

Supplemental Digital Content is available in the text.
Keywords: body mass index, cardiovascular diseases, lipids, lipoproteins, HDL, metabolism
Rationale:
Evidence suggests that the gut microbiome is involved in the development of cardiovascular disease, with the host–microbe interaction regulating immune and metabolic pathways. However, there was no firm evidence for associations between microbiota and metabolic risk factors for cardiovascular disease from large-scale studies in humans. In particular, there was no strong evidence for association between cardiovascular disease and aberrant blood lipid levels.
Objectives:
To identify intestinal bacteria taxa, whose proportions correlate with body mass index and lipid levels, and to determine whether lipid variance can be explained by microbiota relative to age, sex, and host genetics.
Methods and Results:
We studied 893 subjects from the LifeLines-DEEP population cohort. After correcting for age and sex, we identified 34 bacterial taxa associated with body mass index and blood lipids; most are novel associations. Cross-validation analysis revealed that microbiota explain 4.5% of the variance in body mass index, 6% in triglycerides, and 4% in high-density lipoproteins, independent of age, sex, and genetic risk factors. A novel risk model, including the gut microbiome explained ≤25.9% of high-density lipoprotein variance, significantly outperforming the risk model without microbiome. Strikingly, the microbiome had little effect on low-density lipoproteins or total cholesterol.
Conclusions:
Our studies suggest that the gut microbiome may play an important role in the variation in body mass index and blood lipid levels, independent of age, sex, and host genetics. Our findings support the potential of therapies altering the gut microbiome to control body mass, triglycerides, and high-density lipoproteins.
In recent years, the gut microbiome has emerged as an important player in human health.1,2 Gut microbiota comprise thousands of microbial species that are involved in host metabolism by regulating energy extraction, activation of the immune system, drug metabolism, and other processes.3,4 Association of bacterial composition with many diseases has been observed, including immune, inflammatory, and metabolic phenotypes.5–7 Several mechanisms for the downstream effect of microbiota were discovered which also suggest that they play a role in cardiovascular disease (CVD). The microbiota play an important role in choline diet–induced trimethylamine N-oxide production, which has been implicated in CVD.8 A further mouse study has demonstrated that atherosclerosis susceptibility can be transmitted via gut microbiota transplantation.9 Furthermore, dysbiosis in the gut has been shown to induce increased permeability of the intestine, leading to increased systemic levels of bacterial products causing low-grade chronic inflammation.10 This inflammation may directly affect atherogenesis and has also been hypothesized to lead to the development of insulin resistance with concomitant effects on plasma lipids.11 Gut microbiota have also been linked with lipid metabolism through their role in bile acid metabolism. They can also influence the efficiency of energy harvest from ingested food12,13 and play a crucial role in the metabolic processes and development of obesity.
In This Issue, see p 743
Editorial, see p 750
In line with these observations, altering the gut microbiome in humans and mice has shown improvement in metabolic syndrome.13–15 However, the evidence for a causal relationship between the gut microbiome and the development of CVD has not been firmly established for lack of large-scale human studies. Atherosclerosis, a lipid-driven disease, is the main underlying cause of CVD. However, to date, no studies of sufficient size have been done to assess the association between lipids and microbiota. In this study, we performed a systematic analysis of host genome, gut microbiome, body mass index (BMI), and blood lipids in 893 human subjects from the Dutch LifeLines-DEEP cohort.16 We investigated that which gut bacteria were associated with BMI and blood lipids, and how much of the variation in blood lipids could be explained by the gut microbiome, relative to age, sex, BMI, and host genetics.
Methods
Population Cohort
The LifeLines-DEEP cohort is a subcohort of the LifeLines cohort (167 729 subjects),17 which uses a broad range of investigative procedures in assessing the biomedical, sociodemographic, behavioral, physical, and psychological factors that contribute to the health and disease of the general population. A subset of ≈1500 participants also took part in LifeLines-DEEP: for these participants, additional biological materials were collected, including genome-wide genotyping and analysis of the gut microbiome composition. A full description of the LifeLines-DEEP data set is given in the article describing the study design.16
Lipid Measurements
We had lipid measurements available for all 1500 LifeLines-DEEP samples. Total cholesterol (TC) was measured with an enzymatic colorimetric method, high-density lipoprotein (HDL) cholesterol with a colorimetric method, and triglycerides with a colorimetric UV method (Modular P analyzer, Roche Diagnostics, Burgdorf, Switzerland). The low-density lipoprotein (LDL) cholesterol concentration was calculated using the Friedewald equation. More details were reported previously.18 The triglyceride level was further log2 transformed.
Genotype Information
All LifeLines-DEEP samples were genotyped using the HumanCytoSNP-12 BeadChip and ImmunoChip, a customized Illumina Infinium array. The data were harmonized,19 merged, and subsequently imputed using the Genome of the Netherlands (GoNL) data set.20,21 Further details and information on the quality control are described by Tigchelaar et al.16 We removed ethnic outliers and genetically related participants from our study.
