

Abstract
The E3 ubiquitin ligase and tumor suppressor SCFFbw7 exists as three isoforms that govern the degradation of a host of critical cell regulators, including c-Myc, cyclin E, and PGC-1α. Peroxisome proliferator activated receptor-gamma coactivator 1α (PGC-1α) is a transcriptional coactivator with broad effects on cellular energy metabolism. Cellular PGC-1α levels are tightly controlled in a dynamic state by the balance of synthesis and rapid degradation via the ubiquitin-proteasome system. Yet, isoform-specific functions of SCFFbw7 are yet to be determined. Here, we show that the E3 ubiquitin ligase, SCFFbw7, regulates cellular PGC-1α levels via two independent, isoform specific, mechanisms. The cytoplasmic isoform (SCFFbw7β) reduces cellular PGC-1α levels via accelerated ubiquitin-proteasome degradation. In contrast, the nuclear isoform (SCFFbw7α) increases cellular PGC-1α levels and protein stability via inhibition of ubiquitin-proteasomal degradation. When nuclear Fbw7α proteins are redirected to the cytoplasm, cellular PGC-1α protein levels are reduced through accelerated ubiquitin-proteasomal degradation. We find that SCFFbw7β catalyzes high molecular weight PGC-1α-ubiquitin conjugation, whereas SCFFbw7α produces low molecular weight PGC-1α-ubiquitin conjugates that are not effective degradation signals. Thus, selective ubiquitination by specific Fbw7 isoforms represents a novel mechanism that tightly regulates cellular PGC-1α levels. Fbw7 isoforms mediate degradation of a host of regulatory proteins. The E3 ubiquitin ligase, Fbw7, mediates PGC-1α levels via selective isoform-specific ubiquitination. Fbw7β reduces cellular PGC-1α via ubiquitin-mediated degradation, whereas Fbw7α increases cellular PGC-1α via ubiquitin-mediated stabilization.
Keywords: Fbw7, PGC-1α, protein degradation, proteasome, ubiquitin, energy metabolism
INTRODUCTION
Fbw7 (F-box, WD repeat domain containing 7) is an E3 ubiquitin ligase and a component of SCF (complex of SKP1, CUL1, and F-box protein) that regulates via ubiquitination a network of proteins with central roles in cell growth control and cell cycle progression, as well as tumor suppression. It is evolutionarily conserved from yeast, nematode, fly and man (Cdc4, lin-12, Archipelago, and Fbw7, respectively) (Welcker and Clurman, 2008).
Within the ubiquitin-proteasome pathway, the conjugation of ubiquitin to its protein substrate proceeds via a three-step cascade mechanism involving ubiquitin activation, transfer to a ubiquitin-conjugating enzyme, and ultimately covalent linkage to a substrate via a ubiquitin-protein ligase, such as Fbw7. Fbw7 functions as a homodimer and recruits its own E2 ubiquitin-conjugating enzymes (Welcker and Clurman, 2008). In successive reactions, a polyubiquitin chain is synthesized by progressive transfer of additional activated ubiquitin moieties to the previously conjugated ubiquitin molecule. This polyubiquitin chain is a recognition marker for the 26S proteasome. Following conjugation of ubiquitin, the target protein is therefore degraded by the proteasome, and free ubiquitin is released and recycled (Schwartz and Ciechanover, 2009). Substrates of Fbw7 include c-Myc, c-Jun, Notch, and cyclin E (Welcker and Clurman, 2008).
Human Fbw7 encodes three transcripts (Fbw7α, Fbw7β, and Fbw7γ) that are generated through alternative splicing. Each isoform contains a unique, isoform-specific first exon, which determines distinct subcellular localizations. Fbw7α is nuclear, whereas Fbw7β is cytoplasmic and Fbw7γ nucleolar. The majority of cellular Fbw7 is the alpha isoform, with a relative isoform mRNA abundance of ~1:0.07:0.01 (α: β: γ) (Grim et al., 2008). Isoform expression is differentially regulated by cell cycle and various stimuli (Sionov et al., 2013). Studies of cells lacking specific Fbw7 isoforms suggest that Fbw7α accounts for almost all Fbw7 activity toward cyclin E and c-Myc (Grim et al., 2008), whereas Fbw7β degrades Notch/NICD (Sancho et al., 2013). However, the outcomes of substrates targeted by the various Fbw7 isoforms are not well understood, nor are the mechanisms of Fbw7 isoform action and regulation. One such bona fide substrate is PGC-1α.
Members of the PPAR-γ (peroxisome proliferator activated receptor-gamma) coactivator-1 (PGC-1) family of transcriptional coregulators serve as inducible coactivators of nuclear receptor and non-nuclear receptor transcription factors involved in the control of mitochondrial biogenesis and cellular energy metabolic pathways. Dysregulation of PGC-1α has been implicated in the pathogenesis of diabetes and insulin resistance (Lin et al., 2005; Handschin and Spiegelman, 2006). PGC-1α was identified through its functional interaction with the nuclear receptor PPAR-γ in brown adipose tissue (Puigserver et al., 1998). PGC-1α and its homologue, PGC-1β, are preferentially expressed in tissues with high oxidative capacity such as heart and skeletal muscle, and serve critical roles in the regulation of mitochondrial function and cellular energy metabolism, as does a naturally occurring 270 amino acid 3′ splice variant (NT-PGC-1α) recently described by Zhang et al. (2009). The docking of PGC-1 coactivators to specific transcription factors provides a key platform for the recruitment of regulatory protein complexes that exert powerful effects on gene transcription triggering biological responses that equip the cell to meet the energy demands of the changing environment (Finck and Kelly, 2006). PGC-1α is a potent co-activator of multiple nuclear receptor transcription factors including PPAR-γ, PPAR-α, PPAR-β, RXRα, and the glucocorticoid receptor. In addition, several non-nuclear receptor PGC-1 partners have been identified, including nuclear respiratory factors 1 and 2 (NRF-1 and 2) and Foxhead Box (01) (Kelly and Scarpulla, 2004; Puigserver and Spiegelman, 2003). A variety of physiological and dietary factors induce PGC-1α mRNA expression. For example, PGC-1α expression is stimulated by exercise and by fasting (Lin et al., 2005; Finck and Kelly, 2006) and PGC-1α has recently been linked to regulation of angiogenesis (Arany et al., 2008). PGC-1α thus serves as an inducible regulator of metabolism and tissue homeostasis.
