

Abstract
Glucosinolates are a group of sulfur-containing glycosides found in many plant species, including cruciferous vegetables such as broccoli, cabbage, brussels sprouts, and cauliflower. Accumulating evidence increasingly supports the beneficial effects of dietary glucosinolates on overall health, including as potential anti-cancer agents, because of their role in the prevention of the initiation of carcinogenesis via the induction of cellular defense detoxifying/antioxidant enzymes and their epigenetic mechanisms, including modification of the CpG methylation of cancer-related genes, histone modification regulation and changes in the expression of miRNAs. In this context, the defense mechanism mediated by Nrf2-antioxidative stress and anti-inflammatory signaling pathways can contribute to cellular protection against oxidative stress and reactive metabolites of carcinogens. In this review, we summarize the cancer chemopreventive role of naturally occurring glucosinolate derivatives as inhibitors of carcinogenesis, with particular emphasis on specific molecular targets and epigenetic alterations in in vitro and in vivo human cancer animal models.
Keywords: cancer, chemoprevention, DNA methylation, epigenetics, histone modifications, isothiocyanates, indoles, MicroRNA
Introduction
Cancer chemoprevention is a major cancer preventive strategy that utilizes naturally occurring dietary phytochemicals or therapeutic drugs with relatively low toxicity to inhibit the malignant transformation of initiated cells at the promotion or progression stages [1, 2]. Thus, chemoprevention can involve preventing carcinogens from reaching target sites, undergoing metabolic activation, or subsequently interacting with crucial cellular macromolecules (e.g., DNA, RNA, and proteins) at the initiation stage [3, 4]. Interestingly, prevention and/or protection from chemical carcinogens by phytochemicals present in glucosinolate-containing cruciferous vegetables is of great interest because they may provide a safe and cost-effective strategy for combating cancer [5, 6**]. In this context, numerous epidemiological and pharmacological studies have revealed that the consumption of cruciferous vegetables has substantial potential for human cancer chemoprevention [7].
Isothiocyanates (ITCs) and indoles are biologically active molecules formed from glucosinolate precursors present in a large number of edible species existing in sixteen families of dicotyledonous angiosperms [8]. It has been described more than 200 different naturally-occurring glucosinolates isolated from plants, with a relatively high content in cruciferous vegetables such as broccoli, cabbage, cauliflower, turnip, horseradish, watercress, and brussels sprouts [9, 10]. Some naturally occurring glucosinolates and their breakdown products have received considerable attention as chemopreventive agents, including the ITCs 4-methylsulphinylbutyl isothiocyanate (sulforaphane, SFN) and phenethyl isothiocyanate (PEITC); and the indoles indole-3-carbinol (I3C) and 3.3′-diindolylmethane (DIM) [7, 11**, 12]. The glucosinolates have undergone several human clinical trials for treatment evaluation for various diseases, including cancer (www.clinicaltrials.gov). Thus, the protective role of dietary glucosinolates has been extensively studied using in vitro and in vivo approaches in cancer and cardiovascular and neurological diseases using rodent and human models [13, 14]. These studies have shown that glucosinolates and their derivatives may modulate many relevant processes, such as the induction of cytoprotective enzymes, inhibition of inflammatory processes, modulation of cancer signaling pathways including cellular proliferation, angiogenesis, the epithelial– mesenchymal transition, cancer stem cell self-renewal and suppressing diverse oncogenic signaling pathways, including nuclear factor-κB, hormone receptor, and signal transducer and activator of transcription [15-17]. More recently, increasing evidence has also shown that glucosinolate derivatives have the potential to modulate epigenetic alterations, such as DNA methylation, histone modifications, non-coding microRNAs (miRNAs), regulation of polycomb group proteins and epigenetic cofactor modifiers, which all may contribute to carcinogenesis [18, 15]. Here, we review the cancer chemopreventive role of naturally occurring glucosinolate derivatives as inhibitors of carcinogenesis, particularly emphasizing specific molecular and epigenetic alterations in in vitro and in vivo animal models of human cancers.
Biosynthesis and metabolism of glucosinolates
Glucosinolates are a group of sulfur-containing glycosides found in the plant order Brassicales, which includes the Brassica or Cruciferous vegetables such as broccoli, cabbage, brussels, and cauliflower [19]. These plants have been used for food or medicinal purposes, with the latter partially due to their relatively high content of glucosinolates, which distinguish them from other plant species [20]. Thus far, nearly 200 different glucosinolates with different substituents have been reported, which can be classified into three groups based on the structure of different amino acid precursors: aliphatic glucosinolates, indole glucosinolates, and aromatic glucosinolates [10] (Figure 1A). The content of glucosinolate in plants depends on many factors, such as plant variety, growing conditions, climate and the tissue-specific distribution in a plant [21]. For example, in Brassica vegetables, 0.5-28 μmol aliphatic/aromatic glucosinolates per gram of dry weight and 0.7–8 μmol indole glucosinolates per gram of dry weight have been reported [20]. Glucosinolates are relatively biologically inert glucosides; however, their hydrolysis by myrosinase (b-thioglucosidase) enzymes after chopping vegetables, chewing of raw vegetables or insect attack leads to the conversion of biologically active compounds, such as ITCs, thiocyanates, nitriles and epithionitriles, depending on glucosinolate substrate, pH, temperature, presence of ferrous ions, and level and activity of specific protein factors, including thiocyanate-forming protein (TFP) and epithiospecifier protein (ESP) [21, 19] (Figure 1B). Nevertheless, when the plant myrosinase enzyme is inactivated by heat during the cooking process, the action of myrosinase originated from gastrointestinal tract bacteria allows the formation and absorption of dietary ITCs and indoles in mammals [22].
Figure 1.
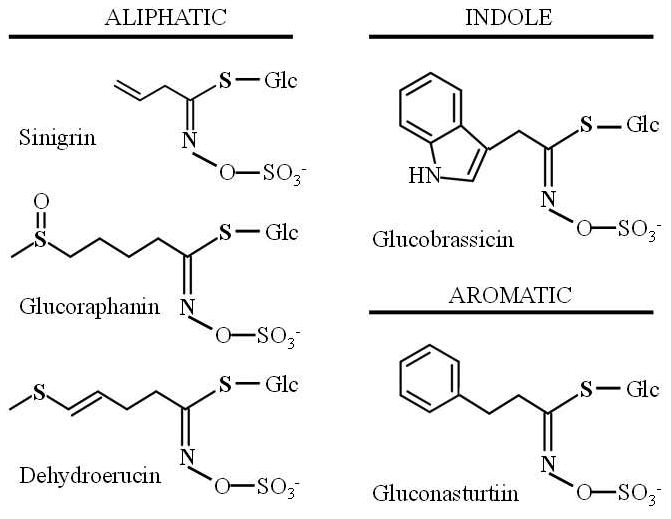
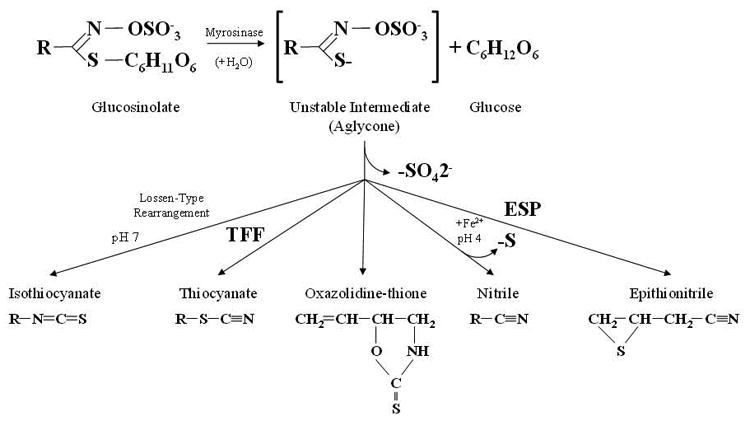
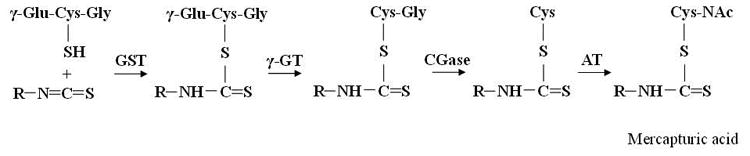
(A) Examples of aliphatic, indole and aromatic glucosinolates found in Brassicaceae vegetables. (B) General model of glucosinolate hydrolysis by myrosinase and specifier proteins indicated as TFP (thiocyanateforming protein) and ESP (epithiospecifier protein). (C) Metabolism of isothiocyanates by the mercapturic acid pathway. Isothiocyanates are conjugated to glutathione by glutathione S-transferases (GSTs) and successively cleaved by γ-glutamyltranspeptidase (γ-GT), cysteinylglycinase (CGase), and N-acetyltransferase (AT) to create N-acetylcysteine conjugates (mercapturic acids).
Several epidemiological and pharmacological studies have demonstrated that dietary glucosinolates and their breakdown products, isothiocyanates, may reduce the risk of carcinogenesis and particular human diseases [14]. Isothiocyanates from dietary vegetables currently investigated for use as chemopreventive agents include SFN from broccoli, cauliflower, and kale, PEITC from watercress, radish and turnip, allyl isothiocyanate (AITC) from cabbage, mustard, and horseradish, benzyl isothiocyanate (BITC) from lepidium cress, 3-methylsulfinylpropyl (iberin) from broccoli, brussels sprouts and cabbage, 4-methylthiobutyl from arugula, and 3-methylthiopropyl from cabbage [13]. Similarly, the indole I3C, which upon exposure to gastric acid undergoes self-condensation to form DIM, is also present in cruciferous vegetables, including broccoli, cabbage, cauliflower, brussels sprouts, collard greens and kale and is used as a chemopreventive agent [23].
After ingestion, isothiocyanates are absorbed from the gastrointestinal tract by passive diffusion into the capillary blood network, reversibly binding to free plasma protein thiols (protein thiocarbamoylation) and crossing the plasma membrane into the cells of tissues [20]. Thus, isothiocyanates are metabolized by the mercapturic acid pathway and initially conjugated to glutathione by glutathione S-transferases (GSTs) and successively cleaved by γ-glutamyltranspeptidase (γ-GT), cysteinylglycinase (CGase), and N-acetyltransferase (AT), creating N-acetylcysteine conjugates (mercapturic acids), which are transported into the kidney and actively secreted in urine for elimination from the body [14] (Figure 1C).
