

Abstract
Whole body knockout (KO) of the P2Y2 receptor (P2Y2R) results in enhanced vasopressin V2 receptor activity and increased renal Na+ conservation. We hypothesized that P2Y2R KO mice would be less sensitive to lithium-induced natriuresis and kaliuresis due to attenuated downregulation of one or more of the major renal Na+ or K+ transporter/channel proteins. KO and wild-type (WT) mice were fed a control or lithium-added diet (40 mmol/kg food) for 14 days. Lithium-induced natriuresis and kaliuresis were significantly (∼25%) attenuated in KO mice. The subunits of the epithelial Na+ channel (ENaC) were variably affected by lithium and genotype, but, overall, medullary levels were decreased substantially by lithium (15–60%) in both genotypes. In contrast, cortical, β-, and γ-ENaC were increased by lithium (∼50%), but only in WT mice. Moreover, an assessment of ENaC activity by benzamil sensitivity suggested that lithium increased ENaC activity in WT mice but in not KO mice. In contrast, medullary levels of Na+-K+-2Cl− cotransporter 2 and cortical levels of the renal outer medullary K+ channel were not downregulated by lithium and were significantly (15–76%) higher in KO mice under both dietary conditions. In addition, under control conditions, tissue osmolality of the inner medulla as well as furosemide sensitivity were significantly higher in KO mice versus WT mice. Therefore, we suggest that increased expression of these proteins, particularly in the control state, reduces Na+ delivery to the distal nephron and provides a buffer to attenuate collecting duct-mediated natriuresis and kaliuresis. Additional studies are warranted to explore the potential therapeutic benefits of purinergic antagonism.
Keywords: vasopressin, aldosterone, sodium transporters, epithelial sodium channel, potassium channels, ATP, nephrogenic diabetes insipidus
despite the advent of newer medications, lithium continues to be the main choice for the treatment of bipolar disorder due to its ability to prevent suicidal tendencies (12, 24). However, the adverse effects of chronic lithium treatment on the kidney leading to nephrogenic diabetes insipidus (NDI) is a major limiting factor in the use of this effective medication (for a review, see Ref. 11). In addition, lithium has been shown to have a myriad of other effects in the kidney (for a review, see Ref. 15). Clinically, lithium-induced NDI is characterized by polyuria, natriuresis, and kaliuresis as well as the loss of other vital substances in the urine. Arginine vasopressin (AVP)-resistant polyuria causes dehydration, hypovolemia, and hypernatremia, leading to hemodynamic instability in elderly patients (23). On the other hand, lithium-induced natriuresis and kaliuresis activate mechanisms that result in renal lithium retention and lithium intoxication, which can become a vicious cycle (1, 34). Currently used therapeutic measures for lithium-induced NDI, such as the use of cyclooxygenase (COX) inhibitors or thiazides, can also cause lithium intoxication, thus limiting their clinical use (25, 35). Although administration of amiloride ameliorated lithium-induced polyuria, it markedly enhanced lithium-induced natriuresis in animal models (2, 19).
The underlying cause for lithium-induced polyuria and natriuresis is likely due to significant alterations in the activity of AVP- and/or aldosterone-regulated proteins involved in renal water and Na+ transport (11, 20). Signaling mediated via the extracellular ATP/UTP-activated P2Y2 receptor (P2Y2R) modulates renal water and Na+ transport by antagonizing the effects of AVP and aldosterone on the kidney (16, 36, 37). P2Y2R deletion has been shown to alter the abundances of key proteins involved in the transport of water and Na+, such as aquaporin-2 (AQP2), Na+-K+-2Cl− cotransporter 2 (NKCC2), and urea transporter (UT)-A1 in the renal medulla (30, 43). In addition, genetic deletion of P2Y2R alters the expression and function of subunits of the epithelial Na+ channel (ENaC) under basal conditions or in response to a high-Na+ diet with or without aldosterone infusion (30, 43).
Recently, we (41, 42) have shown that mice with genetic deletion of P2Y2R have significant resistance to the development of lithium-induced polyuria. This was found to occur without any alterations in serum or renal medullary lithium levels (41, 42). The mechanisms are not completely understood, but our study suggested reduced PGE2 signaling through the EP3 receptor might play a role by preserving cAMP generation and thus protecting collecting duct AQP2 protein in the renal medulla (42).
Based on these novel findings, we hypothesized that P2Y2R deletion might also attenuate lithium-induced natriuresis and kaliuresis by preserving the expression of cAMP-regulated epithelial Na+ transporters and/or channels in either the collecting duct or thick ascending limb (TAL). Using P2Y2R knockout (KO) and wild-type (WT) mice, we documented that genetic deletion of P2Y2R does indeed ameliorate natriuresis and kaliuresis induced by lithium treatment. To gain insights into the potential mechanisms, using semiquantitative immunoblot analysis, we analyzed the protein abundances of Na+ as well as K+ transporters/channels expressed along the nephron and determined the effect of lithium treatment on medullary interstitial osmolality and urinary excretion of AVP and aldosterone, the key hormones that regulate water and Na+ transport in the kidney. In addition, we assessed the in vivo activity of ENaC, Na+-Cl− cotransporter (NCC), and NKCC2 and their contribution to lithium-induced natriuresis and kaliuresis in WT and KO mice by determining acute responses to diuretics that specifically block these transporters or channels. Our findings suggest that the protection afforded in KO mice does not appear to involve an increase in systemic aldosterone activity or the preservation of ENaC subunit protein abundances but may involve greater capacity of the TAL to reabsorb Na+ via increased NKCC2, potentially reducing distal Na+ load, thus ameliorating both kaliuresis and natriuresis due to the damaging effects of lithium on the collecting duct.
METHODS
Animal experiments.
All experimental procedures and breeding strategies were approved by the Institutional Animal Care and Use Committee of the Veterans Affairs Salt Lake City Health Care System. WT and KO mice (B6D2 genetic background) were bred and genotyped at the Veterans Affairs Salt Lake City Health Care System as previously described (43). After baseline 24-h urine samples were collected in metabolic cages for 2 consecutive days, young adult mice were transferred to regular cages and fed ad libitum either the control diet or the same diet supplemented with 40 mmol lithium/kg diet (MP Biomedicals, Solon, OH) for 14 or 21 days. Mice were again housed in metabolic cages for 2–3 days before euthanasia; except for these short periods, mice were group housed. Throughout the study, mice had ad libitum access to plain drinking water. Mice were euthanized by rapid decapitation under isoflurane anesthesia, and blood was collected for analysis. Kidneys were removed, and cortices and medullas were dissected out, flash frozen in liquid nitrogen, and then stored at −80°C for analysis.
