilent Technologies) and quantified by real-time PCR. The sequenced paired-end read length was 49 bp. Raw sequencing data were processed by an Illumina base-calling pipeline. Clean reads were aligned to the modified pig reference genome (Sscrofa10.2) in an unbiased way for bisulfite sequencing data. After removing adaptor sequences, clean reads were aligned to two modified references with BS-seeker 2 v. 2.0.851 using Bowtie 2 v. 2.1.052 in local alignment mode and allowing no more than four mismatches per read. Methylation status was determined using the bs_seeker2-call_methylation.py script and only uniquely aligned reads. For the simulation analysis of pig genome RRBS, we performed an in silico restriction analysis of the current pig reference genome (Sus scrofa 10.2) for MspI or MspI/ApeKI and set the fragment size to 40–220 bp.
2.4. RRBS data analysis
Methylation levels of cytosines were analysed by methylKit.53 Briefly, the number of methylated and unmethylated CpG and non-CpG (CHG and CHH, H representing A/C/T) sites was counted for each region. CGIs were defined as regions >200 bp with a GC fraction >0.5 and an observed-to-expected ratio of CpG >0.6. CGI shores were defined as regions 2 kb in length adjacent to CGIs.18 The porcine CGIs annotation was downloaded from USCS (http://genome.ucsc.edu/, susScr3). The porcine gene annotation was downloaded from the Ensembl (http://www.ensembl.org/, protein-coding genes). To define the differentially methylated cytosines (DMCs), multiple pairwise comparisons were performed against CpG methylation information of six samples and filtered (q < 0.01) using methylKit.53 Tissue-specific differentially methylated CpG sites (tDMC) were identified by a script which we developed to select the CpGs showing tissue-specific methylation levels in comparison to other samples. The result of each comparison among different tissues was compiled. Filtered DMCs were annotated based on genes and CGI features including gene bodies and upstream 2, 5 and 10 kb regions from the TSS. The number of DMCs was visualized by Venn Diagrams using the Jvenn (http://bioinfo.genotoul.fr/jvenn/index.html).54
2.5. Functional annotation of tDMC and correlation analysis between the methylation level and gene expression
Gene ontological (GO) analysis was carried out for tDMC using an in-house developed script. The swine GO terms were downloaded from the Ensembl (http://www.ensembl.org/) and the gene sets obtained from tDMC analysis for each tissue. Gene expression profiles were downloaded from the Gene Expression Omnibus (GEO). The gene expression profiles created with 27k cDNAs probes and 23 healthy porcine tissues (GEO accession ID: GSE4918)55 were downloaded. The tissues and accession numbers used in this study are frontal cortex (GSM110567 and GSM110545), liver (GSM110543 and GSM110569), biceps femoris (GSM110549 and GSM110577) and spleen (GSM110571 and GSM110546), respectively. Differentially expressed genes (DEGs) across four tissues were identified using the GEO2R plugin (P < 0.05) after the adjustment for multiple testing. The correlation between the levels of gene expression and methylation in tDMCs was calculated by the Pearson's correlation test.
2.6. Statistical analysis
All statistical analyses were performed using R programming language (version 3.1.1).56 Statistical difference in methylation rates of CpG and non-CpG sites across tissues was determined using the Wilcoxon signed-rank test. The Pearson's correlation test was used to compare the global methylation patterns and the specific methylation patterns for CGI and CGI shore across tissues which were implemented using methylKit package (version 0.9.2).53 The clustering analysis with the methylation levels and patterns of different samples was carried out using Ward's minimum variance method implemented in methylKit package.53 The statistical significance of enrichments and/or depletions of GO was calculated by the hypergeometric test57 and multiple test correction using R language.
3. Results
3.1. In silico analysis of pig genome to predict possible coverage of CpG sites from RRBS
In silico restriction analysis of the pig genome using enzymes MspI was carried out to predict the possible coverage of CpG sites from the RRBS analysis of the pig genome. The in silico pig genome RRBS library consisting of DNA fragments in a size range of 40–220 bp resulted in a total of 2,371,480 fragments for MspI digestion.
We performed in silico analyses of the distribution pattern of restricted fragments of the pig genome in the window of 200 bp with a 20-bp increment, starting from 200 and up to 500 bp for MspI digestion. In addition, 40–220 bp window was analysed as a comparison to the result of humans and mice (Supplementary Fig. S1). The results were similar to those of humans and mice in which the 40- to 220-bp window covers 37.8% of total fragments of MspI-digested libraries.58
Our in silico analysis showed that 28% of generated fragments digested by MspI was expected to be distributed in the 40- to 220-bp window, which corresponds to 2.6% of the pig genome. For CpG sites, 11.9% of the total CpG sites of the pig genome were predicted in MspI libraries (Table 1 and Supplementary Fig. S1). The size of the pig reduced representation (RR) genome was the closest to that of the human.
Table 1.
Comparison between porcine and other reduced representation (RR) genomes
| Genome |
MspI |
|||
|---|---|---|---|---|
| Human | Mouse | Zebrafish59 | Pig | |
| Size of genome (Gb) | 3.2 | 2.8 | 1.41 | 2.8 |
| Size of RR genome (Mb) | 74 | 38 | 31 | 73 |
| Per cent of whole genome | 2.3 | 1.4 | 2.2 | 2.6 |
| GC content of whole genome (%) | 40.9 | 41.7 | 36.5 | 42.3 |
| Number of fragments (RR genome) | 647,626 | 333,104 | 264,598 | 664,080 |
| Number of CpG sites (RR genome) | 4,068,947 | 1,506,712 | 1,430,390 | 5,586,410 |
| Per cent of total genomic CpG sites | 13.5 | 7 | 5.3 | 18.30 |
All RR genomes were simulated by selecting DNA fragment in a 40- to 220-bp window after in silico restriction enzyme digestion.
3.2. Mapping and analysis of RRBS reads from five tissues and a cell line
We constructed MspI single restriction RRBS libraries as an initial study of the RRBS analysis of the pig genome. According to the in silico analysis, the coverage of nearly 54.71% of total CpGs within CGI (1,295,120 of 2,367,139 total CpGs within CGI) and ∼83.49% of CpGs within the upstream 2 kb region from the transcription start sites (TSS) (250,631 of 300,187 protein-coding genes) was expected in the MspI-digested pig RRBS library.