Dietary glucosinolate derivatives and modulation of phase I and phase II biotransformation enzymes
The generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) is an essential metabolic process for maintaining cellular chemical homeostasis; however, their production at low to moderate concentrations is essential for normal physiological processes [24]. Consequently, the oxidative stress produced by high levels of ROS/RNS during normal cell metabolism leads to potential damage, causing oxidative damage to large biomolecules, such as lipids, proteins, and DNA, which may eventually lead to mutations and ultimately, cancer development [25]. Similarly, oxidative stress also has a significant association with many other chronic diseases, such as neurodegenerative diseases (e.g., Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis), cardiovascular disease, diabetes, and inflammatory diseases [26-28]. In this context, the major chemoprevention mechanisms mediated by dietary glucosinolate derivatives include modulation of phase I drug metabolic enzymes (e.g., cytochrome P450 family, CYP), which prevent procarcinogenic molecule formation and the induction of phase II/detoxifying enzymes (e.g., GST; UDPglucuronosyl transferases, and UGT), which catalyze conjugation reactions to inactivate or detoxify exogenous (e.g., carcinogens and other xenobiotics) and endogenous compounds (e.g., sex steroid hormones) related to cancer development [29-31].
Most evidence suggests that dietary glucosinolate derivatives upregulate phase II/detoxifying enzymes through interaction with the cytoplasmic-anchoring protein Kelch-like ECH-associated protein 1 (Keap 1), which represses the transcription factor NF-E2-related factor 2 (Nrf2), a basic-region leucine zipper (bZIP) transcription factor that binds in combination with small Maf proteins to antioxidant response elements (AREs) in the promoter regions of many antioxidant and phase II biotransformation enzymes, including GST, UGT, heme oxygenase-1 (HO-1), NADP(H):quinone oxidoreductase 1 (NQO1), glutamate cysteine ligase (GCL) and gamma glutamylcysteine synthetase (γGCS) (Figure 2) [32, 33]. Thus, the effects of dietary glucosinolate derivatives upregulating phase II enzymes have been extensively reported using different in vivo and in vitro approaches [34, 13]. For example, sulfur-containing dietary glucosinolate derivatives, such as SFN and PEITC, are potent phase II gene inducers, and these inductions are Nrf2-dependent [35, 13]. SFN attenuates Nrf2 degradation by modifying the Keap1-Nrf2 interaction, which results in the translocation of Nrf2. SFN can react with thiols within Keap1 by forming thionoacyl adducts, thereby releasing Nrf2 from Keap1 binding [36]. Similarly, PEITC can induce the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) and subsequently, phosphorylate Nrf2 and induce its nuclear translocation [37, 38]. Indole glucosinolate hydrolysis products, such as I3C and DIM, also induce both phase I drug metabolic and phase II/detoxifying enzymes by direct interaction with aryl-hydrocarbon receptor (AhR) or increasing the binding affinity of AhR to xenobiotic response elements (XREs) in target genes [31]. Upon binding chemical ligands, cytosolic AhR translocates into the nucleus and dimerizes with its nuclear protein partner Ah receptor nuclear translocator (ArnT), and then, the AhR complex binds to specific DNA sequences and activates transcription [32] (Figure 2).
Figure 2.
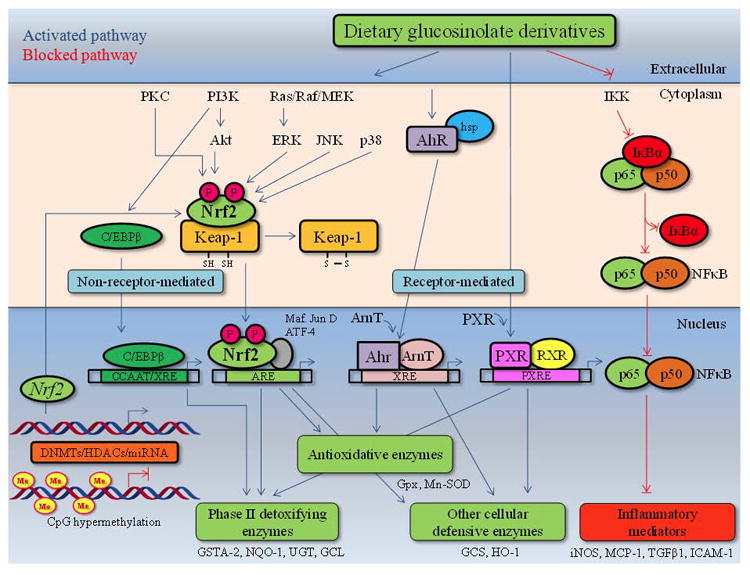
Chemopreventive effects of natural dietary glucosinolate derivatives in cancers induced by Nrf2-mediated antioxidative stress and anti-inflammatory signaling pathways.
The activation of phase II gene expression and enzyme activity by dietary glucosinolate derivatives has been well documented in in vitro and in vivo studies. For example, different studies have reported that SFN significantly induces phase II enzyme expression and activity in human and mouse cells lines, including LNCaP, PC-3, TSU-Pr1, MDA PCa 2a, MDA PCa 2b, MDAMB-231, TRAMP C1, HeLa, HT-29, Caco2, HepG2, Hepa1c1c7 and MCF-7 [39-43]. In contrast, we found that SFN is capable of inhibiting 7,12-dimethylbenz(a)anthracene (DMBA)/12-O-tetradecanoylphorbol-13-acetate (TPA)-induced skin tumorigenesis in C57BL/6 mice mediated by Nrf2 [44]. More recently, we also demonstrated that re-expression of Nrf2 and the subsequent induction of Nrf2 downstream target genes are involved in the cellular protection mediated by SFN during TPA-induced tumor transformation in mouse skin epidermal JB6 (JB6 P+) cells, suggesting the anti-cancer effects of SFN against the TPA-induced neoplastic transformation of mouse skin [45]. Similarly, we also demonstrated that PEITC enhances the expression of various genes, including drug detoxifying enzymes, through the Nrf2 signaling pathway in in vivo and ex vivo studies [46, 47]. Furthermore, PEITC has been demonstrated to stimulate tissue differences in the modulation of rat cytochrome P450 and phase II conjugation systems, showing increased hepatic GST activity, although not in the lung or kidney [48]. Similarly, the expression of the antioxidant enzyme HO-1 has also been shown to be strongly increased by PEITC treatment in PC-3 cells [38]. Interestingly, differences in the basal expression level of Nrf2 and resultant changes in GSH levels in human breast cancer cell lines may be an important determinant of sensitivity to PEITC-induced apoptosis [49]. Indole-containing compounds, such as I3C and DIM, have also been described to possess potent cancer chemopreventive effects, potentially through multi-targets [50], including the induction of endogenous Nrf2, phase II genes (e.g., GSTm2, UGT1A1 and NQO1) and antioxidant genes (e.g., HO-1 and SOD1), as reported in a human liver hepatoma cell line (HepG2-C8) [51].
Dietary glucosinolate derivatives and inflammation modulation
Sustained generation of ROS/RNS has been shown to contribute to the pathological consequences of chronic inflammation, which is believed to be the cause of many human diseases, including cancer [52]. If this crosstalk between inflammation and oxidative stress is prolonged, excessive cellular ROS/RNS will be produced, resulting in genetic changes and/or epigenetic alterations, which lead to the deregulation of oncogenes and tumor suppressor genes [53, 6**]. Cytokines, chemokines, nuclear factor (NF)- κB, nitric oxide synthase-2 (NOS2), cyclooxygenase-2 (COX2), hypoxia inducible factor-1α (HIF1-α), signal transducer and activator of transcription 3 (STAT3), Nrf2 and nuclear factor of activated T cells (NFAT) are key molecular players linking inflammation to cancer [54]. In this context, Nrf2 is a crucial regulator that has been shown to modulate the innate immune response and survival during experimental sepsis using Nrf2-deficient mice and Nrf2-deficient mouse embryonic fibroblasts [55]. Some findings have suggested that there is crosstalk between Nrf2 and inflammation [56]. Interestingly, the Nrf2 pathway has been connected to the inflammatory response in studies using the TRAMP mouse model of prostate carcinogenesis [57]. Similarly, lower induction of phase II antioxidant and detoxification enzymes, such as HO-1, NQO1, UGT1A1, and GSTM1, and higher induction of proinflammatory biomarkers, such as interleukin IL-1β, IL-6, tumor necrosis factor alpha (TNF-α), inducible nitric oxide synthase (iNOS), and COX2, were observed in Nrf2-KO mice [58].
NF-κB is a transcription factor and a key molecular link between inflammation and cancer that regulates several genes whose products inhibit apoptosis and enhance cell cycle progression, angiogenesis and metastasis [59, 52]. Additionally, a considerable number of NF-κB target genes encode mediators of the innate immune response and inflammation, which include cytokines, chemokines, proteases, NOS2 and COX2 [60, 52]. In this context, dietary glucosinolate derivatives have been shown to inhibit NF-κB-mediated processes in vitro and in vivo, playing an important role because NF-κB is involved in the expression of over 500 genes involved in human diseases, including cancer [15, 61]. Thus, glucosinolate derivatives are capable of inhibiting NF-κB regulated pathways triggered by these activators by blocking pro-inflammatory signals at various levels; however, the molecular mechanisms by which these interactions are exerted are complex and poorly understood [15, 61].
Several cellular targets of glucosinolate derivatives have been investigated for modulating the NF-κB signaling pathway. For example, SFN is capable of suppressing the TLR4 signaling cascade by affecting the downstream effectors MyD88, p38 mitogen-activated protein kinase (MAPK) and JNK by interacting with glutathione or other redox regulators, such as thioredoxin or Ref-1, which are indirectly capable of impairing NF-κB DNA binding ability and directly binding the essential thiol groups of p50, affecting NF-κB DNA binding with the potential involvement of Akt regulation [62, 15, 63]. In addition, DIM, PEITC and SFN have been described to repress IKK/IkB phosphorylation and p65 NF-κB nuclear translocation, inhibiting the transcriptional activity of NF-κB and affecting important mediators, such as IL-6, iNOS, TNF-α and COX-2 [64, 15, 65]. Similarly, PEITC is also capable of decreasing the iNOS and COX-2 protein expression levels, leading to reduced expression of both pro-inflammatory mediators, and has also been reported to suppress the phosphorylation of interferon regulatory factor 3 (IRF3) induced by stimulation of the Toll-like receptor that decreases the activation of type I interferons (IFNs) and IFN-inducible genes [13, 66].