Analysis of urine and serum samples.
Urine osmolality was determined by the vapor pressure method (Wescor, Logan, UT). Urine Na+ and K+ were measured by EasyElectrolyte or EasyLyte (Medica, Bedford, MA) instruments. Urine aldosterone and AVP levels were quantified by ELISA (Enzo Life Sciences, Farmingdale, NY) and were used as an index of their circulating levels over 24 h. Serum urea concentrations were determined by an enzymatic method using a commercial kit (BioVision, Milpitas, CA).
Analysis of kidney tissue samples for protein abundances.
Cortical and medullary tissue samples from WT and KO mice (n = 5 mice/treatment group per genotype) were analyzed for protein abundances of Na+ and/or K+ transporters/channels by semiquantitative immunoblot analysis. For immunoblot analysis, cortical and medullary tissue samples were processed separately, as previously described, and shipped to Georgetown University for analysis (40). Briefly, samples were prepared by homogenizing the frozen tissues in a buffer containing protease inhibitors. After the protein concentrations were determined, homogenates were solubilized in Laemmli sample buffer. The quality of the tissue sample preparation was assessed by staining loading gels with Coomassie blue (Gelcode Blue, Pierce Endogen, Rockford, IL) and then examining the sharpness of the bands. For immunoblot analysis, 10–30 μg protein from each sample was loaded into individual lanes of minigels of 7%, 10%, or 12% polyacrylamide (precast, Bio-Rad, Hercules, CA). After electrotransfer of size-fractionated proteins to nitrocellulose membranes, blots were probed with our own rabbit peptide-derived polyclonal antibodies against Na+/H+ exchanger type 3 (NHE3), NKCC2, thiazide-sensitive NCC, Na+-Pi cotransporter type 2 (NaPi-2), and the three subunits of ENaC (α, β, and γ), as previously described (40). We used commercially available antibodies against the α1-subunit of Na+-K+-ATPase (no. 05-369, Millipore, Temecula, CA), the renal outer medullary K+ channel (ROMK; P0856, Sigma-Aldrich, St. Louis, MO), and the large-conductance K+ (BK) channel (maxi-K or KCa1.1, no. 1184-1200, Alamone Laboratories, Jerusalem, Israel). Loading accuracy was evaluated by probing the blots with β-actin monoclonal antibody (A1978, Sigma-Aldrich). Band densities of transporter/channel proteins were determined and normalized to the densities of the respective β-actin bands.
Determination of renal tissue osmolality.
Groups of WT and KO mice were fed either control diet or lithium-added diet (n = 6 mice/treatment per genotype) for 14 days and then euthanized. Twenty-four-hour urine samples were collected before euthanasia, and bladder urine samples were collected at the time of euthanasia. For the determination of tissue osmolality, we followed the method described by Schmidt-Nielsen et al. (31) for rat kidneys, with slight modifications to suit mouse kidneys. After euthanasia, both kidneys were quickly removed and chilled in cold PBS. Kidneys were then blotted to remove adhering water, and the whole inner medulla (IM) and outer stripe of the outer medulla (OSOM) were rapidly excised and separated on a petri dish kept on ice. The dissected tissue samples were blotted with a filter paper, pressed gently to expel residual urine, and then put into preweighed and labeled 1.7-ml microtubes. Each tube contained IM or OSOM of both kidneys from one mouse only. The entire process from euthanasia to transfer of tissue samples into microtubes took <2 min. The microtubes containing the tissue samples were weighed to the nearest 0.01 mg. The caps of the tubes were then opened, and tissue samples were dried on a dry bath at 60°C for 5–6 h. After being dried, the tubes were cooled to room temperature, capped, and weighed. Ultrapure water (80 μl) was then added to each tube, and tubes were gently tapped to ensure complete immersion of the dried tissue sample in the water. The tubes were capped, sealed, and placed in a 95°C dry bath for 3 min. After that, the tubes were cooled to room temperature, spun briefly for a few seconds in a microcentrifuge, and kept at 4°C for 18–24 h for diffusion of solutes. The tubes were then centrifuged, and the supernatants were used for the determination of osmolality by the vapor pressure method (Wescor). Tissue osmolality was calculated using the following formulas published by Schmidt-Nielsen et al. (31).
The water content of the tissue was determined as the difference between wet weight and dry weight of the tissue. Because the specific gravity of water is 1 kg/l, each milligram of water content of the tissue corresponds to 1 μl. Thus, fractional water content (FH2O) = (wet weightt − dry weight)/wet weight.
The apparent solute concentrations in tissue water (CT) were calculated by multiplying the concentration measured in the supernatant (CS) by the dilution factor. Thus, CT = CS[80(= water in the tube)/FH2O × tissue wet weight].
We have validated this method by determining renal tissue osmolality in another cohort of mice under basal conditions or after an acute water loading by intraperitoneal injection of 2 ml sterile water. The measured tissue osmolalities in water-loaded mice were 64% (OSOM) and 49% (IM) of their respective values in mice under basal conditions (n = 3 mice/condition).
Determination of natriuretic and kaliuretic responses to diuretics.
To assess the in vivo activity of ENaC, NCC, and NKCC2 and their contribution to lithium-induced natriuresis and/or kaliuresis in WT and KO mice, we determined the acute responses to diuretics that specifically block these transporters or channels. Groups of WT and KO mice were fed either a control diet or lithium-added diet for up to 21 days (n = 6 mice/genotype per diet). Between days 12 and 18, the effects of diuretics were tested in mice with a 2-day interval between two consecutive tests in the same group of mice, as previously described by us (21). Briefly, in the morning, mice were weighted and then injected intraperitoneally with either benzamil hydrochloride hydrate (Sigma, >98% pure, 1.4 mg/kg body wt) or hydrochlorothiazide (HCTZ; Sigma, meets USP testing specifications, 7.5 mg/kg body wt) or furosemide (Hospira, Lake Forest, IL, USP grade, 24 mg/kg body wt) in sterile water (100 μl). Immediately after the injection, mice were kept in small metabolic cages (1 mouse/cage) without access to food and water for 4 h. Urine voided during this period was collected under light mineral oil in the cages. Volumes of urine samples collected were measured, and their Na+ and K+ contents were determined as described above. Mice were offered food and water immediately after the 4-h period.
Statistical analysis.