A total of six libraries were constructed with 40- to 220-bp insert fragments, which is identical to RRBS studies in humans and mice,58 from five tissues including the neocortex, spleen, liver, muscle, olfactory epithelium and a 3D4/2 (PAM) cell line. We generated a minimum of 3.25Gb clean reads from each RRBS library with the maximum of 5.48Gb from the liver (Table 2). The sequences were submitted to European Nucleotide Archive (ENA) under the study accession number, PRJEB9561. An average of 59.56% of total paired-end reads were aligned to the reference genome according to the criteria described in Materials and methods (Table 2). The reads with a single placement, minimum number of mismatches (4 mismatches per read) and clear strand assignment were defined as uniquely mapped reads. The percentage of uniquely mapped reads was 56.89, 57.16, 50.77, 51.76, 53.56 and 55.08% for the neocortex, spleen, liver, muscle, olfactory epithelium and 3D4/2 cells, respectively, with the average of 54.2%. The frequency of multi-mapped reads ranged from 4.92 to 5.74% for different samples (Table 2). The coverage of CpGs detected within CGIs and the upstream 2 kb region from TSS ranged from 38.3 to 43.32% and 58.75 to 67.8% of total CpGs in the genome, respectively. This is somewhat lower than the theoretical predicted value (54.71% for CGI and 83.49% for upstream 2 kb from TSS) of the simulation because of the efficiency difference between the experiment and theoretical analysis (Supplementary Table S1). The results from our experimental analysis are in accord with the expected values from the simulation, indicating the validity of our data.
Table 2.
Details of mapping of porcine RRBS libraries
| Clean read (M) | Clean read (Gb) | Mapped rate (%) | Uniquely mapped rate (%) | Multiple mapping (%) | CpG methylation (%) | Non-CpG methylation (%) | |
|---|---|---|---|---|---|---|---|
| 3D4/2 | 66.3 | 3.25 | 60.83 | 55.08 | 5.74 | 53.05 | 1.71 |
| Neocortex | 76.8 | 3.76 | 62.29 | 56.89 | 5.40 | 50.87 | 2.30 |
| OE | 67.6 | 3.31 | 58.48 | 53.56 | 4.92 | 49.19 | 1.63 |
| Spleen | 72.6 | 3.56 | 62.38 | 57.16 | 5.22 | 52.84 | 1.66 |
| Liver | 111.84 | 5.48 | 56.17 | 50.77 | 5.40 | 47.83 | 1.67 |
| Muscle | 85.1 | 4.17 | 57.20 | 51.76 | 5.45 | 48.06 | 1.69 |
OE: olfactory epithelium.
Subsequently, we selected CpG sites that were present >10 times in the mapping, namely CpG10, and subjected them to further analyses. The mean coverage of CpG10 was 37- to 56.5-fold among tissues (Supplementary Table S2). The frequency of CpG10 was similar across the six samples, and the average coverage rates of CpG10 for gene-related regions including exon, intron and 10 kb upstream of TSS were 50.88, 44.38 and 42.58%, respectively (Supplementary Fig. S2).
3.3. Global methylation profile of the pig RR genome
RRBS analysis of mammalian genome showed the enrichment of CpG-rich regions2,60 and the presence of a large amount of unmethylated CpG sites in CGIs.19,61 The percentage of methylated CpG sites in the pig RR genome ranged from 47.83 to 53.05% in the six different samples (Table 2). The distribution of read coverage per base for each sample shows that the results did not much suffer from PCR duplication bias, which forms a secondary peak towards the right side of the histograms (Supplementary Fig. S3).53
Methylated CpG sites can be classified by the methylation levels to hypo- (<10%), intermediate (20–80%) and hyper-methylated CpGs (>90%), and the frequencies of three differently methylated CpG10 groups ranged from 34.5 to 40.2%, 18.3 to 23.8% and 21.4 to 28.5%, respectively, of total CpG sites, depending on the type of tissue, showing that hypo-methylated CpG sites were the largest group (Supplementary Table S3). Interestingly, relatively higher hyper- and heavily methylated CpGs (>95%) were detected from 3D4/2 cells than from other tissues (P = 0.0035 and 0.0008 for hyper- and heavy methylation, respectively; Supplementary Fig. S4 and Table S3). The distribution of percentage methylation from our study using five tissues and a cell line showed a pattern analogous to the bimodal distribution (Supplementary Fig. S4), which is similar to the previous mammalian RRBS studies.19,62,63
Our analysis showed that the global CpG methylation is similar among the neocortex, spleen, liver, femoral muscle and olfactory epithelium (Pearson's correlation score: 0.95–0.96). However, comparisons of tissues versus 3D4/2 cell line showed significant difference in the distribution of the CpG methylation (Pearson's correlation score: 0.69–0.7, Fig. 1A). The distance among different tissues based on the global methylation level is shown in Fig. 2. CpG methylation among six different samples showed that the levels of methylation of 2 kb upstream of TSS and CGIs of the PAM cell line (3D4/2) were significantly higher than those in other tissues (Fig. 1 and Supplementary Fig. S5). Regarding the results of the PAM cell line, we manually confirmed the methylation patterns of several randomly selected CpG sites detected in our RRBS by independent bisulfite sequencing. A total of 7 DMRs were amplified from the bisulfite-treated genomic DNA template and sequenced. The seven DMRs were located on four hypo-methylated (ABLIM2, CBFA2T3, SDK1 and TBCD)- and three hyper-methylated genes (CD14, DKC1 and GPC3)-associated regions in the PAM cell line. All results were consistent with those from the RRBS analysis of 3D4/2 (data not shown).
Figure 1.

Correlation analysis of the CpG methylation patterns among different tissues. Scatter plots of percentage methylation values for pairwise comparisons in six libraries [pulmonary alveolar macrophage (PAM) cells, neocortex, spleen, liver, muscle and olfactory epithelium (OE)]. Colours in the scatter plot indicate the number of CpG sites with identical methylation pattern (methylated or non-methylated); yellow denotes large number of correlations, blue denotes lack of correlation and green denotes different methylation patterns. Numbers in the upper right side represent the pairwise Pearson's correlation scores. Histograms on the diagonal are methylation distribution of CpG sites for each sample. Comparisons of global CpG methylation level (A), methylation level of CpG in CGI regions (B) and CGI shore (C).
Figure 2.

Hierarchical cluster analysis according to methylation patterns across different tissues. Distance between tissues according to their methylation patterns was estimated by the Ward's minimum variance method.