Recent evidence suggests an important crosstalk between NF-κB and Nrf2 signaling, and the strong mechanism by which glucosinolate derivatives affect NF-κB may be partially mediated by their ability to activate the Nrf2-ARE signaling cascade (Figure 2). For example, Nrf2 knockout mice subjected to pro-inflammatory stimuli simultaneously demonstrated decreased levels of anti-oxidant/phase 2 enzymes and upregulation of NF-κB pro-inflammatory mediators, such as COX-2, iNOS, IL-1, IL-6, cPLA2 and TNF-α [67-69, 58]. Modulation of Nrf2 and NF-κB crosstalk is not well characterized, but it appears to occur through a common MAPK network because common regulatory sequences in the transactivation domains of Nrf2 and NF-κB have been described [15]. In addition, NF-κB can antagonize Nrf2 activity at the transcriptional level by interacting with the co-activator CREB-binding protein (CBP), which is required for translocation, and concomitant recruitment of histone deacetylase (HDAC). In contrast, ARE-mediated gene activation by Nrf2 can inactivate NF-κB by different mechanisms. For example, upregulation of HO-1, one of the key target genes of the Nrf2 signaling pathway, has been suggested to inhibit NF-κB nuclear translocation [70]. Additionally, GSH/Grx-1-dependent S-glutathionylation of p65 NF-κB produces NF-κB inactivation [71]. Accordingly, C57BL/6 mice pre-treated with SFN in the presence of dextran sodium sulfate (DSS) demonstrated significantly reduced expression of inflammatory markers, such as IL-6 and interferon γ, with increased expression of Nrf2-dependent genes [72]. Similarly, SFN treatment of WT but not Nrf2 KO mice restored the number of sunburn cells to their basal level post-UVB irradiation, demonstrating decreased inflammatory biomarker activity in SFN-treated WT compared with Nrf2 KO mice, revealing a protective role for Nrf2 when activated by SFN against UVB-induced skin inflammation [73]. Moreover, SFN has also been reported to induce significant downregulation of pro-inflammatory microRNA-155 by epigenetic mechanisms that together with the regulation of other target NFκB coactivators, such as CCAAT-enhancer binding proteins, cAMP response element binding protein, and activator protein-1 (AP-1), open new frontiers in the complex activities exerted by glucosinolate derivatives [74, 13].
Dietary glucosinolate derivatives and epigenetic mechanisms modulating carcinogenesis, inflammation and reactive oxygen species
Epigenetic regulation comprises DNA modifications without changes in sequence that result in changes in gene expression or phenotype [31]. Recently, a large amount of evidence has demonstrated that epigenetic alterations, such as DNA methylation, histone modifications and non-coding miRNAs, consistently contribute to carcinogenesis, and constituents in the diet, including dietary glucosinolate derivatives, have the potential to alter a number of these epigenetic events [18, 15, 26]. Although most research on the cellular effects of dietary glucosinolate derivatives has primarily focused on detoxifying enzyme effects, increasing evidence has demonstrated the chemopreventive effects of dietary glucosinolate derivatives on the regulation of silenced genes in cancer.
DNA methylation was the first epigenetic alteration to be observed in cancer cells, and it represents the most common molecular alteration in the origin of many cancers [75, 76]. DNA methylation occurs at the 5′ position of cytosine residues within CpG dinucleotides through addition of a methyl group by DNA methyltransferases (DNMTs), which include DNMT1, DNMT3A and DNMT3B, leading to transcriptional silencing of tumor suppressors and other genes with important biological functions. Conversely, global hypomethylation causes genome instability and inappropriate activation of oncogenes and transposable elements [77, 78, 26]. In this context, dietary glucosinolate derivatives, such as SFN, PEITC and DIM, have been shown to inhibit the carcinogenic process, enhance xenobiotic metabolism, induce cell cycle arrest and apoptosis and affect the cancer epigenome in various human cancers and cancer mouse models, demonstrating relevance as chemopreventive agents [79, 13, 23]. In different studies, the treatment of human and mouse cells with different dietary glucosinolate derivatives has resulted in the downregulation of DNMT activity, with concomitant promoter demethylation and re-expression of genes such as GSTP1 (glutathione S-transferase pi 1), Nrf2, hTERT (telomerase reverse transcriptase), TGFBR1 (transforming growth factor, beta receptor I) and CYR61 (Cysteine-rich angiogenic inducer 61) (Table 1) [40, 45, 80, 81, 23, 82, 83, 11**].
Table 1. Examples of the effect of dietary glucosinolate derivatives on DNA methylation, histone modifications and miRNAs mechanisms.
| Epigenetic mechanism | Dietary agent | Molecular mechanism | Validated target(s) | In vitro model | In vivo model | Concentration | Treatment duration | References | |
|---|---|---|---|---|---|---|---|---|---|
| DNA methylation | Sulforaphane | ↓methylation in promoter region, ↓ DNMT1, ↓ DNMT3a | Nrf2 | TRAMPC1 mouse prostate cells | 2.5 μM | 5 days | [40] | ||
| Sulforaphane | ↓methylation in promoter region, ↓ DNMT1, ↓ DNMT3a, ↓ DNMT3b | Nrf2 | JB6 P+ mouse skin cancer cells | 2.5 μM | 5 days | [45] | |||
| Sulforaphane | ↓ DNMT1 expression | Caco-2 human colon cancer cells | 50 μM | 5 days | [80] | ||||
| Sulforaphane | ↓methylation in promoter region, ↓ DNMT1 and ↓DNMT3a | hTERT | MCF-7 and MDA-MB-231 human breast cancer cells | 10 μM | 6 days | [23, 81] | |||
| Phenethyl isothiocyanate | ↓methylation in promoter region | GSTP1 | LNCap (androgen-dependent/independe nt) human prostate cancer cells | 2.0 μM | 5 days | [82] | |||
| 3.3′-diindolylmethane | ↓methylation in promoter region, ↓ DNMT1, ↓ DNMT3a, ↓ DNMT3b | Nrf2 | TRAMPC1 mouse prostate cells | TRAMP mice prostate tumors | 5 μM / 1% DIM diet | 5 days / 24 weeks | [83] | ||
| 3.3′-diindolylmethane (formulation with higher bioavailability) | miR-34a | LNCaP and C4-2B human prostate cancer cells | 6 μM | 5 days | [100] | ||||
| 3.3′-diindolylmethane | ↓methylation in promoter region, ↓ DNMT1, ↓ DNMT3b | TGFBR1, CYR61 | LNCap human prostate cancer cells | 15 μM | 48 hours | [11] | |||
|
| |||||||||
| Histone modifications | Sulforaphane | ↓HDAC1, ↓HDAC4, ↓HDAC5, ↓HDAC7, ↑H3Ac | Nrf2 | Mouse prostate TRAMPC1 cells | 2.5 μM | 5 days | [40] | ||
| Sulforaphane | ↓HDAC1, ↓HDAC2, ↓HDAC3, ↓HDAC4 | Nrf2 | Mouse skin JB6 P+ cells | 2.5 μM | 5 days | [45] | |||
| Sulforaphane | ↓ HDAC activity, ↑ H3Ac, ↑H4Ac | p21, bax | Apcmin mice colon tumors | Single oral dose of 10 μM / ∼6 μM | 6 hours / 10 weeks | [87] | |||
| Sulforaphane | ↓ HDAC activity, ↑ H3Ac, ↑H4Ac | p21 | Human colorectal HCT116 cells | 15 μM | 47 hours | [119] | |||
| Sulforaphane | ↓ HDAC activity, ↑ H3Ac, ↑H4Ac | p21, bax | Human prostate BPH-1, LnCaP and PC-3 cells | 15 μM | 48 hours | [120] | |||
| Sulforaphane | ↓ HDAC activity, ↓HDAC1 | Human embryonic kidney 293 cells | 15 μM | 47 hours | [119] | ||||
| Sulforaphane | ↓ HDAC activity, ↑ global histone acetylation | Human PC-3 prostate cancer xenografts in nude mice | 7.5 μmol per animal | 21 days | [90] | ||||
| Sulforaphane | ↓ HDAC activity, ↑H3Ac, ↑H4Ac | Human peripheral blood mononuclear cells (PBMC) | 68 g BroccoSprouts | 3 and 6 hours following consumption | [90] | ||||
| Sulforaphane | ↓ HDAC activity | Human breast MDA-MB-231, MDA-MB-468, MCF-7, and T47D cell lines | 25 μM | 48 hours | [121] | ||||
| Sulforaphane | ↓ HDAC activity, ↑H3Ac, ↑H4Ac, ↑H3K9Ac, ↓H3K9, ↓H3K27, ↑RBP2 | hTERT | MCF-7 and MDA-MB-231 human breast cancer cells | 10 μM | 6 days | [23, 81] | |||
| Sulforaphane | ↓H3K27 | Human SCC-13 skin cancer cells | 20 μM | 48 hours | [91] | ||||
| Phenethyl isothiocyanate | ↓HDAC expression, ↑H3Ac, ↑H3K4, ↓H3K9, ↓ HDAC1 | Human prostate LNCap cells | 0.1-20 μM | 36 hours | [82] | ||||
| Phenethyl isothiocyanate | ↑H3Ac, ↑H3K4, ↓H3K9 | p21 | Human prostate LNCap cells | 10 μM | 30 hours | [88] | |||
| Phenylhexyl isothiocyanate | ↓HDAC activity, ↑H3Ac, ↑H4Ac, ↑H3K14, ↑H3K4, ↓H3K9 | p21 | Human leukemia HL-60 cells | 40 μM | 7 hours | [122] | |||
| 3.3′-diindolylmethane | ↓HDAC1, ↓HDAC2, ↓HDAC3, ↓HDAC4 | Human colon HT-29 cells | 60 μM | 24 hours | [123] | ||||
| 3.3′-diindolylmethane | ↓HDAC1, ↓HDAC2, ↓HDAC3, ↓HDAC4 | p21, p27 | Human colon HT-29 and SW620 cells | Human HT-29 colon cancer xenografts in nude mice | 60 μM / 300 mg/kg/day | 24 hours / 2 days | [93] | ||
| 3.3′-diindolylmethane | ↑ histone acetyl transferase p300, ↑H4Ac | COX-2 | Human breast MCF-7 cells | 10 μM | 15 minutes | [89] | |||
| 3.3′-diindolylmethane | ↓HDAC1, ↓HDAC2, ↓HDAC3, ↓HDAC4, ↓HDAC8 | Mouse prostate TRAMPC1 cells | 5 μM | 5 days | [83] | ||||
| 3.3′-diindolylmethane | ↑H3K4 | TGFBR1, CYR61 | LNCap human prostate cancer cells | 15 μM | 48 hours | [11] | |||
|
| |||||||||
| microRNAs | Sulforaphane | miR-140, miR-29a, and miR-21 | MCF10DCIS, MCF-7, MDA-MB-231 breast cancer cells | 10 μM | 7 days | [103] | |||
| Sulforaphane | miR-let7-a | K-ras | BxPc-3, MIA-PaCa2 pancreatic cancer cells | 10 μM | 72 hours | [102] | |||
| 3.3′-diindolylmethane | miR-200b, miR-200c, let-7b, let-7c, let-7d, and let-7e | ZEB1 | MiaPaCa-2, Panc-1, and Aspc-1 pancreatic cancer cells | 25 μM | 48 hours | [97] | |||
| 3.3′-diindolylmethane | miR-146a | EGFR, IRAK-1, and MTA-2 | Colo357 and Panc-1 pancreatic cancer cells | 25 μM | 48 hours | [124] | |||
| 3.3′-diindolylmethane | miR-21 | Cdc25A | MCF-7 and MDA-MB-468 breast cancer cells | Human MCF-7 breast cancer xenografts in nude mice | 30-60 μM / 5 mg/kg | 24-96 hours / 7 weeks after cell injection | [99] | ||
| 3.3′-diindolylmethane (formulation with higher bioavailability) | let-7a, let-7b, let-7c, and let-7d | EZH2 | LNCaP, C4-2B and PC3 human prostate cancer cells | Human prostate samples from patients with stage I or stage II Pca undergoingradical prostatectomy | 25 μM / 225 mg orally twice daily x 14-72 days (based on scheduling of surgery) | 24 hours / 2–4 weeks prior to surgery | [101] | ||
| Indole-3-carbinol | miR-21, miR-31, miR-130a, and miR-146 | Human lung carcinoma A549 cells | Vinyl carbamate-induced female A/J mice lung tumors | 100 - 150 μM | 24 hours / 15 weeks | [98] | |||
| Indole-3-carbinol | miR-10a, miR-26a, miR-34b, miR-125a-prec | Sprague-Dawley rats lung cancer samples induced by environmen tal cigarette smoke (ECS) | 2,500 mg/kg diet | 28 days | [12] | ||||
| Phenethyl isothiocyanate | let-7a, let-7c, miR-26b, miR-99b, miR-123-prec, miR-125b, miR-146-prec, miR-192, miR-222-prec | Sprague-Dawley rats lung cancer samples induced by environmen tal cigarette smoke (ECS) | 500 mg/kg diet | 28 days | [12] | ||||
Interestingly, Wong et al. described the genome-wide effects of SFN and DIM on promoter methylation in normal prostate epithelial cells and prostate cancer cells [11**]. Accordingly, both SFN and DIM treatment decreased the expression of DNMTs in normal prostate epithelial cells (PrEC) and androgen-dependent (LnCAP) and androgen-independent (PC3) prostate cancer cells. Specifically, SFN and DIM altered promoter methylation in different sets of genes in normal prostate epithelial cells and prostate cancer cells; however, they shared similar gene targets in a single cell line, reversing many of the cancer-associated methylation alterations, including aberrantly methylated genes that are dysregulated during cancer progression (e.g., cell migration, cell adhesion, cell-cell signaling, and transcriptional regulation).