Quantitative data are expressed as means ± SEM. Differences due to the main factors, genotype and dietary lithium, and their interactions were determined by two-way ANOVA using Sigma Stat Software (Chicago, IL) or GraphPad Instat software (La Jolla, CA). Differences between individual pairs of means were determined by a multiple-comparisons test (Holm-Sidak or Kruskal-Wallis ANOVA on ranks) after a significant (P < 0.05) one-way ANOVA. The Kruskal-Wallis test was used when data were not normally distributed or variances were not equal. Where applicable, an unpaired t-test was used to directly compare the means of two groups.
RESULTS
Water and electrolyte homeostasis.
Urine osmolality was markedly reduced by 14 days of lithium treatment (Fig. 1A). This reduction was attenuated in KO mice so that there was a significant (P < 0.05) difference in urine osmolality at this point. Likewise, urine volume (Fig. 1B) and water intake (Fig. 1C) were increased by lithium to a much larger extent in WT mice compared with KO mice. Both urinary Na+ (Fig. 2A) and K+ (Fig. 2B) were increased by lithium treatment; however, the effect on both electrolytes was significantly blunted in KO mice. To determine whether differences in two major Na+/K+ homeostatic hormones, i.e., AVP or aldosterone, might play a role in the observed differences, the urinary excretion of both hormones was measured in 24-h urine samples (Fig. 3). Urine AVP was significantly increased (P < 0.05) by lithium in both genotypes; however, there were no significant differences between genotypes in either pre- or postlithium collections (Fig. 3A). Surprisingly, urine aldosterone (Fig. 3B) was significantly higher in KO mice compared with WT mice in the pretreatment collection (P = 0.038). With lithium therapy, urine aldosterone fell in KO mice so that it was significantly different from their own pretreatment levels (P = 0.038) but not from that of WT mice (P = 0.14). Finally, urine aldosterone in WT mice after lithium treatment rose numerically, but it was not significantly different from their own pretreatment values (P = 0.063).
Fig. 1.
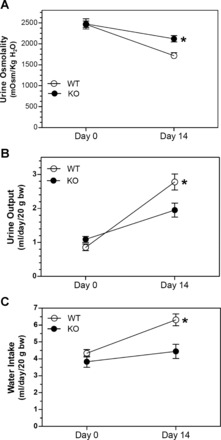
P2Y2 receptor knockout (KO) mice are significantly resistant to lithium (Li)-induced polyuria and polydipsia. KO mice were less sensitive to Li than wild-type (WT) mice with regard to a reduction in urine osmolality (A), increases in urine volume (B), and increases in water intake (C). n = 9 mice/genotype per time point. *Significant difference between genotypes by an unpaired t-test (P < 0.05).
Fig. 2.
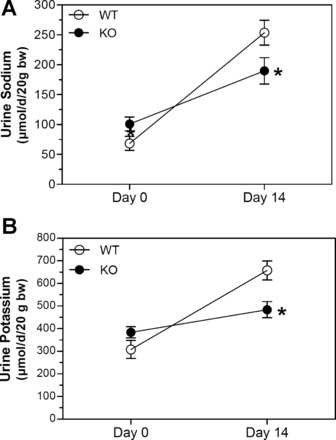
KO mice are significantly resistant to Li-induced natriuresis and kaliuresis. A and B: 24-h urinary excretion of Na+ (A) and K+ (B) on days 0 and 14 of the study. n = 9 mice/genotype per time point. *Significant difference between genotypes by an unpaired t-test (P < 0.05).
Fig. 3.
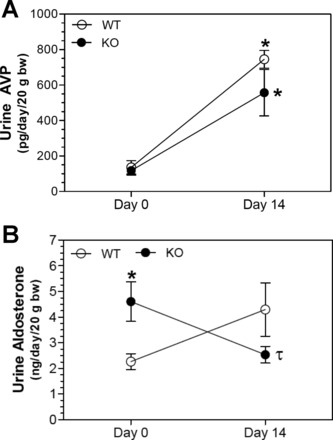
Effect of Li on the urinary excretion of aldosterone and arginine vasopressin (AVP) in WT and KO mice. A and B: 24-h excretion of aldosterone (A) and AVP (B) on days 0 and 14 of the study. n = 5 mice/genotype per time point. *Significant difference between genotypes (P < 0.05); τsignificant difference between pre- and post-Li collections by an unpaired t-test (n = 4–5 mice/group).
Expression of Na+ transporters/channels.
Semiquantitative immunoblot analysis was used to analyze differences in the protein abundance of Na+ transporters/channels along the nephron. Figures 4 and 5 show representative immunoblots (A) of the cortex and medulla, respectively, and the corresponding plots (B) of densitometric means (±SE) for each of the four groups. Symbols above the bars in Figs. 4 and 5 indicate the results of the multiple-comparisons tests that were performed when the one-way ANOVA indicated that a significant difference existed between two or more groups.
Fig. 4.
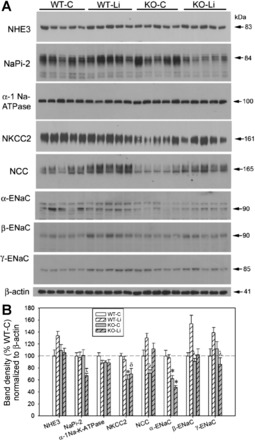
Effect of Li on the protein abundance of major renal Na+ transporters in the cortex (CTX) of WT and KO mice. Top: representative Western blots of cortex homogenates. Blots were loaded with equal amounts of protein per lane, and band densities were normalized by probing for β-actin on the lower portion of the membrane (n = 5 mice/treatment per genotype). A representative β-actin blot is also shown. NHE, Na+/H+ exchanger; NaPi-2, Na+-Pi cotransporter 2; NKCC2, Na+-K+-2Cl− cotransporter 2; NCC, Na+-Cl− cotransporter; ENaC, epithelial Na+ channel. Bottom: bar graph summary of densitometry (means ± SE). Symbols above the bars show the results of a multiple-comparisons test after significant (P < 0.05) one-way ANOVA. The following groups are shown: control WT (WT-C), Li-treated WT (WT-Li), control KO (KO-C), and Li-treated KO (KO-Li). *Significant difference from both WT-C and WT-Li groups (both WT groups); τsignificant difference from both WT-C and KO-C groups (both control treatments); δsignificant difference from the WT-C group only; λsignificant difference from the WT-Li group only.
Fig. 5.