The rates of methylation for non-CpG sites (CHG and CHH; H representing A/C/T) varied from 1.63 to 1.71% of the total non-CpGs except for the neocortex, which showed slightly higher amount of methylated non-CpGs (2.3% of the total) compared with other tissues (Table 2 and Fig. 3). This result is consistent with that in a previous report in humans.64 We also observed differences in the level of methylation of 2 kb upstream of TSS and CGI regions of non-CpG sites (CHG and CHH) from the PAM and neocortex (Fig. 3B, C and D). Also, the percentage of methylated non-CpG sites in the 2 kb upstream region of TSS of the liver was higher over the spleen, muscle and olfactory epithelium, which was contributed by higher CHG methylation rate of the 2 kb upstream region of TSS in the liver than others.
Figure 3.

Comparison of the percentage methylation level of CpG and non-CpG sites among different tissues. Methylation level was indicated according to different criteria including global, 2 kb upstream of TSS (up2 kb) and CGI. Non-CpG methylation was separately evaluated for mCHG and mCHH. PAM, pulmonary alveolar macrophage; OE, olfactory epithelium. Percentage methylation of CpG sites (A), non-CpG (B), CHG (C) and CHH (D) for global, up2 kb and CGI. ** denotes P < 1 × 10−5, * denotes P < 1 × 10−3.
3.4. Methylation of CGI and CGI shore
Correlation of methylation levels among tissues was performed at CGIs and CGI shores (2 kb regions on either side of CGIs), which constitute the largest portion of CpG10 with its CGI features such as CGI, CGI shore and CGI shelf (regions 2 kb further from CGI shores) (Supplementary Fig. S6). More than half of CpG10 (60.2–62.4%) were mapped to CGIs and the CGI shores. Non-CGI regions within CpG10 ranged from 31.59 to 33.47% among the tested tissues. The rest of CpG10 were mapped to the CGI shelf. The density of CpG10 gradually decreased when moving away from CGIs as reported in humans and mouse.19,65 The per cent methylation of CpG10 in CGI features was compared across tissues. The CGI shores showed a much higher degree of methylation than CGI (Fig. 1B and C), which is consistent with previous reports that the methylation level of CGI was lower than that of the CGI shore and global regions.19,65 The Pearson's correlation values among the methylation rates of the neocortex, spleen, liver, muscle and olfactory epithelium within CGI shore were from 0.91 to 0.93, which is somewhat lower than those of CGI (0.96–0.97), indicating the presence of tissue-specific methylation despite the small difference (Fig. 1). Results of individual DMCs were summarized in Supplementary Table S4.
3.5. CpG association to gene features in pig methylome
The current annotation of the Ensembl gene set for the pig genome shows 21,607 protein-coding genes (www.ensembl.org/Sus_scrofa/). Excluding genes on unplaced scaffolds and mitochondrial DNA, 19,429 genes with chromosomal assignment were used to define the gene features including exon, intron and upstream 2, 5 and 10 kb of TSS (Supplementary Fig. S2). On average, 24.1% of total CpG10 was mapped to the gene body (8.08% to exons and 16.2% to introns), and this covered 77.74% of the expected gene body regions which according to the RRBS simulation (Supplementary Table S5). Depending on the type of tissues, 6.29–6.52% and 10.24–10.49% of CpG10 were localized 5 and 10 kb upstream of TSS, respectively. The patterns of CpG10 association to gene features were consistent across different tissues (Supplementary Fig. S2).
3.6. Comparison of gene number and methylation rate in pig chromosomes
To address the influence of gene density on methylation rates of pig chromosomes, we compared the observed methylation rates with the number of expected CpG sites and the number of genes for each chromosome (Fig. 4 and Supplementary Fig. S5). The percentage methylation rate was similar across chromosomes except for chromosome Y. In most of the chromosomes, there was a strong positive correlation between the total number of CpG sites and genes. The exception was the ratio of CpG sites to gene numbers in chromosomes 3, 6 and 12, which was higher than the ratio in other chromosomes and was reversed for chromosome 13. However, as expected, the GC percentage of the chromosomes was correlated with the CpG methylation for each chromosome (Fig. 4 and Supplementary Fig. S5). We hypothesized that chromosomes with higher gene numbers could result in lower methylation level than chromosomes with lower gene density, because promoter and CGI regions showed lower levels of CpG methylation than other genomic features (Fig. 4 and Supplementary Fig. S5) and the CpG density of those regions was higher than the average. However, no correlation was observed between gene number and CpG methylation levels of chromosomes in our analysis. The observed methylation rates were similar among different tissues except for 3D4/2 cells, especially for the upstream 2 and 5 kb regions of TSS (Fig. 4).
Figure 4.

Comparison of the methylation levels in gene-related regions in relation to the numbers of genes and CpGs on individual pig chromosomes. The gene annotation data of the pig genome were obtained from Ensemble. The total number of CpG for each chromosome was obtained from the current pig genome assembly (Sscrofa10.2). Gene body (A), 2 kb (B) and 5 kb (C) upstream regions of TSS. This figure is available in black and white in print and in colour at DNA Research online.
3.7. Methylation patterns of imprinted genes from RRBS analysis of the pig genome
We analysed the RRBS reads corresponding to 22 out of 30 genes known for genomic imprinting in pigs (http://www.geneimprint.com/), which had annotation of genomic location in Sscrofa10.2 (data not shown). Among them, eight genes, DIO3, DIRAS3, MEG3, MEST, NAP1L5, NECD, NNAT, PHLDA2, satisfied our criteria to be selected as candidates for the analysis of imprinted genes, which includes clear genomic locations including gene bodies and the upstream 2 and 2–10 kb regions from TSS in the current pig genome assembly (Supplementary Table S6). Interestingly, the levels of CpG methylation of the eight known imprinted genes were significantly higher than those of randomly selected non-imprinted genes (GAPDH, COX5B and ACTA1) at gene bodies and upstream 2 kb of TSS regions in pig tissue samples (Fig. 5). The difference was less consistent in the upstream 2–10 kb region of TSS. In addition, we found a difference in the level of methylation among different tissues. Especially, the difference was the largest in 3D4/2 cells. For example, CpGs in the gene bodies of DIO3, DIRAS3, MEG3 and NNAT were highly methylated comparing with other tissues. The methylation patterns of upstream 2 and 2–10 kb regions from TSS were also similar (Fig. 5).
Figure 5.

The pattern of CpG methylation of imprinted genes from six tissues in pigs. The result was visualized by heat map. Gene body, exon and intron; up2 kb and up2–10 kb, upstream 2 kb and 2–10 kb regions from TSS, respectively.