Histone modifications have been broadly recognized as critically important triggers of gene silencing via post-translational modifications of histones at amino-terminal tails [26]. For example, the open chromatin state and gene activation is mediated by histone acetyltransferases (HATs), which transfer acetyl groups to the ε-amino group of lysine residues in histone tails, whereas the condensed chromatin state and the respective gene silencing is commonly regulated by HDAC enzymes, which remove histone acetyl groups by catalyzing their transfer to coenzyme A (CoA) [84, 26]. Similarly, the histone methylation of lysine and arginine residues mediated by histone methyltransferases (HMTs) and demethylases (HDMs) has also been described as a mechanism activating or repressing the gene expression in various forms of cancer [85, 15]. For example, methylation of H3K4, H3K36 and H3K79 has been associated with transcriptionally active chromatin, whereas methylation of H3K9, H3K27 and H4K20 has been associated with transcriptionally repressed chromatin, constituting two of the important silencing mechanisms in mammalian cells [86, 26].
Thus far, several studies have demonstrated the effects of dietary glucosinolate derivatives on histone modification mechanisms in in vitro and in vivo animal cancer models [18, 6**]. Thus, the HDAC inhibitory activity of different dietary glucosinolate derivatives has been widely reported to alter the tumorigenesis processes, with a concomitant increase in the expression of tumor suppressor, pro-apoptotic, anti-oxidant and anti-inflammatory genes [45, 87-89]. Dietary glucosinolate derivatives, such as SFN, PEITC and DIM, have been specifically associated with HDAC inhibitory activity in the peripheral blood mononuclear cells of human patients who consumed broccoli sprouts, different cancer cell lines (kidney, colon, prostate, leukemia and breast) and in vivo and in vitro cancer mouse models, such as APCmin/+ and TRAMPC1 mice and the JB6P+ skin cell line (Table 1) [45, 40, 87, 90]. Specifically, SFN and PEITC treatments in human breast and prostate cancer cell lines have been shown to increase H3Ac, H3K9Ac and H4Ac acetylation and H3K4 methylation and decrease the methylation of H3K9 and H3K27 [23, 82, 88]. Similarly, DIM treatments in the androgen-dependent LnCAP prostate cancer cell line have also been demonstrated to increase H3K4me3 in the promoter regions of the TGFBR1 and CYR61 genes, as revealed by ChIP assays [11**]. Interestingly, analysis of the impact of SFN on the level and function of polycomb group (PcG) proteins in SCC-13 skin cancer cells revealed that SFN treatment causes a concentration-dependent reduction in PcG protein (Bmi-1, Ezh2) expression and reduced histone H3 lysine 27 trimethylation, which is correlated with the accumulation of cells in G2/M phase, reduced levels of cyclin B1, cyclin A, cyclin dependent kinases 1 and 2, and increased p21Cip1 expression [91].
These results were also observed in other skin-derived immortalized cells and transformed cell lines. In contrast, DIM has been reported to significantly decrease HDAC2 protein expression but not HDAC1, HDAC3, HDAC4, HDAC6 or HDAC8 protein expression in androgen-insensitive PC-3 and androgen-sensitive LNCaP prostate cancer cell lines [92]. Interestingly, the same study was observed that I3C treatment slightly inhibits HDAC activity in LNCaP cells with no HDAC inhibition in PC-3 cells. Similarly, DIM has been shown to suppress the expression of the HDAC2 and HDAC3 proteins in TRAMP-C1 cells, with a concomitant increase in apoptosis, decrease in cell proliferation and enhanced Nrf2 and Nrf2-target gene NQO1 expression in prostate tissues [83]. DIM can selectively induce the proteasome-mediated degradation of class I histone deacetylases (HDAC1, HDAC2, HDAC3, and HDAC8) without affecting class II HDAC proteins in human colon cancer cells in vitro and in vivo in tumor xenografts [93]. Thus, the HDAC depletion was associated with DNA damage induction, which triggered apoptosis.
miRNAs have become an important component of epigenetic gene regulation in mammals [6**]. Typically, miRNAs are a class of endogenous small non-coding RNA molecules 20-25 nucleotides in length cleaved from approximately 70-100 nucleotide hairpin pre-miRNA precursors that regulate gene expression by inhibiting translation and/or triggering the degradation of target messenger RNAs (mRNAs) [94, 95]. Different studies in cancer have shown that miRNAs interact with genes in many different cellular pathways, displaying a differential gene expression profile between normal and tumor tissues and between tumor types [78, 96]. For example, the overexpressed miR-17-92 oncogenic cluster may function as an oncogene and promote cancer development by negatively regulating tumor suppressor genes and/or genes that control differentiation or apoptosis, such as E2F1 (a cell cycle and apoptosis regulator), BIM (a pro-apoptotic gene that counteracts BCL2) and PTEN (a negative regulator of the oncogenic pro-survival PI3K/AKT signaling pathway) [96]. In contrast, downregulation of the let-7 and miR-15/miR-16 miRNAs, which target the RAS and BCL2 oncogenes, respectively, has been previously described [78]. For example, altered expression of a number of miRNA molecules in the lung following the exposure of rats to environmental cigarette smoke (ECS) can be attenuated by dietary agents, such as PEITC and I3C [12]. Thus, the ECS-downregulated miRNAs affected by PEITC have a variety of functions, such as the stress response, TGF-β expression, NF-κB activation, Ras activation, cell proliferation, apoptosis, and angiogenesis. In addition, I3C-regulated miRNAs are involved in p53 function, TGF-β expression, Erbb2 activation, and angiogenesis (Table 1). Similarly, DIM treatment has been reported to cause alterations in the expression of several miRNAs, including miR200 and the let-7 family, which were increased in gemcitabine-resistant pancreatic cancer cells with a concomitant reversal of the mesenchymal phenotype to an epithelial phenotype [97].
Interestingly, I3C is also capable of reducing the effects of vinyl carbamate (a potent carcinogen causing lung tumors) in the lung by modulating the expression of several oncomiRs [98]. Other studies have shown that treatment of breast cancer cell lines with DIM increases the expression of miR-21, exhibiting dose-dependent inhibition of cell proliferation and the development of breast tumors in an in vivo MCF-7 xenograft model [99]. Similarly, in human prostate cancer, interventions including high bioavailability formulations of DIM for 2 to 4 weeks in patients prior to radical prostatectomy demonstrated an association between the re-expression of miR-34a and decreased androgen receptor (AR) signaling, prostate specific antigen (PSA) and Notch-1 [100]. Furthermore, in the same patient group, DIM supplementation increased the expression of let family miRNAs and decreased the expression of the histone methyltransferase EZH2 [101]. More recently, SFN has been shown to mediate the induction of miR-let-7a expression, which in turn inhibits K-ras expression and cancer stem cell (CSC) characteristics during pancreatic ductal adenocarcinoma (PDA) progression [102]. Moreover, SFN can also modulate the expression of several miRNAs, including miR-140, miR-29a, and miR-21, in basal-like ductal carcinoma in situ (DCIS) stem-like cells, inducing significant changes in the exosomal secretion of miRNAs more closely resembling that of non-stem cancer cells, representing a promising chemopreventive strategy in the early stages of non-invasive breast cancer [103]. Taken together, the current studies using both in vitro and in vivo approaches suggest that dietary glucosinolate derivatives may function as miRNA regulators in a number of cancer types and target systems.