Microbiome Data Generation
Sequencing
Microbiome data were generated for 1180 LifeLines-DEEP samples. Fecal samples were collected at home within 2 weeks after collection of blood samples and stored immediately at −20°C. After transport on dry ice, all samples were stored at −80°C. Aliquots were made, and DNA was isolated with the AllPrep DNA/RNA Mini Kit (Qiagen; cat. #80204). Isolated DNA was sequenced at the Broad Institute, Boston, using Illumina MiSeq paired-ends. Hypervariable region V4 was selected using forward primer 515F [GTGCCAGCMGCCGCGGTAA] and reverse primer 806R [GGACTACHVGGGTWTCTAAT]. We used custom scripts to remove the primer sequences and align the paired-end reads. Details are given in the study by Gevers et al.22
OTU Picking
Selection of unique bacterial sequences, so-called operational taxonomic unit (OTU) picking, was performed using the QIIME (the toolbox for Quantitative Insights Into Microbial Ecology) reference optimal picking, which uses UCLUST (an algorithm to cluster sequence reads based on similarity)23 (version 1.2.22q) to perform the clustering. Matching OTUs to bacteria was done using a primer-specific version of the GreenGenes 13.5 reference database.24 Using TaxMan,25 we created the primer-specific reference database containing only reference entries that matched the selected primers. During this process, we restricted probe-reference mismatches to a maximum of 25%. The 16S regions that were captured by our primers, including the primer sequences, were extracted from the full 16S sequences. For each of the reference sequences, we determined the overlapping part of the taxonomy of each of the reference reads in the clusters and used this overlap as the taxonomic label for the cluster. This process is based on, and similar to, the work described by Bonder et al,26 Brandt et al,25 May et al,27 and Ding et al.28 We used QIIME29 for exploratory analysis and for gathering basic statistics on the microbiome data set.
Quality Control
Overall, for 1021 samples, we had lipid measurements, genotype, and microbiome information. We excluded 99 samples from participants who were taking antibiotic or other potential microbiome-modifying drugs or who were on lipid-lowering medication. The library size of microbial sequencing varied greatly among samples, ranging from 3969 to 336 900 reads. The sequence depth can significantly bias the measures of microbial composition, and rarefication was widely used to make the library sizes equal by randomly selecting the same number of reads per sample.30,31 We compared the number of samples at different sequence depths and determined the rarefication depth based on criteria to obtain both the number of reads and the number of samples as high as possible. We rarefied the library size to 15 000 read-depth using the rarefy function in R package vegan (version 2.3-0). At this depth, we only excluded 29 subjects. After these exclusion steps, we had 893 samples (380 men and 513 women) for final analysis. Their characteristics are summarized in Table.
Table.
Summary of Physical Characteristics of 893 LifeLines-DEEP Subjects
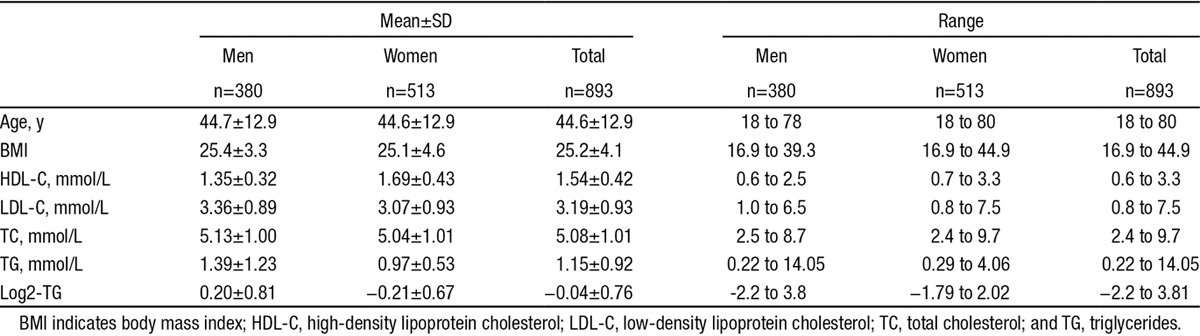
Furthermore, we filtered on the OTU abundance and confined our analysis to 645 OTUs, each of which comprised ≥0.05% of reads and was present in at least 1% of the population. These OTUs accounted for an average of 99% of total reads per sample. The OTUs were assigned to 173 taxonomies that were further truncated to 136 taxonomies after removing identical or highly similar information between different clade levels.
Statistical Analysis
Analysis of Microbial Diversity
The microbial Shannon diversity index was calculated using the diversity function in R package vegan (version 2.3-0).
Two-Part Model for Association Analysis
We observed that the distribution of the abundance of OTUs or taxonomies departed significantly from a normal distribution because of the fact that bacteria were not presented in many samples. Only 50 of 645 OTUs (7.7%) were presented in >90% of samples, whereas 448 OTUs (69.5%) were detected in <50% of samples. At the taxonomic level, 32 of 136 (9.5%) taxonomies were detected in >90% of samples, whereas 60 taxonomies (44.1%) were detected in <50% of samples. There are different explanations for the detection rate: (1) the bacteria are really absent in the samples; (2) the abundance levels of bacteria are lower and not to be detected at the current sequencing and rarefication depth; (3) the abundance levels are similar and it is a random effect because of the sequencing or rarefication procedure. We therefore adopted a novel, 2-part model that was developed to account for both binary (detected/undetected) and quantitative features.32 This approach overcomes the problem of a non-normal distribution, which is a feature of the majority of gut bacteria OTUs or taxa.
The 2-part model is illustrated in Online Figure I. The first part describes a binomial analysis that tests for association of detecting a microbe (represented by an OTU or taxonomy) with a trait. The binary feature (b) of a microbe under study was coded as 0 for undetected or 1 for detected for each sample. The binary model is described as: y = β1b + e, where y refers to the trait level (BMI or lipid level) per individual after adjusting for age and sex, b is a binary feature, β1 is the estimated effect for the binary effect, and e represents the residuals.