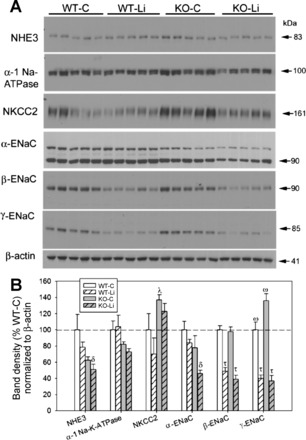
Effect of Li on the protein abundance of major renal Na+ transporters in the medulla of WT and KO mice. Top: representative Western blots of medullary homogenates. Blots were loaded with equal amounts of protein per lane, and band densities were normalized by probing for β-actin on the lower portion of the membrane (n = 5 mice/treatment per genotype). A representative β-actin blot is also shown. Bottom: bar graph summary of densitometry (means ± SE). Symbols above the bars show the results of a multiple-comparisons test after significant (P < 0.05) one-way ANOVA. ωSignificant difference from all other groups; τsignificant difference from both the WT-C and KO-C groups (both control treatments); δsignificant difference from the WT-C group only; λsignificant difference from the WT-Li group only.
In the cortex (Fig. 4), there was a significant effect of group (P < 0.05) on NaPi-2, NKCC2, and α-, β-, and γ-ENaC. The subsequent multiple-comparisons test revealed that NaPi-2 in the lithium-treated KO group was significantly lower than in both control groups of mice (indicated by τ). Moreover, in these lithium-treated KO mice, there was a significant reduction in NKCC2 (lower than control WT mice; indicated by δ), α-ENaC (lower than both WT groups, indicated by *), and γ-ENaC (lower than lithium-treated WT mice; indicated by λ). In addition, mice in the control KO group showed reduced NKCC2 (relative to all other groups) as well as reduced NCC and β-ENaC (lower than the lithium-treated WT group).
In the medulla (Fig. 5), there was a significant effect of group on NHE3, NKCC2, and α-, β-, and γ-ENaC. Lithium-treated KO mice had significantly lower NHE3 and α-ENaC (lower than control WT mice; indicated by δ) as well as β- and γ-ENaC (lower than both groups of control mice). Both groups of lithium-treated mice had significantly lower levels of β- and γ-ENaC compared with both groups of control mice. Control KO mice had significantly higher levels of NKCC2 (higher than lithium-treated WT mice; indicated by λ) and γ-ENaC (higher than all other groups; indicated by ω). Finally, γ-ENaC in control WT mice was also significantly lower than control KO mice and was significantly higher than both of the lithium-treated groups (i.e., different from all other groups; indicated by ω).
Figure 6 shows immunoblots of the cortex (A) and medulla (B) for ROMK (Kir1.1) and BK (KCa1.1, Maxi-K) channels. ROMK exists in three isoforms, which differ at the NH2 end of the molecule with ROMK1a (longest) being ∼3 amino acids longer than ROMK1c, which is ∼15 amino acids longer than ROMK1b (shortest). The antibody we used (against amino acids 342–391) should have been able to detect all major isoforms of ROMK (ROMK1a, ROMK1b, and ROMK1c) expressed in the kidney. For ROMK, we analyzed the density of the dominant, likely glycosylated band region between 60 and 65 kDa (8, 39). The band at 99 kDa may represent a dimer form of the protein (5). One-way ANOVA revealed a significant difference among groups for BK channels in the medulla. Medullary BK channels were significantly reduced in the lithium-treated KO group relative to the lithium-treated WT and control KO groups (indicated by μ).
Fig. 6.
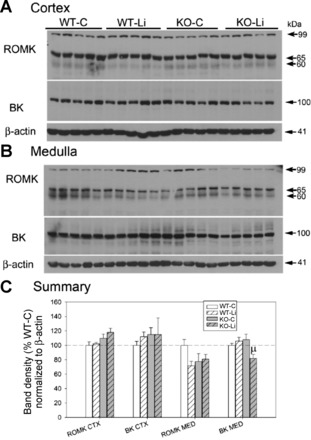
Effect of Li on the protein abundance of major renal K+ transporters in the cortex and medulla of WT and KO mice. A and B: representative Western blots of cortex (A) and medullary (B) homogenates. Blots were loaded with equal amounts of protein per lane, and band densities were normalized by probing for β-actin on the lower portion of the renal outer medullary K+ channel (ROMK) membrane (n = 5 mice/treatment per genotype). BK, large-conductance K+ channel. C: bar graph summary of densitometry (means ± SE). CTX, cortex; MED, medulla. The symbol above the bar shows the result of a multiple-comparisons test after significant (P < 0.05) one-way ANOVA. μSignificant difference from the WT-Li and KO-C groups.
The results of two-way ANOVA (treatment × genotype) along with significant interactions are shown in Table 1. With regard to the effect of P2Y2R KO, medullary NHE3, α1-Na+-K+-ATPase, α-ENaC, and cortical NCC were significantly lower in the KO group relative to the WT group with no significant interactions. In contrast, medullary NKCC2 and cortical ROMK were significantly increased in the KO group. With regard to the effects of lithium, lithium reduced NaPi-2 as well as medullary α-, β-, and γ-ENaC. For γ-ENaC, in the medulla, there was a significant interaction in that the slope of the decrease was greater in the KO group. Lithium increased the expression of NCC as well as cortical β- and γ-ENaC (only in the WT group). β- and γ-ENaC in the cortex were reduced by lithium in the KO group (significant interaction). Finally, there was a significant interaction for the BK channel in the medulla, where it was significantly reduced by lithium in the KO group but not in the WT group.
Table 1.
Summary of the overall effects of genotype and lithium on Na+ and K+ transporters along the nephron
| Results of Two-Way ANOVA |
||
|---|---|---|
| Transporter/Channel | Effect of P2Y2R KO | Effect of lithium |
| NHE3 | ||
| Cortex | ⇆ | ⇆ |
| Medulla | ↓ | ⇆ |
| α1-Na+-K+-ATPase | ||
| Cortex | ⇆ | ⇆ |
| Medulla | ↓ | ⇆ |
| NaPi-2 | ||
| Cortex | ⇆ | ↓ |
| NCC | ||
| Cortex | ↓ | ↑ |
| NKCC2 | ||
| Cortex | ↓ | ⇆ |
| Medulla | ↑ | ⇆ |
| α-ENaC | ||
| Cortex | ↓ | ⇆ |
| Medulla | ↓ | ↓ |
| β-ENaC | ||
| Cortex | ↓ | ↑↓* |
| Medulla | ⇆ | ↓ |
| γ-ENaC | ||
| Cortex | ⇆ | ↑↓* |
| Medulla | ⇆ | ↓* |
| ROMK | ||
| Cortex | ↑ | ⇆ |
| Medulla | ⇆ | ⇆ |
| BK | ||
| Cortex | ⇆ | ⇆ |
| Medulla | ⇆ | ⇆* |
Significant (P < 0.05) two-way ANOVA findings are shown as an increase (↑), a decrease (↓), or no change (⇆).