The molecular aspects that govern PGC-1α turnover are poorly understood. Cellular PGC-1α levels vary dynamically and are tightly controlled by the balance between its synthesis and degradation. Several studies have addressed determinants of PGC-1α synthesis. However, there is relatively little understanding of the mechanism(s) and regulation of PGC-1α protein dynamics and degradation. Cytokines and phosphorylation have been implicated in the control of PGC-1α degradation rates (Puigserver et al., 2001). Puigserver’s group has shown that acetylation is involved in the post-transcriptional control of PGC-1α (Dominy et al, 2010). Recently, Sano et al. (2007), Anderson et al. (2008), and we (Trausch-Azar et al., 2010) proposed that PGC-1α degradation involves the ubiquitin-proteasome pathway.
We recently demonstrated that PGC-1α is localized to the nucleus, where it is rapidly degraded (t½ 0.3–0.5 hours) via the ubiquitin-proteasome system (Trausch-Azar et al., 2010). However, a naturally occurring N-terminal splice variant of 270 residues (NT-PGC-1α) is cytoplasmic and stable (t½ >7 hours). Our results strongly suggest that an N-terminus-dependent ubiquitin-proteasome pathway governs at least some aspects of PGC-1α cellular degradation (Trausch-Azar et al., 2010). Olson et al. (2008) has suggested that Fbw7 may serve as an E3 ubiquitin ligase for phosphorylated-PGC-1α via recognition of a conserved phospho-motif.
Here we investigated the ubiquitin-dependent regulation of PGC-1α by specific Fbw7 isoforms. We show that the E3 ubiquitin ligase, Fbw7, regulates cellular PGC-1α levels via two independent mechanisms. Fbw7β, a cytoplasmic isoform, reduces cellular PGC-1α levels via accelerated ubiquitin-proteasomal degradation. In contrast, Fbw7α, a nuclear isoform increases cellular PGC-1α levels via a decrease in ubiquitin-proteasomal degradation. Fbw7 mutants with distinct subcellular localizations show that cytoplasmic Fbw7 is associated with accelerated ubiquitin-proteasomal degradation. In addition, Fbw7β catalyzes high molecular weight PGC-1α-ubiquitin conjugates as intermediates in degradation, whereas Fbw7α catalyzes low molecular weight PGC-1α-ubiquitin conjugates that are likely not effective degradation intermediates. Thus, Fbw7 isoforms regulate cellular PGC-1α levels via discrete mechanisms via the ubiquitin system.
MATERIALS AND METHODS
Cell culture, reagents and plasmids
The human HeLa cell line, 293T and U20S cells were obtained from the American Type Culture Collection. C2C12 and their differentiation into myotubes has been described earlier (Sun et al., 2005). Cells were maintained under standard conditions using MEM containing 10% FBS. HCT116+/+ (Fbw7+/+) wild type cells and HCT116−/− (Fbw7−/−) cells in which the entire Fbw7 genetic locus has been disrupted were generous gifts of Bert Vogelstein and Kenneth W. Kinzler (Johns Hopkins University and Howard Hughes Medical Institute) (Rajagopalan et al., 2004). Briefly, cells were propagated in McCoy’s 5A medium (Gibco) supplemented with 10% FBS and maintained in a humidified chamber at 37°C with 5% CO2. Isoform-specific Fbw7 targeted cell lines were derived from HCT116 and were generous gifts of Bruce Clurman (University of Washington) (Grim et al., 2008).
Transient transfections of HeLa and Fbw7 cells were performed using X-tremeGENE 9 DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. MG132 was purchased from Peptides International. Primary antibodies used in this study: anti-FLAG (M2; Sigma), anti-actin (Sigma), anti-HA (Clone 16B12) (Covance), and anti-c-Myc (Santa Cruz Biotechnology). Anti-PGC-1α was kindly provided by Dan Kelly (Sanford-Burnham Medical Research Institute at Lake Nona) and HA-ubiquitin was generously provided by Aaron Ciechanover (Technion-Israel Institute of Technology, Haifa, Israel).
Full-length PGC-1α in the pSV-SPORT vector has been previously described (Trausch-Azar et al., 2010; Puigserver et al., 1998). Fbw7 isoforms were cloned into 3pX-FLAG-myc-CMV-24 expression vector (Sigma) as described in (Welcker et al., 2004). All deletions, mutations and insertions to the Fbw7α construct (Fbw7α-NLSmut; Fbw7α-NLSdel; Nβ-Fbw7α, Fbw7−ex1) were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and fully sequenced.
RNA Extraction, cDNA Synthesis and qPCR
Reverse transcriptase qPCR was used for analysis of the mRNA levels. RNA was isolated using the RNeasy Mini Kit (Qiagen). Super-Script III First-Strand Synthesis System for RT-PCR (Life Technologies, Carlsbad, CA) was used to generate cDNA using 2 ug of RNA and oligo dT primer, according to the manufacturer’s instructions. Equal amounts of cDNA were mixed with 10 ul of iTAQ Universal SYBR Green Supermix (Bio-Rad) and 250 nM of both forward and reverse primers. Beta-actin was amplified as an internal control.
qPCR reactions were conducted in triplicate using the MiniOpticon Real-Time PCR Detection System (Bio-Rad). Thermocycling conditions included a denaturation step at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for one minute. Data was processed using Bio-Rad CFX Software Version 1.5 (Bio-Rad). Gene expression was quantified using the comparative Ct method. Primer sequences are available upon request.
Immunofluorescence
Subcellular localization of PGC-1α, Fbw7α, Fbw7β, and Fbw7α mutants was determined by indirect immunofluorescence in HeLa cells. Briefly, cells were seeded onto glass coverslips in 6 cm dishes and transfected the following day using X-tremeGENE 9 Transfection Reagent (Roche). 16–20 hours after transfection, cells were washed with PBSc, a phosphate-buffered saline solution (DPBS, Cellgro) supplemented with 100mM CaCl2 and 50mM MgCl2, fixed with ice-cold methanol:acetone (1:1) for 10 minutes on ice and air dried. After blocking in 1% BSA/PBSc, coverslips were immunostained with primary antibody to determine subcellular localization. After washing with PBSc, coverslips were incubated with the appropriate AlexaFluor 488 or 568 secondary antibodies (Invitrogen/ Molecular Probes) and mounted using Mowiol containing 2.5% 1,4-diazobicyclo-[2.2.2]-octane (DABCO, Sigma). Images were captured using an Olympus FluoView 1000 confocal laser scanning microscope, and Olympus FV10-ASW software v3.0.