The second part of the quantitative analysis tests for association between the lipid level and the abundance of bacteria, but only for the subjects where that microbe is present. The abundance level (q) of a microbe was the log10 transformed read count per individual. The quantitative model is written as: y = β2q + e, where q is the abundance of a microbe, β2 is the estimated effect for the abundance, and e represents the residuals.
To further combine the effect of both binary and quantitative analysis, a meta P value was derived using an unweighted Z method. Then, a final association P value per microbe-trait pair was assigned from the minimum of P values from the binary analysis, quantitative analysis, and meta-analysis. The association Z score was calculated based on the Z distribution. If the association direction was negative, the Z score was assigned a negative value. If the association direction is positive, the Z score was assigned a positive value.
The association P value was set as the minimum value of 3 P values and the distribution of the association P values could be skewed, so we therefore performed 1000× permutation tests to control the false discovery rate (FDR). For each permutation, we randomized the gut microbial composition across individuals and performed the 2-part analysis on permuted data. At a certain P cutoff, the average number of the detected significance (N0) in 1000× permutations was defined as the false positive, and its ratio to the detected positive (N1) in the real analysis was the FDR. We controlled the FDR at 0.05.
This method accounts for the complicated features of the microbial data and maximizes the power. If the association P value comes from the binary model, indicating the effect is only because of the presence/absence of the microbe, the abundance of the microbe in the samples does not matter. If the association P value comes from the quantitative model, this indicates the abundance level of the microbe associates with the trait, and the absence of the microbe has no influence. The explanation would be another microbe takes its place and has a similar function. If the association P value comes from the meta-analysis, indicates that both the presence/absence and the abundance of microbes can influence the trait.
Estimating the Variance Explained by the Gut Microbiome
To estimate the proportion of variation in BMI and lipids that could be explained by the gut microbiome, we performed a 100× cross validation. Each time we split the data randomly into an 80% discovery set and a 20% validation set. In the discovery set, a total of n number of significantly associated OTUs was identified at a certain P value, and the effect sizes of binary and quantitative features of each OTU (β1 and β2) were estimated. Then, the risk of the gut microbiome on BMI or lipids (rm) for each individual in the validation set was calculated using an additive model:
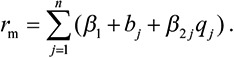 |
The variation in BMI and blood lipids explained by the gut microbiome was represented as the squared correlation coefficient (R2) between the traits and rm, after correcting for age and sex. To ensure the robustness of our estimation, we repeated the cross-validation 100× and calculated the average value of the explained variation. We hypothesized that many microbes may contribute a small effect but may not be confidently detected at an FDR of 0.05. Therefore, we did this analysis at different significant P levels ranging from 1×10−5 to 0.1.
Genetic Risk Score Calculation
A total of 157 lipid-associated single nucleotide polymorphisms (SNPs)33 and 97 BMI-associated SNPs34 were extracted from the literature. The risk alleles and their effect sizes were extracted for each SNP and each lipid type. We excluded 3 SNPs for which genotypes could not be successfully imputed in the LifeLines cohort: rs9411489 at the ABO locus, rs3177928 at the HLA locus, and rs12016871 at the MTIF3 locus. Thus, our final study included genetic information for 96 BMI-associated SNPs and 155 lipid-associated SNPs, including 71 for HDL, 56 for LDL, 40 for triglycerides, and 72 for TC. We then computed weighted genetic risk scores (rg) for BMI and lipids, as described previously.18
The association analysis between individual SNPs and the gut microbiome was performed using the analysis pipeline developed in house for quantitative trait loci analysis.35 We further tested whether the explained variation in the gut microbiome was independent of genetic factors by testing the association between the gut microbiome and genetic risk scores.36 The associations between microbes and the genetic risk score of BMI and lipid levels were assessed using our 2-part model. The significance was controlled at FDR<0.05 by 1000× permutation tests.
Significance of the Microbial Contribution
To test whether the gut microbiome contributes significantly to variation in BMI and blood lipids, we compared the performance of 3 different risk models, in particular the risk models with and without microbial risk:
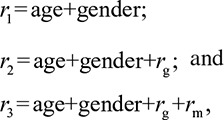 |
where rg is the calculated genetic risk and rm is the highest microbial risk we determined. The variation explained by each risk model was calculated in 100× cross validation, as described above. To evaluate the significance of microbial contribution, the ANOVA test was used to compare the performance of the risk models r3 and r2: the average of F values of the ANOVA test from 100× cross validation was calculated, and the P value was determined based on the F-distribution. As BMI and lipids are highly correlated, we also investigated whether the gut microbiome can contribute to lipid levels independent of BMI. To do so, we tested 4 risk models of lipids, including BMI as a risk factor:
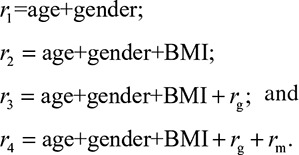 |
Results
Microbial Diversity in the LifeLines-DEEP Cohort
After quality control, our study included 893 human subjects. The study cohort had a wide range of age, BMI, and blood lipids levels (Table). We assessed how variable the gut microbial composition was in the cohort in terms of microbial richness and diversity. The microbial richness reflects the number of OTUs per individual. The cohort had on average 238 OTUs per individual, ranging from 44 to 355. When individuals were grouped into different bins based on their richness, we observed that age and the proportion of women were higher in the richer OTU groups (Figure 1). The Spearman correlation showed that the richness was significantly higher in women (P=0.0055) and increased with age (P=5.87×10−12; Online Table I). Given the abundance of OTUs, we computed the microbial diversity (Shannon’s diversity index) and observed similar significant correlations for age and sex (Online Table I). We then investigated whether bacterial richness and diversity were correlated with BMI and lipid levels. After correcting for age and sex, OTU richness was negatively correlated with BMI (P=3.8×10−4) and triglycerides (P=1.37×10−4), but positively correlated with HDL (P=8.3×10−4). We did not observe significant correlations between microbial richness and LDL or TC levels (Online Table I).