Dietary glucosinolate derivatives: in vivo studies
Naturally occurring glucosinolates and their breakdown products have been extensively used as chemopreventive agents in in vivo studies, including chemically induced rodent cancer models and oncogenedriven cancer development in transgenic mice [104]. Key studies documenting cancer chemoprevention by glucosinolates in chemically induced rodent cancer and transgenic mouse models are summarized in Table 2. For example, we have shown that topical application of SFN decreases the incidence of DMBA/TPA-induced skin tumors in Nrf2 (+/+) wild type (Nrf2 WT) mice but not in sulforaphane-treated Nrf2 KO mice, demonstrating that the chemopreventive effects of SFN in DMBA/TPA-induced skin tumors is mediated by Nrf2 [44]. Similarly, inhibition of skin tumorigenesis was also observed using SFN in chemically induced skin cancer in CD-1 mice during the promotion stage [105]. In contrast, we have also reported that SFN treatments in ApcMin/+ mice lead the regulation of different sets of genes involved in apoptosis, cell growth/maintenance and inflammation in small intestinal polyps, as revealed by gene expression profile analysis using Affymetrix microarrays [106].
Table 2. Chemopreventive effect of dietary glucosinolate derivatives in rodents in vivo models.
| Compound | Animal model | Experimental protocol | Effect | Reference | |
|---|---|---|---|---|---|
| Sulforaphane | DMBA/TPA - CD-1 mice | Topical application of 1, 5 or 10 μmol/mouse in anti-promotion protocol (SFN from 1 week after carcinogen until the end of the study) or a combined anti-initiation, anti-promotion protocol (SFN 7 days prior to carcinogen until the end of the study) | Inhibition of skin tumorigenesis | [105] | |
| DMBA/TPA-induced skin tumorigenesis in C57BL/6 mice | Topical application of 100 nmol of sulforaphane once a day for 14 days prior to DMBA/TPA applications | Decreasing the incidence of skin tumor | [44] | ||
| C57BL/6J wild type and C57BL/6J/Nrf2(-/-) knock-out mice | Topical application of 100 nmol of SFN in 100 μL acetone for 4 and 5 d and irradiated with a single dose of UVB (300 mJ/cm2) during 10 min | Decreasing of inflammation and restored sunburn | [73] | ||
| Apcmin/+ mouse model of gastrointestinal cancer | Dietary feeding of 600 ppm SFN/day during 1 to 5 days | Regulation of different set of genes involving apoptosis, cell growth/maintenance and inflammation in the small intestinal polyps | [106] | ||
| Apcmin/+ mouse model of gastrointestinal cancer | Dietary feeding of 300 and 600 ppm of SFN for 3 weeks | Suppression of polyps in the small intestine with higher apoptotic and lower proliferative indices | [108] | ||
| Apcmin/+ mouse model of gastrointestinal cancer | Dietary feeding of 300 ppm SFN during 10 weeks | Reduction of colon tumor numbers, decreasing levels of prostaglandin E2 or leukotriene B4 in intestinal polyps and inhibition of cell survival and growth-related signaling pathways | [107] | ||
| Apcmin/+ mouse model of gastrointestinal cancer | Dietary feeding of ∼6 μmol SFN/day for 10 weeks | Suppression of polyps formation | [87] | ||
| Apcmin/+ mouse model of gastrointestinal cancer | Dietary feeding of 300 or 600 ppm of SFN for 3 weeks | Suppression of polyps in the small intestine | [108] | ||
| Transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer | Oral gavage of 6 μmol SFN thrice a week for 17 to 19 weeks | Inhibition of prostate intraepithelial neoplasia and pulmonary metastasis | [109] | ||
| Transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer | Feeding with 240 mg broccoli sprouts/mouse/day for 16 weeks | Inhibition of prostate tumor growth | [110] | ||
| C57BL/6J and C57BL/6J/Nrf2(-/-) knock-out mice | Oral gavage of 90 mg/kg SFN (0.2 ml) for 3 and 12 hours | Increasing the expression of Nrf2-dependent detoxification phase I, II drug metabolizing enzymes and phase III transporters genes | [111] | ||
| Phenethyl isothiocyanate | AOM/DSS - C57BL/6 mice colon cancer model | Dietary feeding of 0.05% PEITC and 1% DBM during 20 weeks | Inhibition of colon tumor multiplicity | [112] | |
| Apcmin/+ mouse model of gastrointestinal cancer | Dietary administration of 0.05% PEITC for 3 weeks | Inhibition of intestinal polyp development and reduced intestinal tumor size | [125] | ||
| Polyoma middle-T antigen (PyMT) transgenic mouse model of breast cancer | Dietary feeding of 8 mmol PEITC/kg for 4 to 16 weeks | Reducing size of mammary cancer lesions | [126] | ||
| Transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer | Dietary feeding of 0.05% PEITC for 10 and 16 weeks | Inhibition of prostate tumor incidence | [113] | ||
| Transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer | Dietary administration of 3 mmol PEITC/kg for 19 weeks | Inhibition of incidence and burden of poorly-differentiated prostate tumor | [114] | ||
| Indole-3-carbinol | Transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer | Dietary administration of 1% I3C for 8 and 12 weeks | Inhibition of incidence of palpable tumor and increased expression of Nrf2, NQO-1, as well as cell cycle and apoptosis related biomarkers | [115] | |
| 3.3′-diindolylmethane | Transgenic adenocarcinoma of mouse prostate (TRAMP) model of prostate cancer | Dietary administration of 1% I3C for 12 and 16 weeks | Decreasing of incidence of tumorigenesis and metastasis; increasing of apoptosis, decreasing of cell proliferation and enhanced Nrf2 and Nrf2-target gene NQO1 expression in prostate tissues | [83] | |
Abbreviations: SFN, sulforaphane; PEITC, phenethyl isothiocyanate; I3C, indole-3-carbinol; DIM, 3.3′-diindolylmethane; DMBA, 7,12-dimethylbenz[a]anthracene; AOM, azoxymethane; DSS, dextran sodium sulfate; TPA, 12-O-tetradecanoylphorbol 13-acetate.
These results are in agreement with other studies from our laboratory in which SFN treatments reduced the number of polyps by inhibiting phosphorylated c-Jun N-terminal kinase (p-JNK), phosphorylated extracellular signal-regulated kinase (p-ERK), phosphorylated-Akt (p-Akt), COX-2, and cyclin D1 protein expression in ApcMin/+ mice [107, 108]. In transgenic adenocarcinoma of mouse prostate (TRAMP), an oral gavage of 6 μmol SFN three times per week for 17 to 19 weeks inhibited prostate intraepithelial neoplasia and pulmonary metastasis by reducing cell proliferation and augmenting NK cell lytic activity [109]. In this context, we have reported that TRAMP mice fed with 240 mg of broccoli sprouts/mouse/day for 16 weeks exhibit significant retardation of prostate tumor growth, with a concomitant increase in the expression level of the Nrf2, HO-1, cleaved-caspase-3, cleaved-PARP and Bax proteins and a decrease in the Keap1 and Bcl-XL proteins. Furthermore, the phosphorylation and/or expression level of Akt and its downstream kinase and target proteins (e.g., mTOR, 4E-BP1 and cyclin D1) were also reduced [110]. These data correlate with our previous findings, in which oral administration of SFN was capable of inducing Nrf2-dependent detoxification phase I and II drug metabolizing enzymes and phase III transporters in livers of C57BL/6J and C57BL/6J/Nrf2(-/-) mice using the Affymetrix 39K oligonucleotide microarray [111].
In contrast, ApcMin/+ mice fed with a diet supplemented with 0.05% PEITC for 3 weeks developed significantly less and smaller polyps than those fed with a basal diet (47). We have also reported that PEITC in an azoxymethane (AOM)-initiated and DSS-promoted colon cancer mouse model is capable of lowering tumor incidence and colon tumor multiplicities with smaller polyps compared with mice fed on a basal diet [112]. Thus, in this study, PEITC was associated with an increase in apoptosis (increased cleaved caspase-3 and caspase-7) and cell cycle arrest (increased p21). In the polyoma middle-T antigen transgenic breast cancer mouse model, dietary feeding with an 8 mmol PEITC/kg diet resulted in smaller mammary cancer lesions with a progressive loss of ERα and FOXA1 but persistence of GATA-3 expression (48). In contrast, in TRAMP mice, a diet supplemented with 0.05% PEITC for periods of 10 and 16 weeks decreased the incidence of prostate tumors and was associated with downregulation of the Akt signaling pathway, ultimately decreasing cell proliferation and retarding prostate tumor formation [113]. Similarly, in TRAMP mice, the administration of a 3 mmol PEITC/kg diet suppressed prostate cancer progression by inducing autophagic cell death and overexpressing E-cadherin. Interestingly, PEITC treatment was not associated with a decrease in cellular proliferation, apoptosis induction, or neoangiogenesis inhibition [114]. In studying the chemopreventive efficacy of I3C in TRAMP mice, we observed that I3C suppressed the incidence of palpable tumors and reduced the genitourinary weight [115]. In addition, in this study, I3C induced the expression of Nrf2 and NQO-1 and cell cycle- and apoptosis-related biomarkers in prostate tissue. More recently, the expression of Nrf2 was found to be controlled by epigenetic alterations, such as DNA methylation and histone modifications, and dietary phytochemicals, such as DIM, could decrease the incidence of tumorigenesis and metastasis and increase apoptosis, decrease cell proliferation and enhance the expression of Nrf2 and the Nrf2-target gene NQO1 in prostate tissues [83].
Although there has been extensive research on dietary phytochemicals contributing to the overall understanding of glucosinolate derivatives in terms of their chemical and biological functions and beneficial effects in human health, clinical studies of human participants on the biological effects of dietary glucosinolate are lacking and limited to determining the effects of raw cruciferous vegetables or their extracts under some biological parameters [104, 116]. For example, the inhibitory effects of watercress on the oxidative metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the peripheral blood cells of participants have been described [117]. Similarly, a randomized and placebo-controlled trial utilizing a beverage infused with broccoli sprouts exhibited an inverse association between the excretion of dithiocarbamates and urinary aflatoxin-DNA adducts [118]. Similarly, the consumption of broccoli sprouts decreased histone deacetylase activity in peripheral blood mononuclear cells in humans subjects [90].
Thus far, thirty-one clinical studies have been registered using SFN (www.clinicaltrials.gov; accessed Oct. 22, 2014). Of these studies, ten have been completed and reported data from patients treated with prostate and breast cancer, cardiovascular disease, immune diseases and autism. Similarly, PEITC, which has had fewer registered studies, comprises one of four studies completed for preventing lung cancer in individuals who smoke. Four studies of seven registered for 3IC treatments have been completed for patients with prostate and breast cancer and a specific study on the prevention of cancer in healthy participants. Finally, four studies of ten registered for DIM for patients treated with prostate and cervical cancer as well as specific studies of preventing cancer in healthy participants have been completed. In summary, these findings suggest that dietary glucosinolate derivatives could be extensively utilized in further prospective epidemiological and chemopreventive studies.