3.8. Identification of tDMCs
DMCs were identified using methylKit from six samples (q-value <0.01, Supplementary Fig. S7 and Table S7). Among the DMCs, tissue specifically methylated CpGs were selected. A total of 63,482 and 32,410 tissue-specific DMCs (tDMCs) were identified from gene bodies and upstream 10 kb region from TSS, respectively, under the cut-off value of 30% methylation difference. Excluding the PAM cell library, only 4,780 and 1,680 tDMCs were identified from gene bodies and upstream 10 kb regions from TSS, respectively (Supplementary Fig. S8). Among the tissue samples, spleen-specific DMCs were the largest and occupied 28.7% of the total and followed by the liver (24.1%) and olfactory epithelium (23.1%). Neocortex- and muscle-specific libraries constitute 9.3 and 14.8% of the total tDMCs, respectively (Fig. 6). The tDMC profile including chromosomal positions was summarized in Supplementary Table S8.
Figure 6.
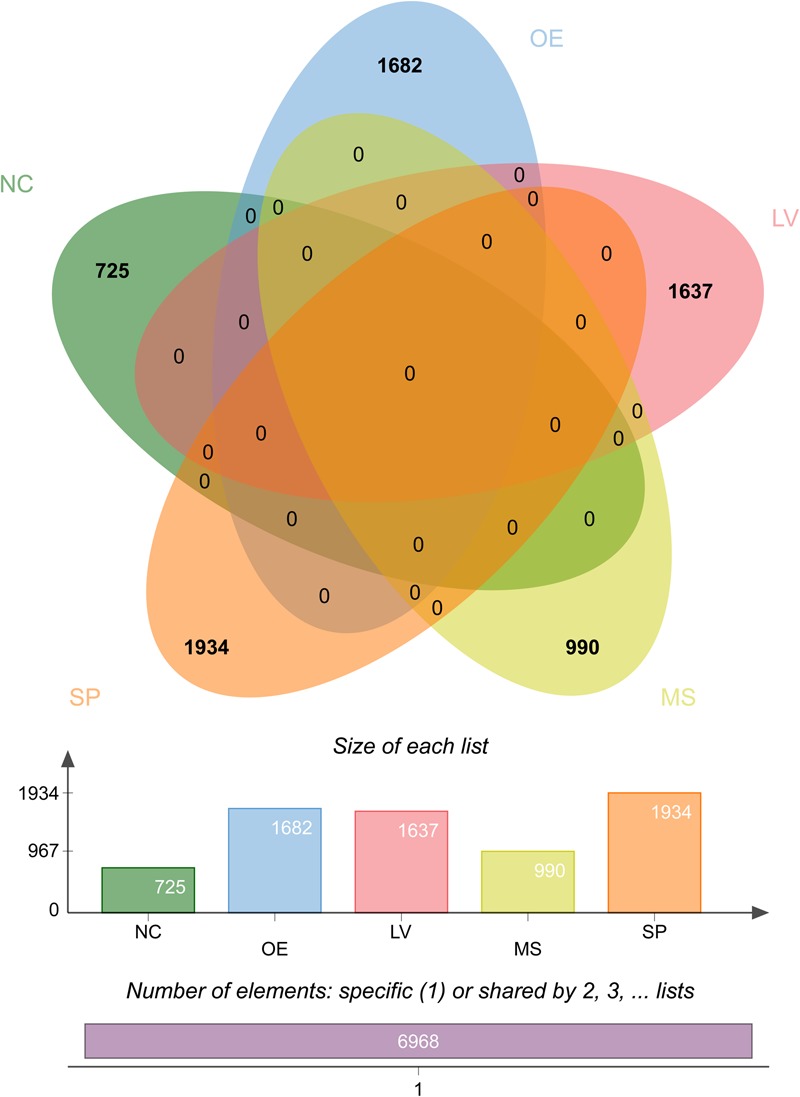
The number of tissue-specific differentially methylated cytosine (tDMCs) from five pig tissues. Venn diagrams showing the number of positions of tDMCs from each tissue. The positions of tDMCs depend on tissue type were visualized by Venn diagram. NC: neocortex; OE: olfactory epithelium; LV: liver; MS: muscle; SP: spleen. The 3D4/2 cells were not represented here. Detailed positions of tDMC were summarized in the Supplementary Table S8. This figure is available in black and white in print and in colour at DNA Research online.
3.9. Functional annotation of tDMCs
Identified tDMCs were mapped to 1,897 genes on the basis of their genomic location (Supplementary Fig. S9). GO analysis was performed for tDMCs in each tissue (Supplementary Tables S9 and S10). Statistical significance of GO terms to tDMCs was evaluated by the hypergeometric test with a multiple correction. The results showed that some of highly ranked GO were matched with characteristics of the tissues. In the neocortex, ‘olfactory behaviour’, ‘positive regulation of neural precursor cell proliferation’, ‘norepinephrine biosynthetic process’, ‘brain development’ and other neuronal signal transduction-related GO terms were significant (q < 0.05). The GO terms for functions of the liver were significantly associated, including ‘enzyme regulator activity’, ‘glycolytic process’, ‘positive regulation of heparan sulfate proteoglycan biosynthetic process’, ‘carbohydrate metabolic process’ and other metabolic activity-related terms. Similarly, ‘actin cytoskeleton’ and ‘mesodermal cell fate specification’ were specific GO terms for the muscle. ‘Leucocyte-mediated cytotoxicity’, ‘focal adhesion’ and ‘cell migration’ were significant for spleen-specific, DMCs-related genes. For PAM cells, GO terms were enriched with transcription regulation-associated terms (Supplementary Table S9). For the olfactory epithelium, we were unable to find significant GO terms matching to the biological characteristics of the tissue, which may be due to the reason that the tissue constitutes of only a small number of olfactory function-related cells together with epithelial cells.
3.10. Correlation between tDMCs and gene expression
To evaluate the influence of DNA methylation on gene expression, we analysed the association between tDMCs and gene expression profiles of the frontal cortex, liver, biceps femoris and spleen. DEGs across four tissues were identified using the GEO2R plugin (P < 0.05) from a microarrary dataset of the GEO database. First, we selected genes (n = 42) present in both tDMCs-associated groups from RRBS and DEG groups from microarrary transcriptome analyses. The Pearson's correlation test showed that a total of 55 tDMCs which associated with 27 genes showed strong negative correlation between the levels of gene expression and CpG methylation (Table 3).
Table 3.