Figure 1.
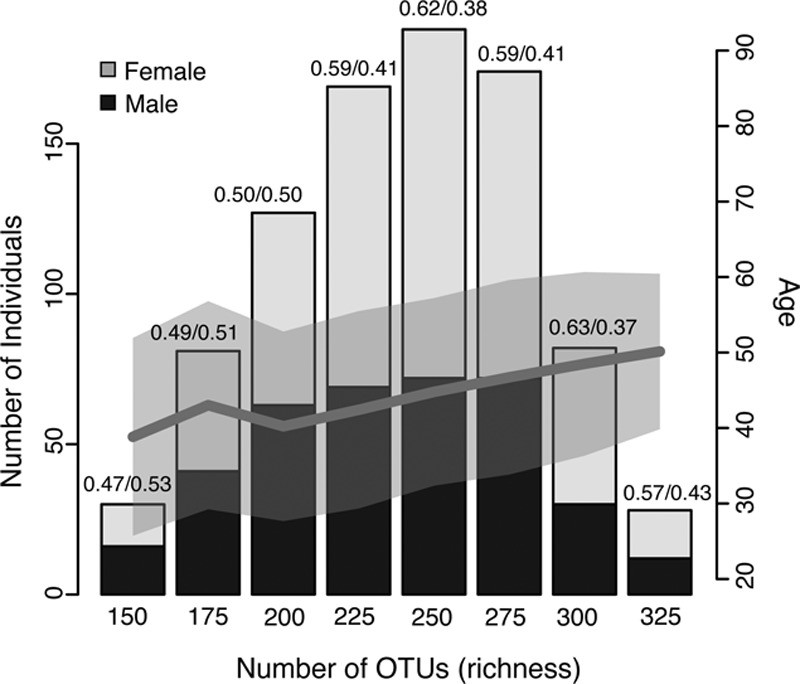
The richness of the gut microbiome. The microbial richness associated with age and sex. The bar plot shows the distribution of individuals binned to different groups of richness. The black and gray colors indicate the proportion of men and women in each group, and the dark gray line indicates the correlation between the average age and richness, whereas the light gray shadow indicates the SD of the age per richness bin. OTU indicates operational taxonomy unit.
Association of Bacteria With Lipid Metabolites
We next tested for association between the individual bacterial OTU, BMI, and blood lipid levels. After adjusting for age and sex, we identified 148 associated OTUs at FDR=0.05: 66 OTUs were associated with BMI, 114 with triglycerides, and 34 with HDL (Online Tables II–IV). We did not detect any significant association at OTU level for LDL or TC. Of the 148 associated OTUs, 12 were shared by all 3 traits (BMI, triglyceride, and HDL), 29 OTUs were shared by BMI and triglycerides, and 4 by BMI and HDL, whereas 21, 64, and 9 OTUs were specifically associated with BMI, triglyceride, and HDL, respectively (Online Figure II). At the taxonomic level, we identified 50 significant associations for 34 unique taxonomies at FDR=0.05: 22 were associated with BMI, 23 with triglycerides, 4 with HDL, and 1 with LDL (Figure 2; Online Table V). We found that 18 associations (36%) were detected by binary analysis (presence/absence); 4 associations (8%) were detected by the quantitative model; and 28 associations (56%) were detected by the meta-analysis of binary and quantitative analyses (Online Table V). Although most of the associated taxonomies were shared across lipid metabolites and BMI, several microbes were predominantly linked to lipids rather than BMI. For example, the family Clostridiaceae/Lachnospiracease (N16 in Figure 2) was specially associated with LDL (P=9.1×10−5; Online Table V) and not detected for BMI nor other lipids. Furthermore, the family Pasteurellaceae (N32; Proteobacteria), genus Coprococcus (N24; Firmicutes), and genus Collinsella species Stercoris (N2) showed strong association with triglyceride levels (P=6.2×10−5, P=4.6×10−5, and P=0.0006, respectively), a nominal significance to other lipids, and no association with BMI (P>0.1).
Figure 2.