NHE3, Na+/H+ exchanger type 3; NaPi-2, Na+-Pi cotransporter type 2; NCC, Na+-Cl− cotransporter; NKCC2, Na+-K+-2Cl− cotransporter 2; ENaC, epithelial Na+ channel; ROMK, renal outer medullary K+ channel; BK, large-conductance K+ channel.
Significant interaction. In the case of cortical β- and γ-ENaC and medullary BK channels, the means increased with lithium in wild-type mice and decreased with lithium in P2Y2 receptor (P2Y2R) knockout (KO) mice. For medullary γ-ENaC, abundance was decreased in both wild-type and KO mice, but to a significantly greater extent in KO mice.
Figure 7 shows renal tissue osmolalities in control or lithium diet-fed mice and their corresponding urine osmolalities. Figure 7A shows tissue osmolalities of OSOMs in the four groups of mice. Lithium treatment caused comparable decreases in both WT and KO mice, with no significant differences between genotypes. On the other hand, IM tissue osmolality was significantly higher (34%, P < 0.02) in WT mice fed the control diet versus KO mice fed the control diet (Fig. 7B). Lithium feeding resulted in decreases in IM tissue osmolalities in both genotypes. Overall, the mean tissue osmolalities in lithium-fed KO mice were numerically higher than the corresponding mean values in WT mice.
Fig. 7.
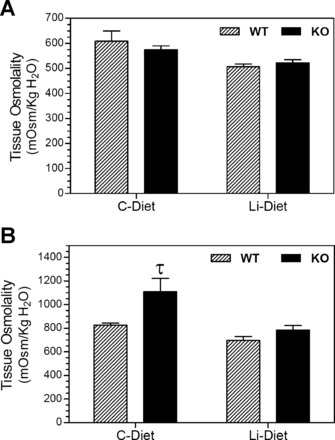
Effect of Li treatment on kidney tissue osmolalities in WT and KO mice. Groups of WT and KO mice were fed control (C) or Li-added diets for 14 days and euthanized (n = 6 mice/treatment per genotype). Inner stripes of outer medulla (A) and inner medullary (B) tissues were collected and processed as described in methods for the determination of tissue osmolalities. τSignificant difference from the corresponding WT group (P < 0.02 by an unpaired t-test).
Figure 8 shows natriuretic and kaliuretic responses to a single injection of benzamil, HCTZ, or furosemide. The acute sensitivity to each of these specific antagonists has been used to estimate or gauge the relative in vivo activation state of ENaC, NCC, and NKCC2, respectively (21). A greater natriuretic response indicates greater relative activity. Under control conditions (Fig. 8A), KO mice had a significantly greater natriuretic response to furosemide than WT, agreeing with higher NKCC2 protein levels. A similar pattern was observed for benzamil (although the number responding in WT mice was too low to conduct a statistical comparison). The response to HCTZ was not different between genotypes. After lithium treatment (Fig. 8B), there were no differences between genotypes. Likewise, there were no differences between genotypes in K+ excretion in the control state (Fig. 8C) or with lithium treatment (Fig. 8D). Kaliuresis was higher in response to HCTZ and furosemide in the control condition compared with after lithium (Fig. 8, C compared with D). Figure 8, E and F, shows a summary of the overall natriuretic or kaliuretic responses, respectively. This provides an estimation of the overall TAL through collecting duct Na+ (or K+) activation of reabsorptive processes. We found that lithium decreased the overall summation of NKCC2, ENaC, and NCC activity in KO mice (Fig. 8E, top two bars); however, the response was opposite in WT mice (Fig. 8E, bottom two bars), with a diminishment of responsiveness. With regard to K+ excretion, control KO mice had the greatest overall kaliuresis in the summed response to the diuretics (Fig. 8F).
Fig. 8.
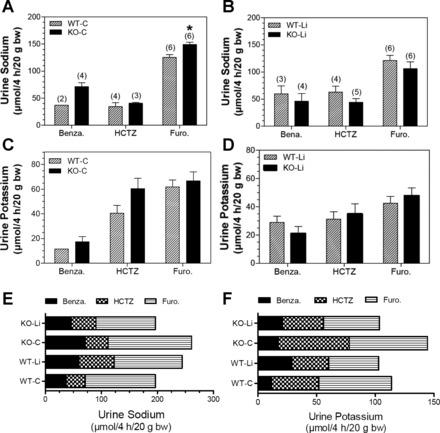
Acute effect of administration of benzamil (Benza), hydrochlorothiazide (HCTZ), or furosemide (Furo) on natriuresis or kaliuresis in WT and KO mice fed control or Li-added diets. Test substances were administered as described in methods, and the Na+ and K+ contents of urine samples collected over 4 h were determined and normalized to body weights of the mice. Although n = 6 mice/group were administered the test substance, however, not all of them voided urine during the test period with each diuretic. The number of mice voided urine during the test period (4 h) in each group with each test substance are shown in parentheses above the bars. A and B: urinary excretion of Na+ (A) and K+ (B) in WT and KO mice fed the control diet. *Significantly different compared with the corresponding treatment in WT mice (P < 0.05). C and D: urinary excretion of Na+ (C) and K+ (D) in WT and KO mice fed the Li-added diet. E and F: Cumulative effect of the three agents used on urinary excretion of Na+ (E) and K+ (F) in WT or KO mice fed control or Li-added diets.
Figure 9 shows the effect of lithium feeding on serum urea levels in WT and KO mice. There were no significant differences in serum urea levels between WT and KO mice fed the control diet. However, lithium caused a modest but significant (P < 0.02) decrease in the serum urea concentration in WT mice. This, in turn, resulted in a significant difference between genotypes (P < 0.04).
Fig. 9.
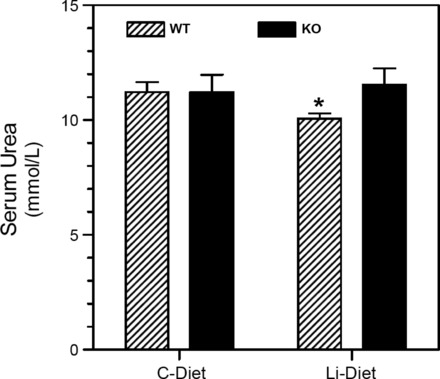
Effect of Li feeding on serum urea levels in WT and KO mice. Groups of WT and KO mice were fed control or Li-added diets for 14 days and euthanized (n = 6 mice/treatment per genotype). Blood was collected by venipuncture at the time of euthanasia. Serum samples were analyzed for urea concentrations as described in methods. *Significantly different from the control diet-fed WT group (P < 0.02 by an unpaired t-test).