Determination of Protein Half Life
As previously described (Trausch-Azar et al., 2010), 16–20 hours after transfection, HeLa or Fbw7 cells were incubated with cycloheximide (100 μg/ml, Sigma) to inhibit further protein synthesis. MG132 (20 μM) was added along with cycloheximide as noted. An equal number of cells was then lysed after 0, 0.5, 1, 2, and 3 hours in PBS containing 5% Igepal, Complete Protease Inhibitor Cocktail (Roche), and 1 mM DTT, for at least 30 minutes after which cells were sonicated, then centrifuged at 14,000 rpm for 10 minutes at 4°C in an Eppendorf microcentrifuge to remove cellular debris. The lysates were mixed with an equal amount of reducing Laemmli sample buffer (Bio-Rad) and equal amounts of sample were run on a 10% TGX gel (Bio-Rad) and electroblotted onto nitrocellulose (Osmonics). Blots were blocked with 5% milk and probed with primary antibody followed by incubation with a secondary horseradish peroxidase-conjugated antibody and detection by chemiluminescence (GE Healthcare). Actin served as an internal control. The resulting bands were quantitated using the Kodak EDAS system and the data was graphed using the Excel graphing program (Microsoft). Protein degradation rate is expressed as half-life (t½), the time for degradation of 50% of the protein. Half-life data reported was evaluated by ≥ 3 independent determinations.
Immunoprecipitation and detection of ubiquitylated proteins
Fbw7−/− cells were transfected with PGC-1α, HA-ubiquitin and Fbw7α or Fbw7β or Fbw7α mutants in a 1:1:1 ratio. 16–20 hours after transfection, cells were lysed in freshly prepared lysis buffer containing 50 mM Tris pH 8.0, 0.5% SDS, 10 mM EDTA, 0.1 mM N-ethylmaleimide, 20 mM DTT and Complete Protease Inhibitor Cocktail (Roche), and boiled for 5 minutes at 95°C. Following brief sonication, cells were spun at 14,000 rpm at 4°C for 10 minutes to remove cellular debris. Supernatants were diluted 10-fold in TNN buffer (50 mM Tris-HCl, 120 mM NaCl, 5 mM EDTA and 0.5% Igepal). Direct immunoprecipitation of ubiquitinylated proteins was performed using 20 ul of EZview Red Anti-HA Affinity Gel (Sigma). The gel was washed, and following the addition of reducing Laemmli sample buffer (Bio-Rad) the immunoprecipitates were run on a 10% TGX gel and analyzed via Western blot. The blots were probed with polyclonal anti- PGC-1α antibody (1:1000 dilution) followed by incubation with a secondary horseradish peroxidase-conjugated antibody and detected by chemiluminescence (GE Healthcare).
Fbw7 in Fbw−/− and Fbw7+/+ Cells
Detection of endogenous and exogenous Fbw7 by Western blots of immunoprecipitates. Fbw7+/+ cells, Fbw7−/− cells and Fbw−/− cells transfected with Fbw7α and Fbw7β were lysed in RIPA buffer containing 50 mM Tris, pH 6.8, 150 mM NaCl, 1% Igepal (Sigma), 0.5% deoxycholate and Complete Protease Inhibitor Cocktail (Roche) for 30 minutes on ice. Following brief sonication, cells were spun at 14,000 rpm at 4°C for 10 minutes to remove cellular debris. 1 mg protein of cleared supernatent was diluted to 1 ml in RIPA buffer and added to PureProteome Protein A Magnetic beads (Millipore) bound with 6 μg of anti-Fbw7 antibody (Bethyl, A310–720A) per manufacturer’s instructions. Following an overnight incubation at 4°C, beads were washed, eluted with reducing Laemmli sample buffer (Bio-Rad) and eluates were run on a 10% TGX gel. Proteins were transferred to nitrocellulose and Western blots were probed with anti-Fbw7 antibody (Bethyl, A310–720A) at 1 μg/ml followed by incubation with a secondary horseradish peroxidase-conjugated antibody and detected by chemiluminescence (GE Healthcare).
RESULTS
As an initial step toward defining the role of Fbw7 isoforms in ubiquitin-proteasome mediated degradation of PGC-1α, we examined the effect of Fbw7α or Fbw7β on endogenous PGC-1α steady state levels in HCT116 in which the entire Fbw7 genetic locus has been disrupted (i.e., Fbw7−/−, 17). As seen in Figure 1A, exogenous expression of Fbw7α substantially increases the steady state levels of endogenous PGC-1α. In contrast, expression of the cytoplasmic isoform, Fbw7β, significantly decreases the steady state levels of PGC-1α. Expression of Fbw7α or Fbw7β has a similar effect on steady state levels of exogenously expressed PGC-1α in all cell types examined (Fbw7−/−, wild type Fbw7+/+, HeLa (Figure 1B), 293 (see below), and U20S (not shown). These observations suggest that cellular PGC-1α protein levels are modulated differently by distinct Fbw7 isoforms, such that Fbw7α increases PGC-1α levels and Fbw7β decreases PGC-1α levels.
FIGURE 1. Differential effect of Fbw7α and Fbw7β on PGC-1α protein steady state levels.

A. To examine endogenous PGC-1α, Fbw7−/− cells were transfected with empty vector, Fbw7α or Fbw7β. 18h later cells were lysed and evaluated via SDS-PAGE and Western blotting for endogenous PGC-1α and actin (upper panel). Quantification of seven independent experiments (lower panel). B. HeLa, Fbw7−/− or Fbw7+/+ cells were transfected with PGC-1α with or without Fbw7α or Fbw7β. 18h later cells were lysed and evaluated via SDS-PAGE and Western blotting for PGC-1α and actin (as a loading control) (upper panel). Quantification of six independent experiments (lower panel).
In order to determine whether the changes in PGC-1α levels are the result of changes in PGC-1α degradation rates, we examined the effect of Fbw7α or Fbw7β on PGC-1α half-life. As seen in Figure 2, in Fbw7−/− cells PGC-1α is rapidly degraded with half-life (t½) ~ 0.6h. Incubation with MG132, a potent and selective inhibitor of the proteasome, markedly slowed the rate of PGC-1α degradation, as we have shown earlier (Trausch-Azar et al., 2010). Cotransfection of Fbw7−/− cells with Fbw7α resulted in a markedly slower rate of PGC-1α degradation (t½ ~ 1.8 hours). This is observed in all cell types studied (Figure 2; Table 1). In contrast, co-expression of Fbw7β resulted in either similar or more rapid degradation (t½ ~ 0.5 hours) (Figure 2, Table 1). In many experiments (see below and subsequent Figures) PGC-1α is often seen as a doublet, the upper band likely phosphorylated, as described earlier (Olson et al., 2008). In all cases, the degradation rate was markedly prolonged by MG132. A summary of numerous PGC-1α degradation studies in HeLa and Fbw7−/− cells is shown in Table 1. In both cell lines, Fbw7α stabilizes PGC-1α, while Fbw7β accelerates ubiquitin-proteasome dependent degradation of PGC-1α. These results correlate with the effects of Fbw7α and Fbw7β on PGC-1α steady state levels.