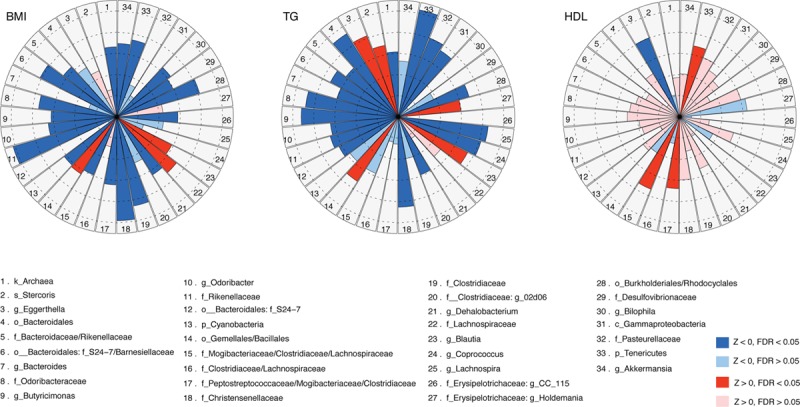
The effect of taxonomies on body mass index (BMI) and lipids. The effects of 34 taxonomies associated with BMI, triglycerides (TG), and high-density lipoprotein (HDL) are shown as Z scores. Red sectors indicate positive associations and blue negative associations. Brighter colors indicate that the association was significant at false discovery rate (FDR) 0.05 level. Dashed circles indicate the scale of Z values from 1 to 5.
We confirmed several previously described bacterial associations with obesity. An increased abundance of genus Akkermansia (N34) has been associated with a decrease in BMI (P=0.0005).37 We also confirmed the association of both the family Christensenellaceae (phylum Firmicutes; N18) and the phylum Tenericutes (mainly represented by order RF-39; N33) with low BMI (P=9.8×10−7 and P=0.0002, respectively), as reported in the TwinsUK cohort.38 In addition, we identified a novel and strong association of these particular bacteria with lower levels of triglyceride (P=2.1×10−5 and P=2.7×10−7, respectively) and higher levels of HDL (P=0.0047 and P=0.0006, respectively). We also observed several new associations with BMI and levels of triglyceride and HDL, such as genus Eggerthella (N3) with increased triglyceride (P=4.1×10−5) and decreased HDL (P=6.3×10−5), and family Pasteurellaceae (N32) with decreased triglyceride (P=6.2×10−5). The genus Butyricimonas (N9) was previously linked to a lean phenotype in mice after fecal transplantation from twins discordant for obesity.14 Our study shows that this genus is strongly associated with decreased triglyceride (P=4.7×10−6) and nominally associated with BMI and HDL in humans.
Variance of Blood Lipid Explained by Microbiota Composition
To anticipate how much BMI and blood lipids can be modulated by the gut microbiome, it is important to estimate what proportion of variation in these metabolic traits can be explained by the microbiome. To do so, we performed a 100× cross-validation analysis by splitting the data set randomly into an 80% discovery set and a 20% validation set. The OTUs identified at P=1×10−5 level in the discovery set explained 2.74% variation in BMI in the validation set, 3.83% in triglycerides, 2.46% in HDL, 0.01% variation in LDL, and 0.01% in TC. When the association significance decreased and the risk model included more (but less-significant) OTUs, the explained variation increased to 4.57% in BMI, 6.0% in triglycerides, 4.0% in HDL, but was only 1.5% in LDL and 0.7% in TC (Figure 3A). To test the robustness of our estimation, we rerarefied the OTU library 100× and repeated the whole analysis. This approach yielded similar results, thereby confirming the robustness of our estimation (Online Figure III).
Figure 3.
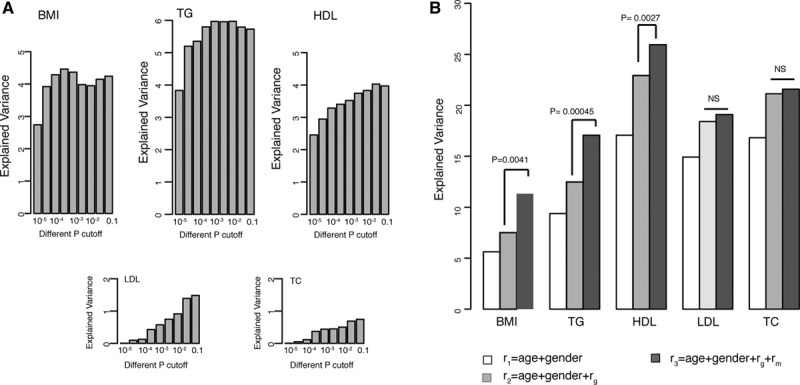
The contribution of the gut microbiome to body mass index (BMI) and lipids. A, The variation explained by gut microbes at different levels of significance. B, The variation explained by different risk models, including age, sex, genetic risk, and microbial risk. The significance of microbial contribution is indicated as the P value of the ANOVA test that compared the performance of the risk models r2 and r3. HDL indicates high-density lipoprotein; LDL, low-density lipoprotein; TC, total cholesterol; and TG, triglycerides.