DISCUSSION
The most salient feature of our study was that two prominent clinical abnormalities induced by lithium treatment, i.e., natriuresis and kaliuresis, were significantly blunted in P2Y2R KO mice compared with WT mice. The protection afforded KO mice did not appear to involve higher levels of AVP or aldosterone since these hormones (in urine) were numerically lower in KO mice versus WT mice after lithium. Moreover, protein changes in the aldosterone-sensitive distal nephron did not show any increases in KO mice that were larger than observed in WT mice. The one aldosterone-sensitive protein that was increased by lithium (NCC) was actually increased to a greater extent in WT mice. Furthermore, Na+-retentive activity as estimated by the summed natriuretic response to three diuretics would suggest that lithium reduced the Na+-retentive capacity of the distal nephron in KO mice (rather than increased it). Taken together, these findings suggest that the P2Y2R KO mouse is indeed less sensitive to both the natriuresis and kaliuresis accompanying lithium, but we do not have any evidence to support that enhanced aldosterone-sensitive distal nephron Na+ retention is the mechanism. On the other hand, we have not ruled out a role for enhanced Na+ reabsorption by the medullary TAL as providing at least some protection. We will expand on these findings below.
The protection in KO mice is reasonable considering that activity through P2Y2R has been demonstrated to be associated with antagonism of V2 receptor-related activity (16, 30, 36, 37, 43). On the other hand, this was not a certainty given the fact that lithium is thought to enter the collecting duct via reabsorption through ENaC (4), and some studies (27, 28, 32) have shown increased ENaC activity in P2Y2R KO mice that might result in greater vulnerability. In our study, ENaC was either equivalently downregulated or in some instances downregulated to a greater extent in KO mice, e.g., γ-ENaC in the medulla, which might suggest the collecting duct was indeed more sensitive in KO mice. In support of this, our estimation of ENaC activity (using benzamil sensitivity) showed that lithium reduces ENaC activity in KO mice but actually enhances it in WT mice (Fig. 8, A compared with B).
On the other hand, the relatively higher levels of medullary NKCC2 may play a significant role. Greater activity of NKCC2 in the basal state was supported by our finding of increased furosemide sensitivity in KO mice versus WT mice. This notion is also supported by previous studies on renal Na+ handling in P2Y2R KO mice. Rieg et al. (30) reported that P2Y2R KO mice have increased NKCC2 protein abundance in the medulla. In addition, in agreement with our findings, they also showed increased sensitivity to furosemide, but not chlorothiazide or amiloride, in KO mice. These observations suggest that Na+ absorption through NKCC2 may be a major route in mice lacking P2Y2R. This increase in medullary NKCC2 may have resulted in reduced delivery of Na+ to the distal tubule for absorption through NCC or ENaC. It is also important to note, however, that cortical NKCC2 levels were lower in KO mice. Despite this, the cumulative response to furosemide was indeed higher in KO mice (vs. WT mice).
This is also not the first time that we have observed noncoordinate regulation of NKCC2 in the cortex versus medulla in these mice and in others (29, 40). Additional studies will be needed to understand the mechanism underlying these differences, although there are at least three different isoforms of NKCC2 with different affinities and expression patterns along the TAL (3).
The greater capacity to reabsorb Na+ in the TAL is consistent with the blunting of lithium-induced natriuresis in KO mice. However, one piece of the puzzle that does not quite fit with regard to the potential protective role of NKCC2 is the fact that furosemide sensitivity and NKCC2 protein abundance in the medulla fell to a greater extent in KO mice with lithium compared with WT mice.
Consistent with reduced macula densa Na+ delivery and subsequent stimulation of the renin-angiotensin-aldosterone system, urinary aldosterone levels in our P2Y2R KO mice were significantly higher than those of WT mice under basal conditions. This is not in agreement with Rieg et al. (30), who found reduced serum aldosterone in P2Y2R KO mice relative to WT mice. Moreover, we found a significant fall in urine aldosterone in KO mice with lithium therapy. This was opposite to what was observed in WT mice, which responded as expected with elevated urine aldosterone and AVP and increased renal NCC levels, an aldosterone-regulated protein (14). Moreover, we found significantly lower NCC protein levels in KO mice regardless of treatment (relative to WT mice by two-way ANOVA), which is not consistent with the idea of an elevated renin-angiotensin-aldosterone system (14). Additional studies will be needed to fully understand aldosterone regulation in P2Y2R KO mice. In addition, there may be other factors involved in P2Y2R KO mice that can potentially affect the expression of distal nephron transporters and channels. For example, we (42) recently reported that the protein abundance of EP3 receptors in the medulla of P2Y2R KO mice was reduced by 70% compared with WT mice. And there is evidence that PGE2 actions through EP receptors may influence the expression and activity of ENaC (7, 10, 38).
The observations on medullary tissue osmolalities render further support to the above presumption that KO mice have the potential to generate higher interstitial osmotic gradients or sustain them in the face of lithium. In fact, the IM tissue osmolality in control diet-fed KO mice was significantly higher compared with that in control WT mice. To the best of our knowledge, this is the first piece of evidence showing that genetic deletion of P2Y2R results in a significant elevation of IM tissue osmolality. It should be noted that the absorption of Na+ in the collecting duct contributes to only 50% of IM tissue osmolality, whereas the rest of the 50% is contributed by the urea recycling through UT-A and UT-B isoforms across the tubular and vascular structures in the IM (18). Previously, we (43) documented that genetic deletion of P2Y2R results in significant increases in UT-A1 and UT-A2 protein abundances in the medulla, which might increase the urea recycling capability in KO mice. In view of this, further studies by approaches beyond the scope of the present work are needed to establish the contribution of Na+ versus urea transport for the high osmolality of IM tissue in KO mice reported here.
We also evaluated the expression of two major K+ channels, the inwardly rectifying, ATP-sensitive ROMK1 channel (KCNJ1) and the BK channel (KCNMA1), to see if they were affected by lithium and P2Y2R KO. The ROMK channel is expressed in the renal tubule as three isoforms with somewhat differential distribution (39). ROMK1a is expressed in the collecting duct (both the cortical collecting duct and outer medullary collecting duct) and is responsible primarily for K+ secretion. ROMK1b is in the TAL through the cortical collecting duct, and ROMK1c is in the TAL and distal convoluted tubule. In the TAL, ROMK has an important role in K+ recycling and generation of the urinary concentrating gradient. The BK channel is important in flow-mediated K+ secretion in the collecting duct. It also can adaptively increase in response to a K+ diet (26). In the cortex, ROMK was slightly but significantly increased in KO mice. What this means in terms of K+ balance is unclear since aldosterone normally leads to K+ excretion by facilitating secretion in the collecting duct. On the other hand, BK protein was decreased by lithium in KO mice but increased by lithium in WT mice. A reduction in this secretory, flux-activated channel might reduce kaliuresis in KO mice. However, it is not possible to definitively assign cause-and-effect relationships to the change in kaliuresis versus the change in BK channel expression, i.e., reduced flow through the collecting duct may have also reduced BK expression and activity.