FIGURE 2. Fbw7 isoform specific impact on PGC-1α degradation rate.
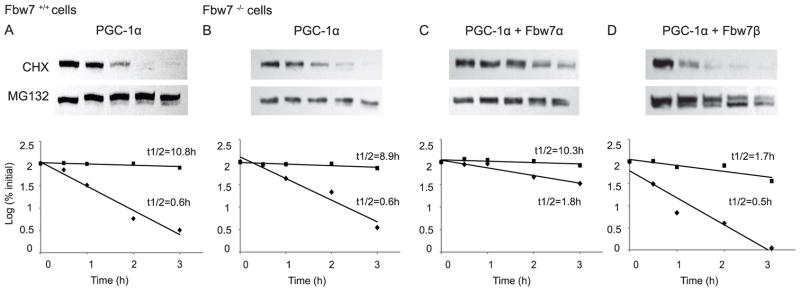
18h after transfection with the indicated plasmids, Fbw+/+ (A) or Fbw7−/− (B, C, D) cells were treated with CHX +/− MG132. Cells were thereafter lysed at 0, 0.5, 1, 2, and 3h and were evaluated for PGC-1α degradation via SDS-PAGE, Western blotting for PGC-1α. The pixels for each band were measured and normalized so that the number of pixels at t=0 was 100%. The log10 of the percent of pixels was plotted versus time for each time point and the t½ was calculated from the log of 50%.
Table 1.
Summary of Fbw7α or Fbw7β on PGC-1α degradation rates.
| PGC-1α | +Fbw7α | +Fbw7β | |
|---|---|---|---|
| HeLa | 0.3 +/− 0.02h (38) | 2.9 +/− 0.4h (17) | 0.2 +/− 0.02h (7) |
| Fbw7−/− | 0.8 +/− 0.09h (18) | 2.0 +/− 0.5h (10) | 0.6 +/− 0.04h (9) |
PGC-1α degradation rates in the presence of Fbw7α or Fbw7β were determined as described in Figure 2. Figures indicate mean +/− SEM (number of independent determinations).
We next examined the effect of Fbw7α or Fbw7β on endogenous PGC-1α or exogenously expressed PGC-1α levels in cells where either Fbw7α or Fbw7β were specifically targeted. Endogenous Fbw7 protein expression is seen in Fbw7+/+ cells, but not in Fbw−/− cells as shown previously by Welcker et al (Welcker et al., 2013; Welcker et al., 2004). Transfection with Fbw7 increases Fbw7 protein expression 102–103-fold (supplemental Figure 1). As seen in Figure 3, Fbw7α markedly increases endogenous PGC-1α levels in cells in which either Fbw7α or Fbw7β is targeted (Figure 3A). In contrast, expression of Fbw7β consistently reduces PGC-1α, albeit to a small degree (Figure 3A). When PGC-1α is exogenously expressed, Fbw7α increases PGC-1α levels in cells in which either Fbw7α or Fbw7β is targeted (Figure 3B). In contrast, Fbw7β expression markedly decreases PGC-1α levels in both cell types (Figure 3B). As seen earlier (Grim et al., 2008), and as expected, in Fbw7α targeted cells, Fbw7α mRNA is decreased and Fbw7β mRNA increased, whereas in Fbw7β targeted cells Fbw7β mRNA is decreased (data not shown). These data are consistent with our other findings that suggest Fbw7α stabilizes PGC-1α and Fbw7β accelerates PGC-1α degradation.
FIGURE 3. Effect of Fbw7α and Fbw7β on PGC-1α steady state levels in isoform-specific Fbw7 targeted cells.

A. Effects of Fbw7α and Fbw7β on endogenous PGC-1α in Fbw7α-targeted and Fbw7β-targeted cells. B. Effects of Fbw7α and Fbw7β on exogenously expressed PGC-1α in Fbw7α-targeted and Fbw7β-targeted cells. Details are provided in legend to Figure 1.
Our observations are distinct from those of Olson et al. (2008) who reported a limited (~5%, see Discussion) effect of Fbw7 on PGC-1α degradation. However, the N-terminus of PGC-1α appears to be important for its regulation and Olson et al. (2008) utilized an N-terminally FLAG-tagged PGC-1α. In order to determine the effect of an N-terminal FLAG-tag on Fbw7 isoform-mediated degradation of PGC-1α, we examined the effect of Fbw7α and Fbw7β on FLAG-PGC-1α steady state levels. As seen in supplemental Figure 2, in this case Fbw7α and Fbw7β each reduced cellular FLAG-PGC-1α levels 10–20%. Fbw7α and Fbw-7β minimally increased the rate of FLAG-PGC-1α degradation (t½~0.4 hours), as seen in supplemental Figure 3. Thus, a distinct free N-terminal region of PGC-1α is important for isoform specific regulation and N-terminal-FLAG-tagged PGC-1α degradation is not modulated in the same manner as non-tagged PGC-1α. Taken together, we conclude that Fbw7α stabilizes while Fbw7β accelerates PGC-1α degradation.
Fbw7α and Fbw7β are nuclear and cytoplasmic, respectively (Welcker and Clurman, 2008). We hypothesized that the different degradation rates of PGC-1α are dependent upon the distinct subcellular localization of Fbw7 isoforms. These isoforms differ in exon 1: Fbw7α contains two nuclear localization sequences (exon 1 aa 11–14 and BD1 aa 2–4). Fbw7β exon 1, in contrast, contains a hydrophobic amino acid sequence which confers membrane association (Welcker et al., 2004). In order to determine if localization of Fbw7 was a significant determinant of PGC-1α degradation rate, we generated three Fbw7α chimeric proteins: Fbw7α-NLSmut (in which the two NLS residues were substituted with alanine), Fbw7α-NLSdel (in which the two NLS were deleted), and Nβ-Fbw7α (in which exon 1 of Fbw7β was placed N-terminal to Fbw7α) (Figure 4A). We determined the subcellular localization of the three mutants, as well as Fbw7α and Fbw7β, by confocal immunofluorescence. As seen in Figure 4B, Fbw7α is nuclear and Fbw7β is cytoplasmic, as expected. Both Fbw7α-NLSmut and Fbw7α-NLSdel were largely nuclear with many cells demonstrating cytoplasmic staining. Importantly, Nβ-Fbw7α was largely cytoplasmic with little nuclear staining. Quantification of Fbw7α chimeric protein localization is seen in Table 2. Thus, these chimeric proteins demonstrate a gradient of nuclear/cytoplasmic localization from Fbw7α to Fbw7β (Figure 4A, 4B). We next examined the colocalization of PGC-1α with each of the five Fbw7 species. Regardless of which Fbw7 isoform was expressed, PGC-1α was localized to the nucleus (Figure 4C). Colocalization of Fbw7 and PGC-1α depended on the degree of nuclear location of each Fbw7 isoform. With nuclear Fbw7α, PGC-1α and Fbw7α colocalized well. With Fbw7β, PGC-1α was nuclear whereas Fbw7β was cytoplasmic with essentially no colocalization. With Fbw7α-NLSmut and Fbw7α-NLSdel, there was substantial colocalization in the nucleus, whereas with Nβ-Fbw7α there was not.