Microbiota Contribute Significantly to Lipid Variation, Independently of Age, Sex, and Genetics
Evidence has already shown that the gut microbiome can be shaped by host genetics.38 We further tested whether the explained variation in the gut microbiome was independent of genetic factors by testing the association between the gut microbiome and genetic risk scores.36 To date, 157 genetic loci have been reported to be associated with blood lipid levels,33 and 97 loci have been associated with BMI.34 In our cohort, these SNPs collectively explained 2.1% variation in BMI (P=1.66×10−5), 3.4% in triglycerides (P=3.22×10−8), 7.5% in HDL (P<2.2×10−16), 4.6% in LDL (P=8.0×10−11), and 5.6% in TC (P=7.7×10−13), after correcting for age and sex. However, we did not observe any significant association between the microbiome and the genetic risk at FDR=0.05. We did not find a significant association for either single SNPs (Online Table VI) or for the combined lipid and BMI genetic risk scores (Online Table VII). Our results indicated that the proportion of variation in BMI and lipid levels explained by the gut microbiome was different from that explained by genetic variation. Therefore, we further assessed whether the microbiome could make a significant contribution to the explained variation beyond age, sex, and genetic factors. Our analysis unambiguously showed that age, sex, genetics, and the gut microbiome collectively explained 11.3% of the variation in BMI, 17.1% in triglycerides, and 25.9% in HDL cholesterol, with the microbiome making a significant contribution to the explained variation in BMI (P=4.1×10−3), triglycerides (P=4.5×10−4), and HDL (P=2.7×10−3; Figure 3B). When we included BMI as a risk factor, the total explained variation in lipids increased to 25% in triglycerides, 37.4% in HDL, 22.3% in LDL, and 22.3% in TC (Online Figure IV). The microbiome made a lesser, but still significant, contribution to triglycerides (P=4×10−3) and HDL (P=0.026). Our study therefore indicates that the gut microbiome can explain a substantial proportion of the variation, independent of age, sex, BMI, and genetics.
Discussion
Obesity and aberrant levels of blood lipids are associated with a high risk of CVD. Studying the effect of the gut microbiome on BMI and blood lipid levels yields insight into the role of the microbiome in the development of CVD. Although animal studies have shown that microbiota can influence lipid metabolism,39 no large-scale studies have been performed in humans thus far. Here, we investigated the impact of the gut microbiome on BMI and blood lipid levels in 893 human subjects from the LifeLines-Deep cohort. The power of our study is reflected by 3 factors. First, to our knowledge, it is the largest association study linking the gut microbiome to blood lipids in humans to date. Second, our cohort represented a wide range of ages, BMI, and blood lipids, as well as microbial composition. We also had detailed medication information per individual and could exclude those taking lipid-lowering or antibiotic medication. Moreover, we adopted a novel and powerful 2-part model to account for both the binary and quantitative features of microbial data. We established associations for 34 taxonomies with BMI and blood lipid levels, and we estimated that gut microbiota composition can explain ≤6% of the variation in lipid levels and that this effect is independent of age, sex, and host genetics.
Our results for the microbiota associated with BMI are in line with a recent study of 416 twin-pairs from the TwinsUK population38; in particular, we confirmed that lower abundances of families Christensenellaceae, Rikenellaceae, class Mollicutes, genus Dehalobacterium, and kingdom Archaea were associated with a high BMI. Of 22 independent taxa associated with BMI by our study, 16 were also accessed in the TwinsUK study: 11 (68.8%) showed significant association with BMI (P<0.05) with the same direction of effect as we found (Online Table V). We also identified a correlation of decreased bacterial diversity with increased BMI, which is in line with previous observations.40
However, many of the taxonomies we identified are novel findings. Several of the identified bacteria are known to be involved in the bile acid metabolic pathway. In particular, order Bacteroidales (phylum Bacteroidetes) and family Clostridiaceae (phylum Firmicutes) are both negatively correlated with BMI and triglycerides and known to be involved in bile acid metabolism.41 Bile acid activity of commensal bacteria is involved in a complex interplay with host hepatic enzymes, and together they promote digestion and absorption of dietary lipids.42 Interestingly, several small-scale studies reported lowered cholesterol on using probiotics with bile salt hydrolytic activity.43,44 Our study found support for the role of bacterial bile acids in lipid metabolism. Another pathway enriched in several associated bacteria is short chain fatty acid metabolism. Both orders Bacteroidales and Clostridiales, identified in our study, are involved in short chain fatty acid metabolism.41 Short chain fatty acids are produced by microbiota from dietary fibers, effect host body energy homeostasis, and are protective against metabolic syndrome, type 2 diabetes mellitus, and atherosclerosis.15,45–47
To firmly establish the gut microbiome as a risk factor for obesity and aberrant levels of blood lipids, we have been able to estimate that the microbiome could explain 4.57% to 6% of the variation in BMI, triglyceride and HDL, respectively. We did not detect any significant association between the gut microbiome and genetic predisposition to obesity and aberrant levels of blood lipids, suggesting the variation explained by the microbiome is independent of that explained by genetic variants. It should be noted, however, that the genetic risk score was limited to our established 157 lipid-associated SNPs33 and 97 BMI-associated SNPs,34 which together only explain a small proportion of the heritability of lipid levels. We might have missed the effect of other, not yet discovered SNPs. Our risk model included age, sex, genetic variation, and gut microbiome and explained 11.3% of the variation in BMI, 17.1% in triglycerides, and 25.9% in HDL, significantly outperforming the risk model without the microbiome. Because blood lipids and BMI are highly correlated with each other and many associated bacteria were shared, we investigated whether the observed effect of the gut microbiome on lipids might just be the confounded effect of BMI. We showed that by including BMI in the risk model, the gut microbiome made a smaller, but significant, contribution to the variation in triglycerides and HDL, suggesting that the microbiome affects blood lipids partly independently of BMI. Our results therefore indicate that the gut microbiome is a potentially important player in blood lipid metabolism. In contrast to genetics, sex, and age (all fixed characteristics), an individual’s microbiota composition can be modified by diet, prebiotics and probiotics, and fecal transplantation. Studies have shown that diet can alter the gut microbiome.48 Our study has not addressed how much of the association we observed between gut microbiome and blood lipids might be explained by diet. A better understanding of this could provide more insights into the role of diet in microbiome and lipid metabolism.