Conclusions and future perspectives
Naturally occurring glucosinolates have been extensively used in in vitro, in vivo, preclinical and clinical studies, supporting the idea that dietary glucosinolates and their derivatives have potential beneficial effects for cancer prevention. In extensive mechanistic studies, robust chemopreventive effects have been observed by glucosinolate derivatives, such as SFN, PEITC, 3IC and DIM, demonstrating that they can modulate oxidative stress and inflammatory damage caused by exposure to various toxicants, such as environmental pollutants, carcinogens, dietary mutagens, and solar radiation, which can result in genetic mutations and molecular alterations that cause the initiation of carcinogenesis in normal cells. In contrast, accumulating evidence has shown that cancer initiation and progression are driven not only by acquired genetic alterations or mutations but also epigenetic disruption of gene expression. Epigenetic alterations and modifications through dietary glucosinolate derivatives can largely restore the expression of many tumor suppressor genes. Although in vitro approaches have greatly contributed to understanding the regulation of the molecular pathways involved in different cancers, including the epigenetic network exerted by glucosinolate derivatives, in vivo data are lacking for most of these dietary compounds. Still, the health effects of dietary glucosinolates in humans are considered promising; however, there are several challenges and limitations to better understanding the molecular mechanisms underlying the chemopreventive effects of these dietary compounds, such as the safety profile of dosage regimens and potential interactions between different glucosinolates and other constituents in the diet. Notably, emerging technologies and research tools, such as RNA interference, microarrays, proteomics and genome-wide DNA methylation/histone modifications/miRNA profiling, have been addressing novel mechanisms through which glucosinolate derivatives may prevent cancer.
Acknowledgments
We thank all the members in Dr. Ah-Ng Tony Kong's lab for their helpful discussion and preparation of this manuscript.
Grant Support: This work was supported in part by institutional funds and by R01-CA118947, R01-CA152826, from the National Cancer Institute (NCI), R01AT007065 from the National Center for Complementary and Alternative Medicines (NCCAM) and the Office of Dietary Supplements (ODS).
Abbreviations
- AP-1
Activator protein-1
- AITC
Allyl isothiocyanate
- AR
Androgen Receptor
- AOM
Azoxymethane
- AREs
Antioxidant response elements
- AhR
Aryl-hydrocarbon receptor
- ArnT
Ah receptor nuclear translocator
- bZIP
Basic-region leucine zipper
- BITC
Benzyl isothiocyanate
- CSC
Cancer stem cells
- JNK
c-Jun N-terminal kinase
- CoA
Coenzyme A
- CBP
CREB-binding protein
- COX2
Cyclooxygenase-2
- CYR61
Cysteine-rich angiogenic inducer 61
- CGase
Cysteinylglycinase
- CYP
Cytochrome P450 family
- HDMs
Demethylases
- DSS
Dextran sodium sulphate
- DNMTs
DNA methyltransferases
- DCIS
Ductal carcinoma In Situ
- ECS
Environmental cigarette smoke
- ESP
Epithiospecifier protein
- ERK
Extracellular signalregulated kinase
- γGCS
Gamma glutamylcysteine synthetase
- GCL
Glutamate cysteine ligase
- GSTs
Glutathione S-transferases
- GSTP1
Glutathione S-transferase pi 1
- HO-1
Heme oxygenase-1
- HATs
Histone acetyltransferases
- HDAC
Histone deacetylase
- HMTs
Histone methyltransferases
- hTERT
telomerase reverse transcriptase
- HIF1- α
Hypoxia inducible factor-1α
- I3C
Indole-3-carbinol
- iNOS
Inducible nitric oxide synthase
- IRF3
Interferon regulatory factor 3
- ITCs
Isothiocyanates
- Keap 1
Kelch-like ECHassociated protein 1
- mRNAs
Messenger RNAs
- miRNAs
microRNAs
- MAPK
Mitogen-activated protein kinase
- AT
N-acetyltransferase
- NQO1
NADP(H): quinone oxidoreductase 1
- Nrf2
NF-E2-related factor 2
- NOS2
Nitric oxide synthase-2
- NF-κB
Nuclear factor-kappa-B
- NFAT
Nuclear factor of activated T cells
- PDA
Pancreatic ductal adenocarcinoma
- PEITC
Phenethyl isothiocyanate
- PcG
Polycomb group
- TNF- α
Tumor necrosis factor alpha
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- STAT3
Signal transducers and activators of transcription 3
- TFP
Thiocyanate-forming protein
- TGFBR1
Transforming growth factor beta receptor I
- TRAMP
Transgenic adenocarcinoma of mouse prostate
- IFNs
Type I interferons
- UGT
UDP-glucuronosyl transferases
- XRE
Xenobiotic response element
- γ-GT
γ-glutamyltranspeptidase
- DIM
3.3′-diindolylmethane
- Iberin
3-methylsulfinylpropyl
- sulforaphane SFN
4-methylsulphinylbutyl isothiocyanate
- DMBA
7,12-dimethylbenz(a)anthracene
- TPA
12-O-tetradecanoylphorbol-13-acetate
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest: Francisco Fuentes, Ximena Paredes-Gonzalez, and Ah-Ng Tony Kong declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
** Of major importance
- 1.Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45(1):1–8. [PubMed] [Google Scholar]
- 2.Fuentes F, Shu L, Lee JH, Su ZY, Lee KR, Kong ANT. Nrf2-target approaches in cancer chemoprevention mediated by dietary phytochemicals. In: Bode AM, Dong Z, editors. Cancer Prevention Methods in Pharmacology and Toxicology. Springer; New York: 2014. pp. 53–83. [Google Scholar]
- 3.Su ZY, Shu L, Khor TO, Lee JH, Fuentes F, Kong AN. A perspective on dietary phytochemicals and cancer chemoprevention: oxidative stress, nrf2, and epigenomics. Top Curr Chem. 2013;329:133–62. doi: 10.1007/128_2012_340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C, Pung D, Leong V, Hebbar V, Shen G, Nair S, et al. Induction of detoxifying enzymes by garlic organosulfur compounds through transcription factor Nrf2: effect of chemical structure and stress signals. Free Radic Biol Med. 2004;37(10):1578–90. doi: 10.1016/j.freeradbiomed.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Pan MH, Ho CT. Chemopreventive effects of natural dietary compounds on cancer development. Chemical Society reviews. 2008;37(11):2558–74. doi: 10.1039/b801558a. [DOI] [PubMed] [Google Scholar]
- 6**.Lee JH, Khor TO, Shu L, Su ZY, Fuentes F, Kong AN. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacology & therapeutics. 2013;137(2):153–71. doi: 10.1016/j.pharmthera.2012.09.008. The paper presents the current advances in the study of Nrf2 signaling; Nrf2-deficient tumor mouse models; the epigenetic control of Nrf2 in tumorigenesis and chemoprevention; Nrf2-mediated cancer chemoprevention by naturally occurring dietary phytochemicals; and the mutation or hyper-expression of the Nrf2–Keap1 signaling pathway in advanced tumor cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdull Razis AF, Noor NM. Cruciferous vegetables: dietary phytochemicals for cancer prevention. Asian Pacific journal of cancer prevention : APJCP. 2013;14(3):1565–70. doi: 10.7314/apjcp.2013.14.3.1565. [DOI] [PubMed] [Google Scholar]
- 8.Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56(1):5–51. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 9.Hecht SS. Chemoprevention by Isothiocyanates. In: Kelloff GJ, Hawk ET, Sigman CC, editors. Cancer Chemoprevention, Volume 1: Promising Cancer Chemoprevention Agents. Totowa, NJ: Humana Press Inc.; 2004. pp. 21–35. [Google Scholar]
- 10.Ishida M, Hara M, Fukino N, Kakizaki T, Morimitsu Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breeding science. 2014;64(1):48–59. doi: 10.1270/jsbbs.64.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Wong CP, Hsu A, Buchanan A, Palomera-Sanchez Z, Beaver LM, Houseman EA, et al. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS One. 2014;9(1):e86787. doi: 10.1371/journal.pone.0086787. The results of this study show that sulforaphane and 3,3′-diindolylmethane are epigenetic modulators that have broad and complex effects on DNA methylation profiles reversing many of the cancer-associated methylation alterations, including aberrantly methylated genes that are dysregulated or are highly involved in cancer progression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izzotti A, Calin GA, Steele VE, Cartiglia C, Longobardi M, Croce CM, et al. Chemoprevention of cigarette smoke-induced alterations of MicroRNA expression in rat lungs. Cancer prevention research. 2010;3(1):62–72. doi: 10.1158/1940-6207.CAPR-09-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12(1):87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinkova-Kostova AT, Kostov RV. Glucosinolates and isothiocyanates in health and disease. Trends in molecular medicine. 2012;18(6):337–47. doi: 10.1016/j.molmed.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Khor TO, Shu L, Su ZY, Fuentes F, Lee JH, et al. Plants vs. cancer: a review on natural phytochemicals in preventing and treating cancers and their druggability. Anti-cancer agents in medicinal chemistry. 2012;12(10):1281–305. doi: 10.2174/187152012803833026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukherjee S, Gangopadhyay H, Das DK. Broccoli: a unique vegetable that protects mammalian hearts through the redox cycling of the thioredoxin superfamily. Journal of agricultural and food chemistry. 2008;56(2):609–17. doi: 10.1021/jf0728146. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, de Rivero Vaccari JP, Wang H, Diaz P, German R, Marcillo AE, et al. Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. Journal of neurotrauma. 2012;29(5):936–45. doi: 10.1089/neu.2011.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Link A, Balaguer F, Goel A. Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem Pharmacol. 2010;80(12):1771–92. doi: 10.1016/j.bcp.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams DJ, Pun S, Chaliha M, Scheelings P, O'Hare T. An unusual combination in papaya (Carica papaya): The good (glucosinolates) and the bad (cyanogenic glycosides) Journal of Food Composition and Analysis. 2013;29(1):82–6. http://dx.doi.org/10.1016/j.jfca.2012.06.007. [Google Scholar]
- 20.Wu X, Zhou QH, Xu K. Are isothiocyanates potential anti-cancer drugs? Acta pharmacologica Sinica. 2009;30(5):501–12. doi: 10.1038/aps.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padilla G, Cartea ME, Velasco P, de Haro A, Ordas A. Variation of glucosinolates in vegetable crops of Brassica rapa. Phytochemistry. 2007;68(4):536–45. doi: 10.1016/j.phytochem.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 22.Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 1998;7(12):1091–100. [PubMed] [Google Scholar]