FIGURE 4. Confocal microscopic localization of Fbw7 isoforms in the absence and presence of exogenous PGC-1α.

18h after transfection with Fbw7 isoforms alone or cotransfected with PGC-1α, HeLa cells were fixed and localization of Fbw7 isoforms and PGC-1α was visualized with anti-FLAG (Fbw7) (B, C) and anti-PGC-1α via double-label confocal immunofluorescence (C). Panel A diagrams the Fbw7 isoforms examined in B and C and indicates the subcellular localization as shown in Table 2.
Table 2.
Subcellular distribution of Fbw7α chimeric proteins.
| % Nuclear | % N≫C | % N~C | % Cyto | |
|---|---|---|---|---|
| Fbw7α | 88 | 12 | 0 | 0 |
| Fbw7α-NLSmut | 0 | 77 | 5 | 18 |
| Fbw7α-NLSdel | 0 | 73 | 4 | 23 |
| Nβ-Fbw7α | 0 | 10 | 6 | 84 |
| Fbw7β | 0 | 3 | 6 | 91 |
Subcellular localization of each Fbw7α chimeric protein was quantified and expressed as % nuclear, % N≫C, % N~C, and % cytoplasmic. Localization of Fbw7α and Fbw7β served as controls.
In order to determine if PGC-1α subcellular localization was altered in cells lacking Fbw7, we examined PGC-1α subcellular distribution in Fbw7+/+ and Fbw7−/− cells in the absence or presence of MG132. As seen in Table 3, PGC-1α localization was unaltered by Fbw7 expression.
Table 3.
Subcellular distribution of PGC-1α in Fbw7+/+ and Fbw7−/− cells.
| % Nuclear | % N≫C | % N~C | % Cyto | |
|---|---|---|---|---|
| Fbw7+/+ | 0 | 93 | 7 | 0 |
| Fbw7+/+ + MG132 | 0 | 95 | 5 | 0 |
| Fbw7−/− | 0 | 90 | 9 | 1 |
| Fbw7−/− + MG132 | 0 | 96 | 3 | 1 |
Subcellular localization of PGC-1α protein +/− MG132 was quantified and expressed as % nuclear, % N≫C, % N~C, and % cytoplasmic. Fbw7α and Fbw7β served as controls.
We next examined if changing the cellular localization of Fbw7α impacts the steady state level of PGC-1α. As seen in Figure 5, cellular PGC-1α level was increased by Fbw7α and markedly decreased by Fbw7β, similar to that seen in Figure 1. Nβ-Fbw7α, a “cytosolic” Fbw7α, had an effect closely similar to that of Fbw7β resulting in the destabilization of PGC-1α. Both Fbw7α-NLSmut and Fbw7α-NLSdel had effects intermediate between those of Fbw7α and Fbw7β but much closer to that of Fbw7α. The steady state levels of PGC-1α correlated strongly with the degree of cellular colocalization with Fbw7. When Fbw7 is cytosolic, PGC-1α was destabilized. When Fbw7 was nuclear, PGC-1α, also nuclear, was stabilized. Thus, Fbw7 isoform localization appears to be a major determinant of its ability to modulate PGC-1α cellular levels.
FIGURE 5. Effect of Fbw7α chimeric protein mutants on PGC-1α steady state levels.

Fbw7−/− cells were transfected with PGC-1α with or without Fbw7α, Fbw7β, or Fbw7α mutants (Fbw7α-NLSmut; Fbw7α-NLSdel; Nβ-Fbw7α). 18h later cells were lysed and evaluated via SDS-PAGE and Western blotting as described in Figure 1.
We also examined the effect of these Fbw7 chimeric proteins on PGC-1α degradation rate. As seen in Figure 6, Fbw7α decreased and Fbw7β increased the rate of PGC-1α degradation, similar to that seen in Figure 2. Nβ-Fbw7α had an effect closely similar to that of Fbw7β (Figure 6F), whereas both Fbw7α-NLSmut and Fbw7α-NLSdel (Figure 6D, 6E) had effects intermediate between those of Fbw7α and Fbw7β but much closer to that of Fbw7α. Thus, as with the results seen at steady state (Figure 5), Fbw7 isoform localization appears to be a major determinant of its ability to modulate PGC-1α degradation.
FIGURE 6. Effect of Fbw7 chimeric protein mutants on PGC-1α degradation rate.

18h after transfection of the indicated plasmids, Fbw7−/− cells were treated with CHX and thereafter lysed at 0 or 2h and evaluated for PGC-1α degradation via SDS-PAGE, Western blotting for PGC-1α as described in Figures 2 and 4.
As Fbw7α and Fbw7β differ only in exon 1, we examined the effect of exon1-less Fbw7α (Fbw7−ex1) on the steady state level of PGC-1α, the PGC-1α degradation rate, and subcellular localization. As seen in supplemental Figure 4, cellular PGC-1α level was markedly decreased by Fbw7−ex1. Under these conditions, PGC-1α degradation rate was t½~0.5 hours. As expected, Fbw7−ex1 was diffusely present throughout the nucleus and cytoplasm, since the nuclear localization sequence of Fbw7α-exon1 was deleted and the minor nuclear localization within exon 2 was still present. Thus, Fbw7 isoform localization appears to be a major determinant of its ability to modulate PGC-1α degradation.
We next evaluated whether the isoform-specific effects of Fbw7 were confined to PGC-1α or were more general. Welcker et al. (Sun et al., 2005; Welcker et al., 2004) and Wei et al. (2005) have shown that Fbw7α and Fbw7β each accelerate ubiquitin-proteasome-dependent degradation of c-Myc and c-Jun, respectively. We examined the degradation of c-Myc (supplemental Figure 5), and find that cellular (c-Myc) levels are reduced by both Fbw7α and Fbw7β, with the Fbw7α effect greater than the Fbw7β effect. Thus, the differential isoform specific impact of Fbw7α and Fbw7β on PGC-1α degradation is substrate-specific and does not appear to be universal among all Fbw7 substrates.