Our study supports the potential of microbiota-modifying intervention to correct lipid disbalance and thereby help prevent CVD. From potential to action, the next steps are to validate the associations we report in independent cohorts and to prove there is a causal axis of gut microbiome–lipids–CVD in functional studies. It is essential to gain more mechanistic insight into the functioning of the gut microbiome, although research in humans is still in its infancy.
The gut microbiomes in our study were profiled by 16s ribosomal RNA gene sequencing. This technology can identify microbial taxonomies and composition, but has limitations in identifying genetically specific species and strains. Furthermore, 16s ribosomal RNA gene sequencing provides little information on bacterial genes and their functions. With the decreasing cost of metagenome sequencing and development of techniques for culturing and for functional studies of gut bacteria, we expect to learn more about the levels of bacterial genes, metabolic pathways, and their functions in the future.
In conclusion, we have observed a strong association between the gut microbial composition and the variation in BMI and blood lipid levels, which is independent of age, sex, and host genetics. This observation provides insight into the microbiome’s role in regulating metabolic processes during the development of CVD. We established associations for a total of 34 intestinal bacteria taxonomies with BMI and blood lipids. We observed that the gut microbiome makes a significant contribution, beyond that of clinical risk factors and genetics, to the individual variance seen in BMI and to the blood levels of triglycerides and HDL, but that it has little effect on LDL or TC levels. Our results highlight the potential of therapies that alter the gut microbiome to control body mass, triglycerides, and HDL in CVD prevention. In moving from potential to action, it will be essential to identify the causal axis of microbiome–lipids–CVD and to gain more mechanistic insight into the gut bacteria functions.
Acknowledgments
We thank the LifeLines-DEEP participants and the LifeLines staff in Groningen for their collaboration. We thank Jackie Senior and Kate Mc Intyre for editing the article and Mathieu Platteel for practical and analytic work.
Sources of Funding
This project was funded by grants from the Top Institute Food and Nutrition, Wageningen, to C. Wijmenga, A. Zhernakova, and E.F. Tigchelaar (GH001), the Netherlands Organization for Scientific Research to J. Fu (NWO-VIDI 864.13.013), CardioVasculair Onderzoek Nederland to M.H. Hofker and A. Zhernakova (CVON 2012-03), and NWO (Netherlands Organization for Scientific Research) grants to L. Franke (ZonMW-VIDI 917.14.374) and R.K. Weersma (ZonMW-VIDI 016.136.308). A. Zhernakova holds a Rosalind Franklin Fellowship (University of Groningen) and M.C. Cenit holds a postdoctoral fellowship from the Fundación Alfonso Martín Escudero. This research received funding from the European Community’s Health Seventh Framework Programme (FP7/2007–2013, grant agreement 259867).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- BMI
- body mass index
- CVD
- cardiovascular disease
- FDR
- false discovery rate
- HDL
- high-density lipoprotein
- LDL
- low-density lipoprotein
- OTU
- operational taxonomy unit
- SNP
- single nucleotide polymorphism
- TC
- total cholesterol
In July 2015, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.38 days.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.115.306807/-/DC1.
Novelty and Significance
What Is Known?
The human gut holds about 100 trillion bacteria, which together can weigh several pounds.
This ecosystem (the microbiome) is shaped by early life events, the host genome, diet, and other lifestyle factors.
This bacterial community is associated with an individual’s susceptibility to many diseases, including cardiovascular diseases.
What New Information Does This Article Contribute?
Healthy lipid levels are associated with increased microbial diversity.
Body mass index and blood lipids are associated with 34 different microbial taxonomies.
A risk model including age, sex, genetic factors, and gut microbiome explains a large part of the variation seen in body mass index, triglycerides, and high-density lipoprotein cholesterol.
The bacterial community in the human gut (known as the microbiome) has been referred to as an extra organ, or the second human genome, because of its important role in an individual’s health. As most of these bacteria cannot be cultured, we knew little about their diversity and function until the recent development of innovative DNA sequencing technology. In our study, we defined the microbial composition found in 893 human subjects by sequencing bacteria-specific 16s ribosomal RNA genes; we observed a large interindividual variation in gut bacteria composition. We show that the bacterial diversity is associated with the lipid blood levels at human population level, especially with the levels of triglycerides and high-density lipoprotein cholesterol, and we report significant associations for 34 bacteria taxonomies. Our findings suggest that microbial intervention therapy will have the potential to help control blood lipid levels and prevent disease.