The potential possibility of reduced renal blood flow or glomerular filtration rate in KO mice as a contributory factor to the observed reduction in the natriuretic response is also unlikely in view of the data presented by Rieg et al. (30) showing comparable renal blood flow and glomerular filtration rate in KO and WT mice. In addition, we showed that under basal conditions, both WT and KO mice had similar serum urea levels, an index of glomerular filtration rate, and lithium feeding had no impact on serum urea in KO mice. However, lithium feeding caused a modest but significant reduction in serum urea in WT mice. It is known that lithium treatment causes reduced UT-A1 and UT-A3 protein abundance in the IM (for a review, see Ref. 17), which might impair the urea recycling ability of the kidney in lithium-treated animals, which, in turn, may contribute to the modest decrease in serum urea levels in WT mice but not in KO mice. The latter have been shown to have significantly higher UT-A1 and UT-A2 protein abundance, which potentially increase the urea recycling capability of the kidney (43). In view of these, the role of UTs in the observed alterations in serum urea levels in lithium-treated WT and KO mice needs to be investigated by approaches beyond the scope of this work.
Finally, it appears that the observed significant amelioration of lithium-induced polyuria, natriuresis, and kaliuresis is unique to the deletion of P2Y2R. At least two other currently used therapeutic modalities for the amelioration of lithium-induced polyuria, namely, the administration of amiloride or COX-2 inhibition, did not have a significant effect on lithium-induced natriuresis and kaliuresis. In fact, amiloride treatment markedly enhanced lithium-induced natriuresis by 2.4-fold, whereas COX-2 inhibition did not have any effect on urinary Na+ or K+ levels in lithium-treated rats (2, 13, 19). Furthermore, the tendency for overactivity of ENaC and greater expression of other transporters, such as NKCC2 in the TAL, has been suggested to be related to the salt sensitivity of blood pressure found in P2Y2R KO mice (37). Since lithium-induced NDI is associated with low blood pressure, P2Y2R antagonism may represent a novel method to treat the polyuria, natriuresis, kaliuresis, and hypotension associated with lithium therapy. Additional studies are needed to explore the potential therapeutic benefits of purinergic antagonism on lithium-induced NDI.
To summarize, in this communication, we showed 1) genetic deletion of P2Y2R significantly attenuates lithium-induced natriuretic and diuretic responses; 2) the attenuated natriuretic response was not due to enhanced aldosterone-sensitive distal nephron Na+ absorption through ENaC; 3) a role for enhanced Na+ reabsorption by the AVP-sensitive medullary TAL could not be ruled out; 4) a reduction in the secretory, flux-activated BK channel protein might reduce lithium-induced kaliuresis in KO mice, although the cause-and-effect relationship cannot be excluded in our model; and 5) the possibility of reduced renal blood flow or glomerular filtration rate in KO mice as a contributory factor to the observed reduction in natriuretic response is unlikely. While additional studies are warranted to fully unravel the mechanisms involved, at this stage the observed significant attenuation of lithium-induced natriuresis and kaliuresis in P2Y2R KO mice is an indication that purinergic signaling has a much wider and deeper role in renal pathophysiology than what we recognize and currently understand.
GRANTS
This work was supported by the Department of Veterans Affairs Merit Review Project (to B. K. Kishore) and a grant from the National Kidney Foundation of Utah and Idaho (to B. K. Kishore). Additional funding sources include an Established Investigator Award from the American Heart Association (to C. M. Ecelbarger).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.Z., C.M.E., and B.K.K. conception and design of research; Y.Z., L.L., C.M.E., and B.K.K. performed experiments; Y.Z., L.L., C.M.E., and B.K.K. analyzed data; Y.Z., D.E.K., C.M.E., and B.K.K. interpreted results of experiments; Y.Z., L.L., D.E.K., C.M.E., and B.K.K. edited and revised manuscript; Y.Z., L.L., D.E.K., C.M.E., and B.K.K. approved final version of manuscript; C.M.E. and B.K.K. prepared figures; C.M.E. and B.K.K. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Beverly Koller (University of North Carolina at Chapel Hill) for generously supplying breeders of P2Y2 receptor-null and wild-type mice and R. Mayuri Garikepati and Susanna Tsukerman for technical assistance.
REFERENCES
- 1. Baer L, Glassman AH, Kassir S. Negative sodium balance in lithium carbonate toxicity. Arch Gen Psychiatry 29: 823– 827, 1973. [DOI] [PubMed] [Google Scholar]
- 2. Bedford JJ, Leader JP, Jing R, Walker LJ, Klein JD, Sands JM, Walker RJ. Amiloride restores renal medullary osmolytes in lithium-induced nephrogenic diabetes insipidus. Am J Physiol Renal Physiol 294: F812– F820, 2008. [DOI] [PubMed] [Google Scholar]
- 3. Castrop H, Schnermann J. Isoforms of renal Na-K-2Cl cotransporter NKCC2: expression and functional significance. Am J Physiol Renal Physiol 295: F859– F866, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christensen BM, Zuber AM, Loffing J, Stehle JC, Deen PM, Rossier BC, Hummler E. αENaC-mediated lithium absorption promotes nephrogenic diabetes insipidus. J Am Soc Nephrol 22: 252– 261, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ecelbarger CA, Kim GH, Knepper MS, Liu J, Tate M, Welling PA, Wade JB. Regulation of potassium channel Kir 1.1 (ROMK) abundance in the thick ascending limb of Henle's loop. J Am Soc Nephrol 12: 10– 18, 2001. [DOI] [PubMed] [Google Scholar]
- 6. Ecelbarger CM, Zhang Y, Garikepati RM, Kishore B. Role of the purinergic P2Y2 receptor in the regulation of the renal epithelial sodium channel and other sodium transporters in lithium-treated rats (Abstract). FASEB J 25: 1039 24, 2011. [Google Scholar]
- 7. Els WJ, Helman SI. Dual role of prostaglandins (PGE2) in regulation of channel density and open probability of epithelial Na+ channels in frog skin (R. pipiens). J Membr Biol 155: 75– 87, 1997. [DOI] [PubMed] [Google Scholar]