Among the possibilities that confer localization-specific effects of Fbw7 on PGC-1α ubiquitin-proteasome dependent degradation are differences in the composition of the E2 and accessory proteins that will determine the pattern of substrate-ubiquitin conjugates (i.e., PGC-1α-ubiquitin conjugates), as well as differences in isopeptidase processing of the substrate-ubiquitin conjugates and subsequently proteasomal recognition and degradation. As an initial step to approach these issues we examined the pattern of cellular PGC-1α-ubiquitin conjugates formed in the presence of Fbw7α, Fbw7β or their derived chimeric proteins. Using immunoprecipitation-Western blot analysis we examined specifically PGC-1α-ubiquitin conjugates. Cellular PGC-1α ubiquitin conjugates formed in cells in the presence of Fbw7β are typical of the “high molecular weight” conjugates formed with substrates as intermediates in their degradation (Finley, 2009; Finley et al., 2012). In contrast, PGC-1α ubiquitin conjugates formed in the presence of Fbw7α are mostly low molecular weight species and far less “high molecular weight” conjugates are observed. Conjugates formed in the presence of Fbw7α-NLSmut or Fbw7α-NLSdel are intermediate between those found with Fbw7β and with Fbw7α with substantial low molecular weight species. Conjugates formed in the presence of Nβ-Fbw7α are also intermediate between Fbw7β and Fbw7α, but with an abundance of “high molecular weight” species. As seen in Figure 7, ubiquitin-protein conjugates were readily detected under all experimental conditions examined; as expected ubiquitin-protein conjugates were more abundant in the presence of MG132. This pattern of PGC-1α-ubiquitin conjugates precisely correlates with the levels of PGC-1α and the nuclear/cytoplasmic localization of the five Fbw7 species. Thus, distinct Fbw7 isoforms localized to specific subcellular compartments provide specificity for ubiquitin-conjugates of PGC-1α. Future experiments will examine these regulatory pathways in physiologically relevant tissues (e.g., skeletal muscle, adipose tissue, and heart).
FIGURE 7. Effect of Fbw7α and Fbw7β on ubiquitin conjugates of PGC-1α.

Fbw7−/− cells, +/− MG132, were transfected with PGC-1α, Fbw7α, Fbw7β, Fbw7α mutants and HA-ubiquitin, as noted. 18h later, cells were lysed, aliquots were Western blotted with anti-HA (panel A, B) to detect total cellular ubiquitin-protein conjugates or with anti-PGC-1α (panel C, D) to detect total cellular PGC-1α. Additional aliquots were immunoprecipitated with anti-HA and thereafter Western blotted for PGC-1α (panel E) to determine specific ubiquitin-PGC-1α conjugates. Ubiquitin-PGC-1α conjugates are marked by bracket. Low molecular weight conjugates are seen below ~225 kDa.
DISCUSSION
PGC-1α is an inducible master regulator of cellular energy metabolism. Cellular PGC-1α levels are controlled in a dynamic state dictated by the balance of its synthesis and degradation. PGC-1α is rapidly degraded (t½ ~ 0.3–0.5 hours) in the nucleus via the N-terminus-dependent ubiquitin-proteasome pathway (Trausch-Azar et al., 2010). Our investigation of the molecular mechanisms underlying PGC-1α degradation revealed a key isoform- and localization-specific role for Fbw7, an E3 ubiquitin ligase. The following lines of evidence support this conclusion: (i) expression of Fbw7β markedly reduces steady state PGC-1α cellular levels; (ii) expression of Fbw7β increases the degradation rate of PGC-1α; (iii) this accelerated degradation rate is proteasome (i.e., MG132)-dependent; (iv) Fbw7β is associated with a “high molecular weight” PGC-1α-ubiquitin conjugate pattern; (v) chimeric protein mutants of Fbw7 which contain the β-specific exon 1 fused to Fbw7α correlate with PGC-1α enhanced degradation. Furthermore, Fbw7α confers stability (i.e., decreased degradation rate) on PGC-1α. The following lines of evidence support this conclusion: (i) expression of Fbw7α increases steady state PGC-1α cellular levels; (ii) expression of Fbw7α decreases the degradation rate of PGC-1α; (iii) Fbw7α is associated with a PGC-1α-ubiquitin conjugate pattern with low molecular weight species; (iv) mutants of Fbw7α, a nuclear protein, which are localized to the cytoplasm (where Fbw7β is localized) increase PGC-1α degradation.
PGC-1α is a ubiquitin-dependent rapidly degraded protein; yet the E3 ubiquitin ligase(s) which govern ubiquitin conjugation to PGC-1α have remained elusive. We have herein described the role of Fbw7 isoforms in PGC-1α degradation. Fbw7, a component of SCF ubiquitin ligases, promotes ubiquitin-proteasome dependent degradation of the nuclear oncoproteins c-Myc and c-Jun, the Notch intracellular domain (NICD) and cyclin E (Welcker and Clurman, 2008). Fbw7 isoform-specific cellular localization is conferred by α- and β-specific first exons with Fbw7α localized to the nucleoplasm and Fbw7β localized to the ER/Golgi within the cytoplasm (Welcker et al., 2004). As described herein we find that Fbw7β is associated with accelerated ubiquitin-proteasome dependent degradation on PGC-1α whereas Fbw7α is associated with stabilized PGC-1α. Yet, both Fbw7α and Fbw7β promote c-Myc and c-Jun ubiquitination and degradation despite the localization of Fbw7α to the nucleus and Fbw7β to the cytoplasm (Welcker et al., 2004) (supplemental Figure 5).
There are many classic PGC-1α targets that are reported to be affected by changes in cellular PGC-1α levels. Others have shown an increase in mRNA levels of downstream targets of PGC-1α (including ERRα and MCAD (medium chain acyl CoA dehydrogenase)) following its expression in C2C12 myotubes and cardiac myocytes using Northern blots (Wu et al., 1999; Huss et al., 2002). Using qPCR, we found that Fbw7α- and Fbw7β-mediated changes in cellular PGC-1α levels induced minor increases in ERRα and MCAD expression in C2C12 myotubes, Fbw7−/− and HeLa cells (not shown). Fbw7α had a greater effect than Fbw7β (supplemental Figure 6).
Nonetheless, the difference in the Fbw7β versus Fbw7α effect on PGC-1α degradation appears to be directly related to the cellular localization of the Fbw7 species. This is seen with Fbw7α, Fbw7β, and the chimeric protein mutants (Figures 4, 5, 6, and 7). Cytoplasmic localization is associated with rapid degradation, whereas nuclear localization is associated with stability.