References
- 1.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 3.Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, Li R, Raes J, et al. MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103. doi: 10.1038/nature12198. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- 7.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, Lusis AJ, Hazen SL. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. doi: 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr. 2011;35:14S–20S. doi: 10.1177/0148607111413772. doi: 10.1177/0148607111413772. [DOI] [PubMed] [Google Scholar]
- 11.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15:635–645. doi: 10.1016/j.cmet.2012.04.001. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 14.Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916.e7. doi: 10.1053/j.gastro.2012.06.031. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 16.Tigchelaar EF, Zhernakova A, Dekens JAM, Hermes G, Baranska A, Mujagic Z, Swertz MA, Munoz AM, Deelen P, Cenit MC, Franke L, Scholtens S, Stolk RP, Wijmenga C, Feskens EJM. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open. 2015;5:e006772. doi: 10.1136/bmjopen-2014-006772. doi: 10.1136/bmjopen-2014-006772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scholtens S, Smidt N, Swertz MA, Bakker SJ, Dotinga A, Vonk JM, van Dijk F, van Zon SK, Wijmenga C, Wolffenbuttel BH, Stolk RP. Cohort Profile: LifeLines, a three-generation cohort study and biobank [published online ahead of print December 14, 2014]. Int J Epidemiol. :pii–dyu229. doi: 10.1093/ije/dyu229. [DOI] [PubMed] [Google Scholar]
- 18.Li N, van der Sijde MR, Bakker SJ, Dullaart RP, van der Harst P, Gansevoort RT, Elbers CC, Wijmenga C, Snieder H, Hofker MH, Fu J LifeLines Cohort Study Group. Pleiotropic effects of lipid genes on plasma glucose, HbA1c, and HOMA-IR levels. Diabetes. 2014;63:3149–3158. doi: 10.2337/db13-1800. doi: 10.2337/db13-1800. [DOI] [PubMed] [Google Scholar]
- 19.Deelen P, Bonder MJ, van der Velde KJ, Westra HJ, Winder E, Hendriksen D, Franke L, Swertz MA. Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res Notes. 2014;7:901. doi: 10.1186/1756-0500-7-901. doi: 10.1186/1756-0500-7-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–825. doi: 10.1038/ng.3021. [DOI] [PubMed] [Google Scholar]
- 21.Deelen P, Menelaou A, van Leeuwen EM, et al. Genome of Netherlands Consortium. Improved imputation quality of low-frequency and rare variants in European samples using the ‘Genome of The Netherlands’. Eur J Hum Genet. 2014;22:1321–1326. doi: 10.1038/ejhg.2014.19. doi: 10.1038/ejhg.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 24.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandt BW, Bonder MJ, Huse SM, Zaura E. TaxMan: a server to trim rRNA reference databases and inspect taxonomic coverage. Nucleic Acids Res. 2012;40:W82–W87. doi: 10.1093/nar/gks418. doi: 10.1093/nar/gks418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonder MJ, Abeln S, Zaura E, Brandt BW. Comparing clustering and pre-processing in taxonomy analysis. Bioinformatics. 2012;28:2891–2897. doi: 10.1093/bioinformatics/bts552. doi: 10.1093/bioinformatics/bts552. [DOI] [PubMed] [Google Scholar]
- 27.May A, Abeln S, Crielaard W, Heringa J, Brandt BW. Unraveling the outcome of 16S rDNA-based taxonomy analysis through mock data and simulations. Bioinformatics. 2014;30:1530–1538. doi: 10.1093/bioinformatics/btu085. doi: 10.1093/bioinformatics/btu085. [DOI] [PubMed] [Google Scholar]
- 28.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;509:357–360. doi: 10.1038/nature13178. doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navas-Molina JA, Peralta-Sánchez JM, González A, et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013;531:371–444. doi: 10.1016/B978-0-12-407863-5.00019-8. doi: 10.1016/B978-0-12-407863-5.00019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes JB, Hellmann JJ. The application of rarefaction techniques to molecular inventories of microbial diversity. Methods Enzymol. 2005;397:292–308. doi: 10.1016/S0076-6879(05)97017-1. doi: 10.1016/S0076-6879(05)97017-1. [DOI] [PubMed] [Google Scholar]
- 31.Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput Biol. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keurentjes JJ, Fu J, de Vos CH, Lommen A, Hall RD, Bino RJ, van der Plas LH, Jansen RC, Vreugdenhil D, Koornneef M. The genetics of plant metabolism. Nat Genet. 2006;38:842–849. doi: 10.1038/ng1815. doi: 10.1038/ng1815. [DOI] [PubMed] [Google Scholar]
- 33.Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Locke AE, Kahali B, Berndt SI, et al. LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nitsch D, Molokhia M, Smeeth L, DeStavola BL, Whittaker JC, Leon DA. Limits to causal inference based on Mendelian randomization: a comparison with randomized controlled trials. Am J Epidemiol. 2006;163:397–403. doi: 10.1093/aje/kwj062. doi: 10.1093/aje/kwj062. [DOI] [PubMed] [Google Scholar]
- 37.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Borén J, Oresic M, Bäckhed F. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–1112. doi: 10.1194/jlr.M002774. doi: 10.1194/jlr.M002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Chatelier E, Nielsen T, Qin J, et al. MetaHIT consortium. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 41.Caspi R, Altman T, Billington R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–D471. doi: 10.1093/nar/gkt1103. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Hepner G, Fried R, St Jeor S, Fusetti L, Morin R. Hypocholesterolemic effect of yogurt and milk. Am J Clin Nutr. 1979;32:19–24. doi: 10.1093/ajcn/32.1.19. [DOI] [PubMed] [Google Scholar]
- 44.Hlivak P, Odraska J, Ferencik M, Ebringer L, Jahnova E, Mikes Z. One-year application of probiotic strain Enterococcus faecium M-74 decreases serum cholesterol levels. Bratisl Lek Listy. 2005;106:67–72. [PubMed] [Google Scholar]
- 45.Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 47.Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, Takahashi T, Miyauchi S, Shioi G, Inoue H, Tsujimoto G. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]