Correlation between tDMCs and the level of gene expression from the neocortex, muscle and spleen
| Tissue type | Gene | Chra | Position | Classb |
Mc | Ed | Pearson's corr.e | |
|---|---|---|---|---|---|---|---|---|
| Neocortex | NRXN2 | 2 | 6587361 | UP10K | − | + | −0.897000684 | |
| 2 | 6635041 | Genebody | − | + | −0.838015353 | |||
| 2 | 6635090 | Genebody | − | + | −0.886097969 | |||
| 2 | 6635002 | Genebody | − | + | −0.828919384 | |||
| 2 | 6634986 | Genebody | − | + | −0.902681422 | |||
| 2 | 6634955 | Genebody | − | + | −0.802729923 | |||
| 2 | 6634980 | Genebody | − | + | −0.874609127 | |||
| 2 | 6634930 | Genebody | − | + | −0.720944026 | |||
| 2 | 6634961 | Genebody | − | + | −0.837225041 | |||
| 2 | 6634934 | Genebody | − | + | −0.785419851 | |||
| 2 | 6634951 | Genebody | − | + | −0.865604511 | |||
| MAPT | 12 | 17133831 | Genebody | − | + | −0.926451406 | ||
| 12 | 17133834 | Genebody | − | + | −0.922834214 | |||
| SYNE1 | 1 | 16124463 | Genebody | − | + | −0.80723621 | ||
| 1 | 16124489 | Genebody | − | + | −0.750224969 | |||
| Muscle | CCDC88C | 7 | 119491407 | Genebody | + | − | −0.936174813 | |
| EXOC2 | 7 | 75292 | Genebody | CGI shore | + | − | −0.851066176 | |
| 7 | 75340 | Genebody | CGI shore | + | − | −0.832158353 | ||
| FGR | 6 | 78329214 | Genebody | CGI shore | + | − | −0.802870198 | |
| 6 | 78336406 | Genebody | + | − | −0.755515526 | |||
| MAP2K7 | 2 | 71712421 | UP10K | + | − | −0.857718538 | ||
| NR5A2 | 10 | 27361479 | Genebody | − | + | −0.957673269 | ||
| 10 | 27361475 | Genebody | − | + | −0.95434547 | |||
| 10 | 27361468 | Genebody | − | + | −0.921209644 | |||
| 10 | 27361450 | Genebody | − | + | −0.960461808 | |||
| PCOLCE | 3 | 7921803 | Genebody | − | + | −0.995207087 | ||
| RAB22A | 17 | 65753356 | Genebody | CGI shore | + | − | −0.884084122 | |
| SLA-7 | 7 | 27608918 | UP10K | + | − | −0.914258584 | ||
| TUBGCP6 | 5 | 110682055 | Genebody | CGI shore | + | − | −0.90531831 | |
| VGLL2 | 1 | 49880654 | Genebody | CGI shore | − | + | −0.753634499 | |
| 1 | 49880666 | Genebody | CGI shore | − | + | −0.805639146 | ||
| XPO4 | 11 | 268954 | Genebody | CGI shore | + | − | −0.909143259 | |
| 11 | 269035 | Genebody | + | − | −0.914121809 | |||
| Spleen | ICAM-1 | 2 | 69496888 | Genebody | CGI shore | − | + | −0.755261243 |
| ALDH3A1 | 12 | 62650896 | UP10K | CGI shore | + | − | −0.788071349 | |
| 12 | 62650898 | UP10K | CGI shore | + | − | −0.752480115 | ||
| KIRREL3 | 9 | 59344774 | Genebody | − | + | −0.957798851 | ||
| GTF2IRD1 | 3 | 11271020 | UP10K | − | + | −0.917047466 | ||
| UBA52 | 2 | 59052151 | UP10K | − | + | −0.953385635 | ||
| CD163 | 14 | 153630638 | Genebody | CGI shore | − | + | −0.860727255 | |
| HMHA1 | 2 | 77641888 | UP10K | CGI shore | + | − | −0.861581753 | |
| RPS21 | 17 | 69165427 | UP10K | CGI shore | − | + | −0.829232698 | |
| 17 | 69165425 | UP10K | CGI shore | − | + | −0.811905734 | ||
| 17 | 69169540 | UP10K | − | + | −0.852778602 | |||
| 17 | 69169556 | UP10K | − | + | −0.743101278 | |||
| SLC9A3R2 | 3 | 42231210 | Genebody | CGI shore | − | + | −0.752184061 | |
| ZNF646 | 3 | 17732629 | Genebody | CGI shore | − | + | −0.828529507 | |
| ADAMTS4 | 4 | 97117737 | Genebody | − | + | −0.802729052 | ||
| 4 | 97117751 | Genebody | − | + | −0.8029031 | |||
| 4 | 97117778 | Genebody | − | + | −0.806509102 | |||
| 4 | 97117512 | UP10K | − | + | −0.763107181 | |||
| 4 | 97117534 | UP10K | − | + | −0.814870957 | |||
| TBKBP1 | 12 | 23838338 | Genebody | − | + | −0.89415394 | ||
| 12 | 23838110 | Genebody | CGI shore | − | + | −0.975048371 | ||
| 12 | 23838349 | Genebody | − | + | −0.821418354 | |||
aChromosome.
bClass represents genomic location of tDMC; exon and intron region, 10 kb upstream region from TSS and CGI Shore were marked as Genebody, UP10K and CGI shore, respectively.
cMethylation pattern, + and − represent higher and lower methylation over other tissues, respectively.
dExpression level, + and − represent higher and lower expression levels over other tissues, respectively.
ePearson's correlation score.
From GO analysis of the neocortex, synapse-related genes (NRXN2 and SYNE1) and regulation of nerve system-related gene (MAPT) showed negative correlation between the levels of gene expression and CpG methylation at 15 tDMCs (average Pearson's correlation score less than −0.843). The expression levels of those three genes were higher in neocortex than in other tissues (adjusted P-value <0.05). Fourteen out of 15 tDMCs were located on gene bodies, but only a single tDMC that associated with NRXN2 was mapped to the upstream 10 kb region from TSS. In the muscle, 18 tDMCs were associated with 11 genes and showed negative correlation between the levels of expression and methylation (average Pearson's correlation score less than −0.884). The tDMCs of NR5A2,66 PCOLCE and VGLL2 which promote muscle development and activities were up-regulated and hypo-methylated. For the spleen, tDMCs of three immune-related genes ICAM1, CD163 and TBKBP1 were hypo-methylated (Table 3).