- 23.Shankar S, Kumar D, Srivastava RK. Epigenetic modifications by dietary phytochemicals: implications for personalized nutrition. Pharmacology & therapeutics. 2013;138(1):1–17. doi: 10.1016/j.pharmthera.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-biological interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Tan AC, Konczak I, Sze DM, Ramzan I. Molecular pathways for cancer chemoprevention by dietary phytochemicals. Nutrition and cancer. 2011;63(4):495–505. doi: 10.1080/01635581.2011.538953. [DOI] [PubMed] [Google Scholar]
- 26.Hun Lee J, Shu L, Fuentes F, Su ZY, Tony Kong AN. Cancer chemoprevention by traditional chinese herbal medicine and dietary phytochemicals: targeting nrf2-mediated oxidative stress/anti-inflammatory responses, epigenetics, and cancer stem cells. Journal of traditional and complementary medicine. 2013;3(1):69–79. doi: 10.4103/2225-4110.107700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarozzi A, Angeloni C, Malaguti M, Morroni F, Hrelia S, Hrelia P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxidative medicine and cellular longevity. 2013;2013:415078. doi: 10.1155/2013/415078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Geng YN, Jiang JD, Kong WJ. Antioxidant and anti-inflammatory activities of berberine in the treatment of diabetes mellitus. Evidence-based complementary and alternative medicine : eCAM. 2014;2014:289264. doi: 10.1155/2014/289264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mansuy D. Brief historical overview and recent progress on cytochromes P450: adaptation of aerobic organisms to their chemical environment and new mechanisms of prodrug bioactivation. Annales pharmaceutiques francaises. 2011;69(1):62–9. doi: 10.1016/j.pharma.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Boddupalli S, Mein JR, Lakkanna S, James DR. Induction of phase 2 antioxidant enzymes by broccoli sulforaphane: perspectives in maintaining the antioxidant activity of vitamins a, C, and e. Frontiers in genetics. 2012;3:7. doi: 10.3389/fgene.2012.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarro SL, Li F, Lampe JW. Mechanisms of action of isothiocyanates in cancer chemoprevention: an update. Food & function. 2011;2(10):579–87. doi: 10.1039/c1fo10114e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu S, Kong AN. Targeting carcinogen metabolism by dietary cancer preventive compounds. Curr Cancer Drug Targets. 2007;7(5):416–24. doi: 10.2174/156800907781386669. [DOI] [PubMed] [Google Scholar]
- 33.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Barrera LN, Cassidy A, Johnson IT, Bao Y, Belshaw NJ. Epigenetic and antioxidant effects of dietary isothiocyanates and selenium: potential implications for cancer chemoprevention. The Proceedings of the Nutrition Society. 2012;71(2):237–45. doi: 10.1017/S002966511200016X. [DOI] [PubMed] [Google Scholar]
- 35.Cheung KL, Khor TO, Kong AN. Synergistic effect of combination of phenethyl isothiocyanate and sulforaphane or curcumin and sulforaphane in the inhibition of inflammation. Pharmaceutical research. 2009;26(1):224–31. doi: 10.1007/s11095-008-9734-9. [DOI] [PubMed] [Google Scholar]
- 36.Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chemical research in toxicology. 2005;18(12):1917–26. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 37.Keum YS, Owuor ED, Kim BR, Hu R, Kong AN. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC) Pharmaceutical research. 2003;20(9):1351–6. doi: 10.1023/a:1025737622815. [DOI] [PubMed] [Google Scholar]
- 38.Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, et al. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Molecular cancer therapeutics. 2006;5(8):1918–26. doi: 10.1158/1535-7163.MCT-05-0497. [DOI] [PubMed] [Google Scholar]
- 39.Brooks JD, Paton VG, Vidanes G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2001;10(9):949–54. [PubMed] [Google Scholar]
- 40.Zhang C, Su ZY, Khor TO, Shu L, Kong AN. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem Pharmacol. 2013;85(9):1398–404. doi: 10.1016/j.bcp.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang ZQ, Chen C, Yang B, Hebbar V, Kong AN. Differential responses from seven mammalian cell lines to the treatments of detoxifying enzyme inducers. Life Sci. 2003;72(20):2243–53. doi: 10.1016/s0024-3205(03)00101-2. doi:S0024320503001012[pii] [DOI] [PubMed] [Google Scholar]
- 42.Nair S, Barve A, Khor TO, Shen GX, Lin W, Chan JY, et al. Regulation of Nrf2- and AP-1-mediated gene expression by epigallocatechin-3-gallate and sulforaphane in prostate of Nrf2-knockout or C57BL/6J mice and PC-3 AP-1 human prostate cancer cells. Acta pharmacologica Sinica. 2010;31(9):1223–40. doi: 10.1038/aps.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svehlikova V, Wang S, Jakubikova J, Williamson G, Mithen R, Bao Y. Interactions between sulforaphane and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells. Carcinogenesis. 2004;25(9):1629–37. doi: 10.1093/carcin/bgh169. [DOI] [PubMed] [Google Scholar]
- 44.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthraceneinduced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006;66(16):8293–6. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 45.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer prevention research. 2014;7(3):319–29. doi: 10.1158/1940-6207.CAPR-13-0313-T. [DOI] [PubMed] [Google Scholar]
- 46.Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life Sci. 2006;79(20):1944–55. doi: 10.1016/j.lfs.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 47.Dingley KH, Ubick EA, Chiarappa-Zucca ML, Nowell S, Abel S, Ebeler SE, et al. Effect of dietary constituents with chemopreventive potential on adduct formation of a low dose of the heterocyclic amines PhIP and IQ and phase II hepatic enzymes. Nutrition and cancer. 2003;46(2):212–21. doi: 10.1207/S15327914NC4602_15. [DOI] [PubMed] [Google Scholar]
- 48.Konsue N, Ioannides C. Tissue differences in the modulation of rat cytochromes P450 and phase II conjugation systems by dietary doses of phenethyl isothiocyanate. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2008;46(12):3677–83. doi: 10.1016/j.fct.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 49.Syed Alwi SS, Cavell BE, Donlevy A, Packham G. Differential induction of apoptosis in human breast cancer cell lines by phenethyl isothiocyanate, a glutathione depleting agent. Cell stress & chaperones. 2012;17(5):529–38. doi: 10.1007/s12192-012-0329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmad A, Biersack B, Li Y, Kong D, Bao B, Schobert R, et al. Targeted regulation of PI3K/Akt/mTOR/NF-kappaB signaling by indole compounds and their derivatives: mechanistic details and biological implications for cancer therapy. Anti-cancer agents in medicinal chemistry. 2013;13(7):1002–13. doi: 10.2174/18715206113139990078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saw CL, Cintron M, Wu TY, Guo Y, Huang Y, Jeong WS, et al. Pharmacodynamics of dietary phytochemical indoles I3C and DIM: Induction of Nrf2-mediated phase II drug metabolizing and antioxidant genes and synergism with isothiocyanates. Biopharmaceutics & drug disposition. 2011;32(5):289–300. doi: 10.1002/bdd.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121(11):2373–80. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 53.Chen C, Kong AN. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Radic Biol Med. 2004;36(12):1505–16. doi: 10.1016/j.freeradbiomed.2004.03.015. doi:10.1016/j.freeradbiomed.2004.03.015 S089158490400276X [pii] [DOI] [PubMed] [Google Scholar]
- 54.Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res. 2006;4(4):221–33. doi: 10.1158/1541-7786.MCR-05-0261. doi:4/4/221 [pii] 10.1158/1541-7786.MCR-05-0261. [DOI] [PubMed] [Google Scholar]
- 55.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–95. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang H, Magilnick N, Ou X, Lu SC. Tumour necrosis factor alpha induces co-ordinated activation of rat GSH synthetic enzymes via nuclear factor kappaB and activator protein-1. Biochem J. 2005;391(Pt 2):399–408. doi: 10.1042/BJ20050795. doi:BJ20050795 [pii] 10.1042/BJ20050795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swanson HI, Njar VC, Yu Z, Castro DJ, Gonzalez FJ, Williams DE, et al. Targeting drug-metabolizing enzymes for effective chemoprevention and chemotherapy. Drug Metab Dispos. 2010;38(4):539–44. doi: 10.1124/dmd.109.031351. doi:38/4/539 [pii] 10.1124/dmd.109.031351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006;66(24):11580–4. doi: 10.1158/0008-5472.CAN-06-3562. doi:66/24/11580 [pii] 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 59.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. doi:nature04870 [pii] 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 60.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–59. doi: 10.1038/nri1703. doi:nri1703 [pii] 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 61.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shan Y, Lin N, Yang X, Tan J, Zhao R, Dong S, et al. Sulphoraphane inhibited the expressions of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 through MyD88-dependent toll-like receptor-4 pathway in cultured endothelial cells. Nutr Metab Cardiovasc Dis. 2012;22(3):215–22. doi: 10.1016/j.numecd.2010.06.013. doi:S0939-4753(10)00167-5 [pii] 10.1016/j.numecd.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 63.WW G, MB L, EW D, HD R, Ho E. Phytochemicals from cruciferous vegetables, epigenetics, and prostate cancer prevention. AAPS J. 2013;15(4):951–61. doi: 10.1208/s12248-013-9504-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim HW, Kim J, Lee S, Choi BR, Han JS, Lee KW, et al. 3,3′-Diindolylmethane inhibits lipopolysaccharide-induced microglial hyperactivation and attenuates brain inflammation. Toxicol Sci. 2014;137(1):158–67. doi: 10.1093/toxsci/kft240. doi:kft240 [pii] 10.1093/toxsci/kft240. [DOI] [PubMed] [Google Scholar]
- 65.Negi G, Kumar A, Sharma SS. Nrf2 and NF-kappaB modulation by sulforaphane counteracts multiple manifestations of diabetic neuropathy in rats and high glucose-induced changes. Curr Neurovasc Res. 2011;8(4):294–304. doi: 10.2174/156720211798120972. doi:BSP/CNR/E-Pub/00061 [pii] [DOI] [PubMed] [Google Scholar]
- 66.Park HJ, Kim SJ, Park SJ, Eom SH, Gu GJ, Kim SH, et al. Phenethyl isothiocyanate regulates inflammation through suppression of the TRIF-dependent signaling pathway of Toll-like receptors. Life Sci. 2013;92(13):793–8. doi: 10.1016/j.lfs.2013.02.012. doi:S0024-3205(13)00117-3 [pii] 10.1016/j.lfs.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 67.Cheung KL, Lee JH, Khor TO, Wu TY, Li GX, Chan J, et al. Nrf2 knockout enhances intestinal tumorigenesis in Apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol Carcinog. 2012;53(1):77–84. doi: 10.1002/mc.21950. [DOI] [PubMed] [Google Scholar]