Interestingly, the yeast ortholog of Fbw7 is Cdc4 which functions within the SCFCdc4 complex and is exclusively localized to the nucleus. Cdc4 is required for the ubiquitin-proteasome-dependent degradation of Far1 (Henchoz et al., 1997). In the nucleus SCFCdc4 ubiquitinates Far1, which is then degraded by nuclear proteasomes while cytoplasmic Far1 is stabilized (Blondel et al., 2000). Thus, as for PGC-1α, subcellular localization of Cdc4 restricts degradation of Far1 to the nucleus. Interestingly, and along this line, in flies mutant for the Drosophila Fbw7 ortholog Archipelago (Ago), dMyc accumulates in the cytoplasm (Moberg et al., 2004).
Nuclear PGC-1α is likely to be tightly regulated by multiple ligases under different physiological conditions, as we find that PGC-1α is rapidly degraded in a ubiquitin-proteasome-dependent manner in Fbw7−/− cells (Figure 2). Furthermore, we have previously identified an N-terminal-dependent ligase with function predominantly within the nucleus, which is involved in targeting active PGC-1α for degradation (Trausch-Azar et al., 2010). Thus, under the normal physiological state Fbw7 is not the major ligase responsible for PGC-1α degradation. Indeed, it is possible that the Fbw7 effects are indirect, whereby Fbw7 could promote the degradation of another protein which either accelerates or stabilizes the degradation of PGC-1α. Furthermore, combining our data with that of Olson et al. (2008), it is likely that the N-terminal region of PGC-1α mediated isoform selectivity. However, their studies were performed with N-terminally FLAG-tagged PGC-1α. In order to determine the effect of an N-terminal-FLAG-tag on Fbw7 isoform-mediated degradation of PGC-1α, we examined the effect of Fbw7α or Fbw7β on FLAG-PGC-1α steady state levels. As seen in supplemental Figure 2, Fbw7α and Fbw7β each reduced cellular FLAG-PGC-1α levels 10–20%. Fbw7α and Fbw7β minimally increased the rate of FLAG-PGC-1α degradation (t½ ~ 0.4 hours) as seen in supplemental Figure 3. Thus, N-terminal-FLAG-tagged PGC-1α degradation is not modulated in the same manner as PGC-1α.
Compartmentalized degradation provides one mechanism whereby specific protein substrates are degraded dependent upon their subcellular localization (Blondel et al., 2000; Lingbeck et al., 2003; Lingbeck et al., 2005; Sun et al., 2008; Putters et al., 2011). Herein, localized degradation may be a general mechanism to regulate various biological processes in a spatial and temporal manner. Our observations with Fbw7β and Fbw7α present an interesting paradigm, wherein PGC-1α’s interaction with Fbw7 appears to be atypical of many other Fbw7 substrates and, thus, may offer additional insights into regulation of the ubiquitin-proteasome system and compartmentalized degradation. Herein, Fbw7α actively stabilizes PGC-1α within the nucleus and this is associated with differential ubiquitination. It should be noted, however, that this effect may not be direct, i.e., some factor in the nucleus may either prevent the formation of high molecular weight conjugates or trim large to small conjugates. This mechanism of enhancing or regulating nuclear protein stability via differential ubiquitination may well impact a variety of cellular processes.
The roles of Fbw7α in substrate degradation are complex. Recently, Welcker et al. (2013) have shown that the dimerization of Fbw7 via multiple interaction sites provides an additional degree of control by regulating Fbw7 stability and thus substrate recognition and degradation. However, while many substrates (e.g., c-Myc, c-Jun) appear to be ubiquitinated by Fbw7α, for other substrates (e.g., cyclin E) Fbw7α serves as a co-factor for subsequent ubiquitination via Fbw7γ. In in vitro reconstitution studies, Bhaskaran et al. (2013) suggest that Fbw7α facilitates isomerization of a non-canonical proline-proline bond in the cyclin E degron by prolyl cis-trans isomerase Pin1. This isomerization is required for subsequent binding and ubiquitination by SCFFbw7γ and proteasome-dependent degradation within the cell nucleolus. Furthermore, Min et al. (2012) provide evidence that Pin1 interacts directly with Fbw7α in a phosphorylation-dependent manner, disrupting Fbw7α dimerization and promoting Fbw7α self-ubiquitination and subsequent degradation. In this regard, it is possible that Pin1 interacts with Fbw7α such that Fbw7α is unable to generate “high molecular weight” ubiquitin conjugates with PGC-1α (Figure 7). Future studies will address the roles of Pin1 and Fbw7γ in Fbw7α-mediated PGC-1α stability.
Thus, while the precise molecular details remain to be established, these studies provide evidence that the mechanisms responsible for Fbw7α interactions with itself and its substrates are regulated at several levels including those of ubiquitination, phosphorylation, and dimer formation.
We observe that while Fbw7β promotes PGC-1α ubiquitination and proteasomal degradation, Fbw7α stabilizes PGC-1α. This is seen not only in Fbw7−/− cells lacking endogenous Fbw7 isoforms, but in Fbw7 expressing Fbw7+/+ and HeLa cells, independent of the level of Fbw7 expression. Thus, while the Fbw7α-dependent machinery is involved in active nuclear stabilization of selected proteins, the mechanism(s) is yet to be elucidated. Until recently, the generally accepted paradigm had been that “high molecular weight,” polyubiquitin-tagged substrates are recognized and degraded by the 26S proteasome. The minimal ubiquitin oligomer required for proteasomal recognition was shown to be a tetra-ubiquitin chain (Thrower et al., 2000). Recently, however, monoubiquitination, as well as multiple monoubiquitination recognition and degradation, has been demonstrated (e.g., PAX3 (Boutet et al., 2007)). Most recently Shabek et al. (2012) have shown that a single ubiquitin moiety fused to a polypeptide of ~150 residues is sufficient for proteasomal targeting and degradation. These observations thus suggest that the ubiquitin signal for proteasomal recognition is adaptable and context dependent, and that chain structure and internal ubiquitin linkage (i.e., Lys48, Lys 63, etc.) are critical determinants for the fate of ubiquitylated proteins. Indeed, our observation that PGC-1α-ubiquitin conjugates generated in vivo with Fbw7α differ dramatically from those generated with Fbw7β (Figure 7) is consistent with this notion. Yet to be resolved is whether these ubiquitin conjugates of PGC-1α are polyubiquitin chains, multiple monoubiquitinations (PGC-1α contains 50 lysine residues) or some combination. These differences may also stem from distinct nuclear interactions of Fbw7α with ubiquitin conjugating proteins (E2s), chain linkage specificity, or lack of chain elongation, such as those catalyzed by HUL5-like enzymes that are required for processive degradation of substrates for the proteasome (Aviram and Kornitzer, 2010) or that enhance nuclear retention of active PGC1α or both. Importantly, these observations have discovered a novel layer of regulation of the tumor suppressor Fbw7 that is both isoform specific and substrate specific. Understanding the molecular details surrounding this selectivity, the ubiquitin machinery involved and the identification of similar substrates have implications for further understanding Fbw7 biology, possibilities that will be evaluated in future studies.