4. Discussion
The completion of the pig genome assembly (Sscrofa10.2) provides a basis for understanding the epigenetic modifications of the pig genome including DNA methylation, which is another important issue in modern genetics. In this study, we generated the global methylation profiles from the porcine neocortex, spleen, liver, muscle, olfactory epithelium and a PAM cell line and analysed the RR genome of the pig. Although we did not analyse the methylation patterns of a complete list of pig tissues and the entire genome of the pig such as bisulfite sequencing, our results provide valuable input to the characteristics of the pig methylome considering that there have not been sufficient studies on the genome level of the profiles of the pig methylome from various tissues in a single nucleotide resolution.
Previously, Li et al.50,67 reported the results on the methylation pattern of the pig genome using MeDIP-seq, which investigated the genome-wide relationship between DNA methylation and fat deposition using adipose tissues of breeds with different phenotypes. Recently, Gao et al.44 presented the RRBS analysis of the pig genome as the first report using the intestinal tissue. However, both studies showed limitations of providing information on only specific tissues or mapping resolution of the study.
In this study, we reported the global methylation patterns of five different tissues (the neocortex, spleen, liver, muscle and olfactory epithelium) and a cell line (PAM; 3D4/2) of the pig, which are part of quite diverse biological systems. The results of the global DNA methylation patterns corroborated the results of the previous studies44,58 on the same regions.
Because the design of our RRBS analysis of the pig genome including the restriction enzyme and fragment size selection was identical to that in the studies of humans and mice,58 it was appropriate to compare our results to the available data from those species. The size of the pig RR genome was similar to that of human (73 vs. 74 M, respectively, for MspI, Table 1). The RRBS reads of the pig exhibited a slightly lower frequency of multiple mapped reads than human (5.35 vs. 7.7%, respectively), and the percentage of total CpG sites in the pig RR genome (18.34%) was 4.84% higher than in the human, which may be attributed to slightly lower amount of repeat elements (∼40% of the genome) in the pig compared with those in the human (nearly 50%).
As expected, we observed the enrichment of CpG sites in the RR genome, and the rate was higher in pig (7.1-fold) than in humans (5.8-fold) and mice (5-fold) (Table 1). A drawback of the RRBS is that CpGs within CpG-poor regions are scarcely covered because of the limitations of the RRBS method, which relies on the CpG-associated restriction enzymes. However, ∼60% of human gene contain a CGI near the TSS.68 Therefore, RRBS studies can provide information of CGI and gene-associated CpG sites. Our results given in Supplementary Fig. S4 were consistent with the average of 44.21% of CpGs that belonged to the CGI observed in our RRBS analysis. CGIs consisted of only 1.11% of the pig genome and contained 8.44% of the total CpGs in the pig genome.
For future RRBS studies of the pig genome, it is necessary to improve the genome coverage to reduce the number of missing CpGs. Comparison of the MspI-digested to the MspI/ApeKI–double-digested RR genomes showed significant improvement in the percentage genome (2.6–12.12 Mb) and the total CpG coverage (18.3–28.4%), which were similar to those of studies in humans and mice.58 Further studies using MspI/ApeKI should significantly improve the genome coverage of RRBS for the pig genome.
For GO analysis of the pig genome, gene enrichment analysis using web-based software is difficult to perform because of unavailability of enough functional annotation data. The amount of information on the functional annotation of the pig genome is still limited. As an attempt to overcome the difficulty, we performed a gene enrichment test against our tDMR-related gene dataset using the GeneMANIA (http://www.genemania.org/) software which allows the use of annotation data of the human genome. Our pilot analysis showed that the results were more informative than just relying on pig GO terms (data not shown).
Recent studies recognized the importance of CGI shores which might be associated with tissue-specific DMRs (tDMRs) and cancer-specific DMRs (cDMRs).69,70 We detected a difference in the per cent methylation of CGI shore among the six samples. Lower correlation values between CpG methylation levels on CGI shore from different tissues might suggest a correlation with tissue-specific differential methylation (Fig. 1). In this study, we detected 18 tDMCs located in the CGI shore region and they showed strong correlation with the level of gene expression using the transcriptome profile of a microarray experiment. The use of RNA-seq data should improve our understanding between tDMCs and the regulation of gene expression in pigs. The global difference in the level of non-CpG methylation was observed in the neocortex, which showed ∼35% more methylated non-CpG sites than other samples (Fig. 3). The amount of methylated non-CpG sites was >1.71% in all samples except in the neocortex (2.3%). The total amount of non-CpG sites (CHG and CHH) was almost four times larger than that of CpG sites (data not shown). Although the difference (0.6%) is small, the actual number of CpGs corresponding to that difference may not be as small and warrants further studies.
Recent studies showed that the SV40 large T antigen gene or hTERT transfected immortalized cell line results in DNA hyper-methylation and large-scale changes in gene expression.17 The result also showed the accumulation of DNA methylation at promoter regions according to doubling times or replicative lifespan. This is consistent with our finding of hyper-methylation in 3D4/2 cells which were established by transfecting the SV40 large T antigen (Supplementary Fig. S4). It was reported that a large part of immortality-associated changes in gene expression were related with RNA processing and regulation of transcription factor activity in immortalized cells.71 The GO analysis of tDMCs among different tissues also showed that many of the 3D4/2 cell-specific tDMCs were related to transcriptional regulation. Thus, the uniqueness of the methlylome profile of 3D4/2 cells in this study is consistent to the results of previous methylome analyses using permanent cell lines. However, it still remains inconclusive whether the difference between 3D4/2 cells and other tissues was due to the characteristics of the permanent cell lines or the cell culture conditions or the natural methylation patters of pulmonary alveolar macrophage cells, because only one cell line was included in this study.
To create a suitable reference map of porcine methylome to biological studies, DNA methylation profiles from diverse tissues and diverse developmental stages are necessary. Although the list of samples and developmental stages is still far from being complete, our study contributes to the knowledge of DNA methylation in pig genome, which could provide information regarding biological processes in response to environmental changes, development and disease. To our knowledge, our results are among the initial studies to extensively evaluate the general methylation profile of the pig genome at a single base resolution.
Authors’ contribution
M.K.C. was responsible for sample preparation, genomic DNA isolation, RRBS data analysis using methylKit and involved in drafting the manuscript. J.I.L. performed the read alignment and overall RRBS data analysis. M.T.L. and D.T.N. collected pig samples. S.P. and N.S. performed the statistical analysis. K.M.S., J.B.K. and J.H.K. provided helpful ideas and discussion for the experiment. C.P. was involved in project planning, discussion and writing of the manuscript as a project principle investigator. All authors read and approved the final manuscript.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the fund from the Cooperative Research Program for Agriculture Science & Technology Development (No. PJ009103), Rural Development Administration, Republic of Korea and by the 2013 KU Brain Pool of Konkuk University. Funding to pay the Open Access publication charges for this article was provided by Konkuk University.