- 68.Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, et al. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008;76(11):1485–9. doi: 10.1016/j.bcp.2008.07.017. doi:S0006-2952(08)00501-7 [pii] 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer prevention research. 2008;1(3):187–91. doi: 10.1158/1940-6207.CAPR-08-0028. doi:1940-6207.CAPR-08-0028 [pii] 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bellezza I, Tucci A, Galli F, Grottelli S, Mierla AL, Pilolli F, et al. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. J Nutr Biochem. 2012;23(12):1583–91. doi: 10.1016/j.jnutbio.2011.10.012. doi:S0955-2863(11)00306-8 [pii] 10.1016/j.jnutbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 71.Liao BC, Hsieh CW, Lin YC, Wung BS. The glutaredoxin/glutathione system modulates NF-kappaB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol Sci. 2010;116(1):151–63. doi: 10.1093/toxsci/kfq098. doi:kfq098 [pii] 10.1093/toxsci/kfq098. [DOI] [PubMed] [Google Scholar]
- 72.Wagner AE, Will O, Sturm C, Lipinski S, Rosenstiel P, Rimbach G. DSS-induced acute colitis in C57BL/6 mice is mitigated by sulforaphane pre-treatment. J Nutr Biochem. 2013;24(12):2085–91. doi: 10.1016/j.jnutbio.2013.07.009. doi:S0955-2863(13)00160-5 [pii] 10.1016/j.jnutbio.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 73.Saw CL, Huang MT, Liu Y, Khor TO, Conney AH, Kong AN. Impact of Nrf2 on UVB-induced skin inflammation/photoprotection and photoprotective effect of sulforaphane. Mol Carcinog. 2011;50(6):479–86. doi: 10.1002/mc.20725. [DOI] [PubMed] [Google Scholar]
- 74.Wagner AE, Boesch-Saadatmandi C, Dose J, Schultheiss G, Rimbach G. Anti-inflammatory potential of allyl-isothiocyanate--role of Nrf2, NF-(kappa) B and microRNA-155. J Cell Mol Med. 2012;16(4):836–43. doi: 10.1111/j.1582-4934.2011.01367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 76.Arasaradnam RP, Commane DM, Bradburn D, Mathers JC. A review of dietary factors and its influence on DNA methylation in colorectal carcinogenesis. Epigenetics. 2008;3(4):193–8. doi: 10.4161/epi.3.4.6508. [DOI] [PubMed] [Google Scholar]
- 77.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 78.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 79.Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer letters. 2008;269(2):291–304. doi: 10.1016/j.canlet.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Traka M, Gasper AV, Smith JA, Hawkey CJ, Bao Y, Mithen RF. Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. The Journal of nutrition. 2005;135(8):1865–72. doi: 10.1093/jn/135.8.1865. [DOI] [PubMed] [Google Scholar]
- 81.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5(7):e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol Carcinog. 2007;46(1):24–31. doi: 10.1002/mc.20258. [DOI] [PubMed] [Google Scholar]
- 83.Wu TY, Khor TO, Su ZY, Saw CL, Shu L, Cheung KL, et al. Epigenetic modifications of Nrf2 by 3,3′-diindolylmethane in vitro in TRAMP C1 cell line and in vivo TRAMP prostate tumors. AAPS J. 2013;15(3):864–74. doi: 10.1208/s12248-013-9493-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gerhauser C. Cancer chemoprevention and nutriepigenetics: state of the art and future challenges. Top Curr Chem. 2013;329:73–132. doi: 10.1007/128_2012_360. [DOI] [PubMed] [Google Scholar]
- 85.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 87.Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20(3):506–8. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008;33(2):375–80. [PubMed] [Google Scholar]
- 89.Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3′-diindolylmethane in breast cancer cells. The Journal of nutrition. 2009;139(1):26–32. doi: 10.3945/jn.108.099259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Experimental biology and medicine. 2007;232(2):227–34. [PMC free article] [PubMed] [Google Scholar]
- 91.Balasubramanian S, Chew YC, Eckert RL. Sulforaphane suppresses polycomb group protein level via a proteasome-dependent mechanism in skin cancer cells. Molecular pharmacology. 2011;80(5):870–8. doi: 10.1124/mol.111.072363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beaver LM, Yu TW, Sokolowski EI, Williams DE, Dashwood RH, Ho E. 3,3′-Diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicology and applied pharmacology. 2012;263(3):345–51. doi: 10.1016/j.taap.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70(2):646–54. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Su ZY, Shu L, Lee JH, Fuentes F, Wang H, Wu TY, et al. Perspective on Nrf2, Epigenomics and Cancer Stem Cells in Cancer Chemoprevention Using Dietary Phytochemicals and Traditional Chinese Medicines. Progress in Chemistry. 2013;25(9):1526–43. doi: 10.7536/PC130717. [DOI] [Google Scholar]
- 95.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10(10):704–14. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brait M, Sidransky D. Cancer epigenetics: above and beyond. Toxicol Mech Methods. 2011;21(4):275–88. doi: 10.3109/15376516.2011.562671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li Y, VandenBoom TG, 2nd, Kong D, Wang Z, Ali S, Philip PA, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69(16):6704–12. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. Alteration of microRNA expression in vinyl carbamateinduced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. 2010;31(2):252–8. doi: 10.1093/carcin/bgp208. [DOI] [PubMed] [Google Scholar]
- 99.Jin Y. 3,3′-Diindolylmethane inhibits breast cancer cell growth via miR-21-mediated Cdc25A degradation. Molecular and cellular biochemistry. 2011;358(1-2):345–54. doi: 10.1007/s11010-011-0985-0. [DOI] [PubMed] [Google Scholar]
- 100.Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, et al. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. American journal of translational research. 2012;4(1):14–23. [PMC free article] [PubMed] [Google Scholar]
- 101.Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, et al. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One. 2012;7(3):e33729. doi: 10.1371/journal.pone.0033729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Appari M, Babu KR, Kaczorowski A, Gross W, Herr I. Sulforaphane, quercetin and catechins complement each other in elimination of advanced pancreatic cancer by miR-let-7 induction and K-ras inhibition. Int J Oncol. 2014;45(4):1391–400. doi: 10.3892/ijo.2014.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Q, Eades G, Yao Y, Zhang Y, Zhou Q. Characterization of a stem-like subpopulation in basal-like ductal carcinoma in situ (DCIS) lesions. The Journal of biological chemistry. 2014;289(3):1303–12. doi: 10.1074/jbc.M113.502278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Singh SV, Singh K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis. 2012;33(10):1833–42. doi: 10.1093/carcin/bgs216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gills JJ, Jeffery EH, Matusheski NV, Moon RC, Lantvit DD, Pezzuto JM. Sulforaphane prevents mouse skin tumorigenesis during the stage of promotion. Cancer letters. 2006;236(1):72–9. doi: 10.1016/j.canlet.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 106.Khor TO, Hu R, Shen G, Jeong WS, Hebbar V, Chen C, et al. Pharmacogenomics of cancer chemopreventive isothiocyanate compound sulforaphane in the intestinal polyps of ApcMin/+ mice. Biopharmaceutics & drug disposition. 2006;27(9):407–20. doi: 10.1002/bdd.522. [DOI] [PubMed] [Google Scholar]
- 107.Shen G, Khor TO, Hu R, Yu S, Nair S, Ho CT, et al. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007;67(20):9937–44. doi: 10.1158/0008-5472.CAN-07-1112. [DOI] [PubMed] [Google Scholar]
- 108.Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27(10):2038–46. doi: 10.1093/carcin/bgl049. [DOI] [PubMed] [Google Scholar]
- 109.Singh SV, Warin R, Xiao D, Powolny AA, Stan SD, Arlotti JA, et al. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009;69(5):2117–25. doi: 10.1158/0008-5472.CAN-08-3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Keum YS, Khor TO, Lin W, Shen G, Kwon KH, Barve A, et al. Pharmacokinetics and pharmacodynamics of broccoli sprouts on the suppression of prostate cancer in transgenic adenocarcinoma of mouse prostate (TRAMP) mice: implication of induction of Nrf2, HO-1 and apoptosis and the suppression of Akt-dependent kinase pathway. Pharmaceutical research. 2009;26(10):2324–31. doi: 10.1007/s11095-009-9948-5. [DOI] [PubMed] [Google Scholar]
- 111.Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Cancer letters. 2006;243(2):170–92. doi: 10.1016/j.canlet.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 112.Cheung KL, Khor TO, Huang MT, Kong AN. Differential in vivo mechanism of chemoprevention of tumor formation in azoxymethane/dextran sodium sulfate mice by PEITC and DBM. Carcinogenesis. 2010;31(5):880–5. doi: 10.1093/carcin/bgp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barve A, Khor TO, Hao X, Keum YS, Yang CS, Reddy B, et al. Murine prostate cancer inhibition by dietary phytochemicals--curcumin and phenyethylisothiocyanate. Pharmaceutical research. 2008;25(9):2181–9. doi: 10.1007/s11095-008-9574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Powolny AA, Bommareddy A, Hahm ER, Normolle DP, Beumer JH, Nelson JB, et al. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. Journal of the National Cancer Institute. 2011;103(7):571–84. doi: 10.1093/jnci/djr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu TY, Saw CL, Khor TO, Pung D, Boyanapalli SS, Kong AN. In vivo pharmacodynamics of indole-3-carbinol in the inhibition of prostate cancer in transgenic adenocarcinoma of mouse prostate (TRAMP) mice: involvement of Nrf2 and cell cycle/apoptosis signaling pathways. Mol Carcinog. 2012;51(10):761–70. doi: 10.1002/mc.20841. [DOI] [PubMed] [Google Scholar]
- 116.Fujioka N, Ainslie-Waldman CE, Upadhyaya P, Carmella SG, Fritz VA, Rohwer C, et al. Urinary 3,3′-diindolylmethane: a biomarker of glucobrassicin exposure and indole-3-carbinol uptake in humans. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2014;23(2):282–7. doi: 10.1158/1055-9965.EPI-13-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Syed Alwi SS, Cavell BE, Telang U, Morris ME, Parry BM, Packham G. In vivo modulation of 4E binding protein 1 (4E-BP1) phosphorylation by watercress: a pilot study. The British journal of nutrition. 2010;104(9):1288–96. doi: 10.1017/S0007114510002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, et al. Effects of glucosinolaterich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People′s Republic of China. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2005;14(11 Pt 1):2605–13. doi: 10.1158/1055-9965.EPI-05-0368. [DOI] [PubMed] [Google Scholar]