Supplementary Material
Acknowledgments
Contract grant sponsor for ALS: National Institutes of Health; Contract grant number: R01-GM067620
Contract grant sponsor for AO: ICRF research project grant; Contract grant number: 800005
Footnotes
ALS, JST-A, and AO conceived and designed the experiments. JST-A and MA performed the experiments. ALS, JST-A, and AO analyzed the data. ALS, JST-A, and AO wrote the manuscript.
Conflict of Interest Disclosure: No conflicts to disclose (JST-A, MA, AO, ALS).
References
- 1.Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1 alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 3.Aviram S, Kornitzer D. The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol Cell Biol. 2010;30:985–994. doi: 10.1128/MCB.00909-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhaskaran N, van Drogen F, Ng HF, Kumar R, Ekholm-Reed S, Peter M, Sangfelt O, Reed SI. Fbw7α and Fbw7γ collaborate to shuttle cyclin E1 into the nucleolus for multiubiquitylation. Mol Cell Biol. 2013;33:85–97. doi: 10.1128/MCB.00288-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blondel M, Galan JM, Chi Y, Lafourcade C, Longaretti C, Deshaies RJ, Peter M. Nuclear-specific degradation of Far1 is controlled by the localization of the F-box protein Cdc4. EMBO J. 2000;19:6085–6097. doi: 10.1093/emboj/19.22.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boutet SC, Disatnik MH, Chan LS, Iori K, Rando TA. Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell. 2007;130:349–362. doi: 10.1016/j.cell.2007.05.044. [DOI] [PubMed] [Google Scholar]
- 7.Dominy JE, Jr, Lee Y, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1alpha’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta. 2010;1804:1676–1683. doi: 10.1016/j.bbapap.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finley D, Ulrich HD, Sommer T, Kaiser P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics. 2012;192:319–360. doi: 10.1534/genetics.112.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Ericsson J, Russell DW, Clurman BE. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol. 2008;181:913–920. doi: 10.1083/jcb.200802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 13.Henchoz S, Chi Y, Catarin B, Herskowitz I, Deshaies RJ, Peter M. Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma.Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 15.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 16.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Lingbeck JM, Trausch-Azar JS, Ciechanover A, Schwartz AL. Determinants of nuclear and cytoplasmic ubiquitin-mediated degradation of MyoD. J Biol Chem. 2003;278:1817–1823. doi: 10.1074/jbc.M208815200. [DOI] [PubMed] [Google Scholar]
- 18.Lingbeck JM, Trausch-Azar JS, Ciechanover A, Schwartz AL. E12 and E47 modulate cellular localization and proteasome-mediated degradation of MyoD and Id1. Oncogene. 2005;24:6376–6384. doi: 10.1038/sj.onc.1208789. [DOI] [PubMed] [Google Scholar]
- 19.Min SH, Lau AW, Lee TH, Inuzuka H, Wei S, Huang P, Shaik S, Lee DY, Finn G, Balastik M, Chen CH, Luo M, Tron AE, Decaprio JA, Zhou XZ, Wei W, Lu KP. Negative regulation of the stability and tumor suppressor function of Fbw7 by the Pin1 prolyl isomerase. Mol Cell. 2012;46:771–783. doi: 10.1016/j.molcel.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moberg KH, Mukherjee A, Veraksa A, Artavanis-Tsakonas S, Hariharan IK. The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol. 2004;14:965–974. doi: 10.1016/j.cub.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 21.Olson BL, Hock MB, Ekholm-Reed S, Wohlschlegel JA, Dev KK, Kralli A, Reed SI. SCFCdc4 acts antagonistically to the PGC-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008;22:252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puigserver P, Rhee J, Lin JZ, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 23.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 24.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 25.Putters J, Slotman JA, Gerlach JP, Strous GJ. Specificity, location and function of βTrCP isoforms and their splice variants. Cell Signal. 2011;23:641–647. doi: 10.1016/j.cellsig.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 27.Sancho R, Blake SM, Tendeng C, Clurman BE, Lewis J, Behrens A. Fbw7 Repression by hes5 creates a feedback loop that modulates Notch-mediated intestinal and neural stem cell fate decisions. PLoS Biol. 2013;11:e1001586. doi: 10.1371/journal.pbio.1001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sano M, Tokudome S, Shimizu N, Yoshikawa N, Ogawa C, Shirakawa K, Endo J, Katayama T, Yuasa S, Ieda M, Makino S, Hattori F, Tanaka H, Fukuda K. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor gamma coactivator 1alpha. J Biol Chem. 2007;282:25970–25980. doi: 10.1074/jbc.M703634200. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;48:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 30.Shabek N, Herman-Bachinsky Y, Buchsbaum S, Lewinson O, Haj-Yahya M, Hejjaoui M, Lashuel HA, Sommer T, Brik A, Ciechanover A. The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol Cell. 2012;48:87–97. doi: 10.1016/j.molcel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Sionov RV, Netzer E, Shaulian E. Differential regulation of FBXW7 isoforms by various stress stimuli. Cell Cycle. 2013;12:3547–3554. doi: 10.4161/cc.26591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L, Trausch-Azar JS, Ciechanover A, Schwartz AL. Ubiquitin-proteasome-mediated degradation, intracellular localization, and protein synthesis of MyoD and Id1 during muscle differentiation. J Biol Chem. 2005;280:26448–26456. doi: 10.1074/jbc.M500373200. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Trausch-Azar JS, Muglia LJ, Schwartz AL. Glucocorticoids differentially regulate degradation of MyoD and Id1 by N-terminal ubiquitination to promote muscle protein catabolism. Proc Natl Acad Sci USA. 2008;105:3339–3344. doi: 10.1073/pnas.0800165105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trausch-Azar J, Leone TC, Kelly DP, Schwartz AL. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1{alpha} via the N-terminal pathway. J Biol Chem. 2010;285:40192–40200. doi: 10.1074/jbc.M110.131615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 38.Welcker M, Larimore EA, Swanger J, Bengoechea-Alonso MT, Grim JE, Ericsson J, Zheng N, Clurman BE. Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes Dev. 2013;27:2531–2536. doi: 10.1101/gad.229195.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14:1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- 40.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Huypens P, Adamson AW, Chang JS, Henagan TM, Boudreau A, Lenard NR, Burk D, Klein J, Perwitz N, Shin J, Fasshauer M, Kralli A, Gettys TW. Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1 alpha. J Biol Chem. 2009;284:32813–32826. doi: 10.1074/jbc.M109.037556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.