Supplementary Material
References
- 1.Elango N., Yi S.V.. 2008, DNA methylation and structural and functional bimodality of vertebrate promoters, Mol. Biol. Evol., 25, 1602–8. [DOI] [PubMed] [Google Scholar]
- 2.Bock C., Tomazou E.M., Brinkman A.B. et al. 2010, Quantitative comparison of genome-wide DNA methylation mapping technologies, Nat. Biotechnol., 28, 1106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber M., Hellmann I., Stadler M.B. et al. 2007, Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome, Nat. Genet., 39, 457–66. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki M.M., Bird A.. 2008, DNA methylation landscapes: provocative insights from epigenomics, Nat. Rev. Genet., 9, 465–76. [DOI] [PubMed] [Google Scholar]
- 5.Carrel L., Willard H.F.. 2005, X-inactivation profile reveals extensive variability in X-linked gene expression in females, Nature, 434, 400–4. [DOI] [PubMed] [Google Scholar]
- 6.Ehrlich M. 2009, DNA hypomethylation in cancer cells, Epigenomics, 1, 239–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hon G.C., Hawkins R.D., Caballero O.L. et al. 2012, Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer, Genome Res., 22, 246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan H.D., Santos F., Green K., Dean W., Reik W.. 2005, Epigenetic reprogramming in mammals, Hum. Mol. Genet., 14 (Spec No), R47–58. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki H., Matsui Y.. 2008, Epigenetic events in mammalian germ-cell development: reprogramming and beyond, Nat. Rev. Genet., 9, 129–40. [DOI] [PubMed] [Google Scholar]
- 10.Igarashi J., Muroi S., Kawashima H. et al. 2008, Quantitative analysis of human tissue-specific differences in methylation, Biochem. Biophys. Res. Commun., 376, 658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang P., Song F., Ghosh S. et al. 2011, Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development, BMC Genomics, 12, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziller M.J., Muller F., Liao J. et al. 2011, Genomic distribution and inter-sample variation of non-CpG methylation across human cell types, PLoS Genet., 7, e1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh S., Yates A.J., Frühwald M.C., Miecznikowski J.C., Plass C., Smiraglia D.. 2010, Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues, Epigenetics, 5, 527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bock C., Beerman I., Lien W.H. et al. 2012, DNA methylation dynamics during in vivo differentiation of blood and skin stem cells, Mol. Cell, 47, 633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lokk K., Modhukur V., Rajashekar B. et al. 2014, DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns, Genome Biol., 15, r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varley K.E., Gertz J., Bowling K.M. et al. 2013, Dynamic DNA methylation across diverse human cell lines and tissues, Genome Res., 23, 555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon K., Clouaire T., Bao X.X. et al. 2014, Immortality, but not oncogenic transformation, of primary human cells leads to epigenetic reprogramming of DNA methylation and gene expression, Nucleic Acids Res., 42, 3529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardiner-Garden M., Frommer M.. 1987, CpG islands in vertebrate genomes, J. Mol. Biol., 196, 261–82. [DOI] [PubMed] [Google Scholar]
- 19.Meissner A., Mikkelsen T.S., Gu H. et al. 2008, Genome-scale DNA methylation maps of pluripotent and differentiated cells, Nature, 454, 766–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartung T., Zhang L., Kanwar R. et al. 2012, Diametrically opposite methylome-transcriptome relationships in high- and low-CpG promoter genes in postmitotic neural rat tissue, Epigenetics, 7, 421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberg A.D., Allis C.D., Bernstein E.. 2007, Epigenetics: a landscape takes shape, Cell, 128, 635–8. [DOI] [PubMed] [Google Scholar]
- 22.Meissner A., Gnirke A., Bell G.W., Ramsahoye B., Lander E.S., Jaenisch R.. 2005, Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis, Nucleic Acids Res., 33, 5868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Down T.A., Rakyan V.K., Turner D.J. et al. 2008, A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis, Nat. Biotechnol., 26, 779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cross S.H., Charlton J.A., Nan X., Bird A.P.. 1994, Purification of CpG islands using a methylated DNA binding column, Nat. Genet., 6, 236–44. [DOI] [PubMed] [Google Scholar]
- 25.Brinkman A.B., Simmer F., Ma K., Kaan A., Zhu J., Stunnenberg H.G.. 2010, Whole-genome DNA methylation profiling using MethylCap-seq, Methods, 52, 232–6. [DOI] [PubMed] [Google Scholar]
- 26.Serre D., Lee B.H., Ting A.H.. 2010, MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome, Nucleic Acids Res., 38, 391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dedeurwaerder S., Defrance M., Calonne E., Denis H., Sotiriou C., Fuks F.. 2011, Evaluation of the Infinium Methylation 450K technology, Epigenomics, 3, 771–84. [DOI] [PubMed] [Google Scholar]
- 28.Gu H., Smith Z.D., Bock C., Boyle P., Gnirke A., Meissner A.. 2011, Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling, Nat. Protoc., 6, 468–81. [DOI] [PubMed] [Google Scholar]
- 29.Smith Z.D., Gu H., Bock C., Gnirke A., Meissner A.. 2009, High-throughput bisulfite sequencing in mammalian genomes, Methods, 48, 226–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson K.D. 2005, DNA methylation and human disease, Nat. Rev. Genet., 6, 597–610. [DOI] [PubMed] [Google Scholar]
- 31.Niculescu M.D., Zeisel S.H.. 2002, Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline, J. Nutr., 132, 2333s–5s. [DOI] [PubMed] [Google Scholar]
- 32.Liu X.J., Wang J.Q., Li R.S. et al. 2011, Maternal dietary protein affects transcriptional regulation of myostatin gene distinctively at weaning and finishing stages in skeletal muscle of Meishan pigs, Epigenetics, 6, 899–907. [DOI] [PubMed] [Google Scholar]
- 33.Altmann S., Murani E., Schwerin M., Metges C.C., Wimmers K., Ponsuksili S.. 2012, Maternal dietary protein restriction and excess affects offspring gene expression and methylation of non-SMC subunits of condensin I in liver and skeletal muscle, Epigenetics, 7, 239–52. [DOI] [PubMed] [Google Scholar]
- 34.Kwak W., Kim J.N., Kim D. et al. 2014, Genome-wide DNA methylation profiles of small intestine and liver in fast-growing and slow-growing weaning piglets, Asian Australas. J. Anim. Sci., 27, 1532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rauch T.A., Wang Z., Wu X., Kernstine K.H., Riggs A.D., Pfeifer G.P.. 2012, DNA methylation biomarkers for lung cancer, Tumour Biol., 33, 287–96. [DOI] [PubMed] [Google Scholar]
- 36.Heyn H., Sayols S., Moutinho C. et al. 2014, Linkage of DNA methylation quantitative trait loci to human cancer risk, Cell Rep., 7, 331–8. [DOI] [PubMed] [Google Scholar]
- 37.Gloss B.S., Patterson K.I., Barton C.A. et al. 2012, Integrative genome-wide expression and promoter DNA methylation profiling identifies a potential novel panel of ovarian cancer epigenetic biomarkers, Cancer Lett., 318, 76–85. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi Y., Absher D.M., Gulzar Z.G. et al. 2011, DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer, Genome Res., 21, 1017–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Jager P.L., Srivastava G., Lunnon K. et al. 2014, Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci, Nat. Neurosci., 17, 1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Couldrey C., Brauning R., Bracegirdle J., Maclean P., Henderson H.V., McEwan J.C.. 2014, Genome-wide DNA methylation patterns and transcription analysis in sheep muscle, PLoS ONE, 9, e101853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su J., Wang Y., Xing X., Liu J., Zhang Y.. 2014, Genome-wide analysis of DNA methylation in bovine placentas, BMC Genomics, 15, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y.Z., Sun J.J., Zhang L.Z. et al. 2014, Genome-wide DNA methylation profiles and their relationships with mRNA and the microRNA transcriptome in bovine muscle tissue (Bos taurine), Sci Rep-Uk, 4, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J.R., Hong C.P., Moon J.W. et al. 2014, Genome-wide analysis of DNA methylation patterns in horse, BMC Genomics, 15, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao F., Zhang J., Jiang P. et al. 2014, Marked methylation changes in intestinal genes during the perinatal period of preterm neonates, BMC Genomics, 15, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tumbleson S. 1997, Advances in swine in biomedical research. Edited by M.E. Tumbleson and L.B. Schook. Plenum Publishing Corporation, 1996. Volume 1, pp. 462. Volume 2, pp. 499, Exp. Physiol., 82, 803–804. [Google Scholar]
- 46.Groenen M.A., Archibald A.L., Uenishi H. et al. 2012, Analyses of pig genomes provide insight into porcine demography and evolution, Nature, 491, 393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paudel Y., Madsen O., Megens H.J. et al. 2013, Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication, BMC Genomics, 14, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen D.T., Lee K., Choi H. et al. 2012, The complete swine olfactory subgenome: expansion of the olfactory gene repertoire in the pig genome, BMC Genomics, 13, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubin C.J., Megens H.J., Martinez Barrio A. et al. 2012, Strong signatures of selection in the domestic pig genome, Proc. Natl. Acad. Sci. USA, 109, 19529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li M., Wu H., Luo Z. et al. 2012, An atlas of DNA methylomes in porcine adipose and muscle tissues, Nat. Commun., 3, 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo W., Fiziev P., Yan W. et al. 2013, BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data, BMC Genomics, 14, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B., Salzberg S.L.. 2012, Fast gapped-read alignment with Bowtie 2, Nat. Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akalin A., Kormaksson M., Li S. et al. 2012, methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles, Genome Biol., 13, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C.. 2014, jvenn: an interactive Venn diagram viewer, BMC Bioinformatics, 15, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hornshoj H., Conley L.N., Hedegaard J., Sorensen P., Panitz F., Bendixen C.. 2007, Microarray expression profiles of 20.000 genes across 23 healthy porcine tissues, PLoS ONE, 2, e1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Team R.C. 2014, R: a language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria. [Google Scholar]
- 57.Cho R.J., Huang M., Campbell M.J. et al. 2001, Transcriptional regulation and function during the human cell cycle, Nat. Genet., 27, 48–54. [DOI] [PubMed] [Google Scholar]
- 58.Wang J., Xia Y., Li L. et al. 2013, Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing, BMC Genomics, 14, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chatterjee A., Ozaki Y., Stockwell P.A., Horsfield J.A., Morison I.M., Nakagawa S.. 2013, Mapping the zebrafish brain methylome using reduced representation bisulfite sequencing, Epigenetics, 8, 979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laird P.W. 2010, Principles and challenges of genomewide DNA methylation analysis, Nat. Rev. Genet., 11, 191–203. [DOI] [PubMed] [Google Scholar]
- 61.Saxonov S., Berg P., Brutlag D.L.. 2006, A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters, Proc. Natl. Acad. Sci. USA, 103, 1412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harris R.A., Wang T., Coarfa C. et al. 2010, Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications, Nat. Biotechnol., 28, 1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan H., Chen L., Dogra S. et al. 2012, Measuring the methylome in clinical samples: improved processing of the Infinium Human Methylation450 BeadChip Array, Epigenetics., 7, 1173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo J.U., Su Y., Shin J.H. et al. 2014, Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain, Nat. Neurosci., 17, 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smallwood S.A., Tomizawa S., Krueger F. et al. 2011, Dynamic CpG island methylation landscape in oocytes and preimplantation embryos, Nat. Genet., 43, 811–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolado-Carrancio A., Riancho J.A., Sainz J., Rodriguez-Rey J.C.. 2014, Activation of nuclear receptor NR5A2 increases Glut4 expression and glucose metabolism in muscle cells, Biochem. Biophys. Res. Commun., 446, 614–9. [DOI] [PubMed] [Google Scholar]
- 67.Li M., Wang T., Wu H. et al. 2012, Genome-wide DNA methylation changes between the superficial and deep backfat tissues of the pig, Int. J. Mol. Sci., 13, 7098–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larsen F., Gundersen G., Lopez R., Prydz H.. 1992, CpG islands as gene markers in the human genome, Genomics, 13, 1095–107. [DOI] [PubMed] [Google Scholar]
- 69.Doi A., Park I.H., Wen B. et al. 2009, Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts, Nat. Genet., 41, 1350–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Irizarry R.A., Ladd-Acosta C., Wen B. et al. 2009, The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores, Nat. Genet., 41, 178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu L., Zhang J., Bates S. et al. 2005, A methylation profile of in vitro immortalized human cell lines, Int. J. Oncol., 26, 275–85. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.