- 8. Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890– F897, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galla JN, Forrest JN, Hecht B, Kashgarian M, Hayslett JP. Effect of lithium on water and electrolyte metabolism. Yale J Biol Med 48: 305– 314, 1975. [PMC free article] [PubMed] [Google Scholar]
- 10. González AA, Céspedes C, Villanueva S, Michea L, Vio CP. E prostanoid-1 receptor regulates renal medullary αENaC in rats infused with angiotensin II. Biochem Biophys Res Commun 389: 372– 377, 2009. [DOI] [PubMed] [Google Scholar]
- 11. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol 5: 270– 276, 2009. [DOI] [PubMed] [Google Scholar]
- 12. Hirschowitz J, Kolevzon A, Garakani A. The pharmacological treatment of bipolar disorder: the question of modern advances. Harv Rev Psychiatry 18: 266– 278, 2010. [DOI] [PubMed] [Google Scholar]
- 13. Kim GH, Choi NW, Jung JY, Song JH, Lee CH, Kang CM, Knepper MA. Treating lithium-induced nephrogenic diabetes insipidus with a COX-2 inhibitor improves polyuria via upregulation of APQ2 and NKCC2. Am J Physiol Renal Physiol 294: F702– F709, 2008. [DOI] [PubMed] [Google Scholar]
- 14. Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 95: 14552– 14557, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kishore BK, Ecelbarger CM. Lithium: a versatile tool to understand renal physiology. Am J Physiol Renal Physiol 304: F1139– F1149, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kishore BK, Nelson RD, Miller RL, Carlson NG, Kohan DE. P2Y2 receptors and water transport in the kidney. Purinergic Signal 5: 491– 499, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klein JD, Gunn RB, Roberts BR, Sands JM. Molecular mechanisms of urea transport in health and disease. Pflügers Arch 474: 561– 572, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koeppen BM, Stanton BA. Renal Physiology (4th ed.). Maryland Heights, MO: Mosby Elsevier, 2007, p. 85. [Google Scholar]
- 19. Kortenoeven MLA, Li Y, Shaw S, Gaeggeler HP, Rossier BC, Wetzels JFM, Deen PMT. Amiloride blocks lithium entry through sodium channel thereby attenuating the resultant nephrogenic diabetes insipidus. Kidney Int 76: 44– 53, 2009. [DOI] [PubMed] [Google Scholar]
- 20. Kwon TH, Laursen UH, Marples D, Maunsbach AB, Knepper MA, Frøkiær J, Nielsen S. Altered expression of renal AQPs and Na+ transporters in rats with lithium-induced NDI. Am J Physiol Renal Physiol 279: F552– F564, 2000. [DOI] [PubMed] [Google Scholar]
- 21. Li L, Grikepati M, Tsukerman S, Kohan D, Wade JB, Tiwari S, Ecelbarger CM. Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. Am J Physiol Renal Physiol 304: F279– F288, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Listhrop R, Nelson RD, Ecelbarger CA, Kohan DE, Kishore BK. Genetic deletion of P2Y2 receptor alters the protein abundances of proximal tubular and thick ascending limb transporters (Abstract). FASEB J 21: 1328A, 2007. [Google Scholar]
- 23. Luckey AE, Parsa CJ. Fluid and electrolytes in the aged. Arch Surg 138: 1055– 1060, 2003. [DOI] [PubMed] [Google Scholar]
- 24. Nivoli AM, Murru A, Vieta E. Lithium: still a cornerstone in the long-term treatment in bipolar disorder? Neuropsychobiology 62: 27– 35, 2010. [DOI] [PubMed] [Google Scholar]
- 25. Phelan KM, Mosholder AD, Lu S. Lithium interaction with the cyclooxygenase-2 inhibitors refecoxib and celecoxib and other non-steroidal anti-inflammatory drugs. J Clin Psychiatry 64: 1328– 1334, 2003. [DOI] [PubMed] [Google Scholar]
- 26. Pluznick JL, Sansom SC. BK channels in the kidney: role in K+ secretion and localization of molecular components. Am J Physiol Renal Physiol 291: F517– F529, 2006. [DOI] [PubMed] [Google Scholar]
- 27. Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinerigic P2Y2 receptor tone. J Biol Chem 283: 36599– 36607, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na+ inhibits the open probability of epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J 24: 2056– 2065, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riazi S, Khan O, Tiwari S, Hu X, Ecelbarger CA. Rosiglitazone regulates ENaC and Na-K-2Cl contransporter (NKCC2) in the obese Zucker rat. Am J Nephrol 26: 245– 257, 2006. [DOI] [PubMed] [Google Scholar]
- 30. Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, Vallon V. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J 21: 3717– 3726, 2007. [DOI] [PubMed] [Google Scholar]
- 31. Schmidt-Nielsen B, Graves B, Roth J. Water removal and solute additions determining increases in renal medullary osmolality. Am J Physiol Renal Fluid Electrolyte Physiol 244: F472– F482, 1983. [DOI] [PubMed] [Google Scholar]
- 32. Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, Peti-Peterdi J, Pochynyuk O. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol 21: 1903– 1911, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thompson L, Morgan T, Carney S. Lithium administration and urinary electrolyte excretion in the rat. Miner Electrolyte Metab 10: 122– 126, 1984. [PubMed] [Google Scholar]
- 34. Thomsen K, Jensen J, Olesen OV. Effect of prolonged lithium ingestion on the response to mineralocorticoids in rats. J Pharmacol Exp Ther 196: 463– 468, 1976. [PubMed] [Google Scholar]
- 35. Timmer RT, Sands JM. Lithium intoxication. J Am Soc Nephrol 10: 666– 674, 1999. [DOI] [PubMed] [Google Scholar]
- 36. Vallon V. P2 receptors in the regulation of renal transport mechanisms. Am J Physiol Renal Physiol 294: F10– F27, 2008. [DOI] [PubMed] [Google Scholar]
- 37. Vallon V, Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol 301: F463– F475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang S, Meng F, Xu J, Gu Y. Effects of lipids on ENaC activity in cultured mouse cortical collecting duct cells. J Membr Biol 227: 77– 85, 2009. [DOI] [PubMed] [Google Scholar]
- 39. Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297: F849– F863, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Listhrop R, Ecelbarger CM, Kishore BK. Renal sodium transporter/channel expression and sodium excretion in P2Y2 receptor knockout mice fed a high-NaCl diet with/without aldosterone infusion. Am J Physiol Renal Physiol 300: F657– F668, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y, Nelson RD, Carlson NG, Kamerath CD, Kohan DE, Kishore BK. Potential role of purinergic signaling in lithium-induced nephrogenic diabetes insipidus. Am J Physiol Renal Physiol 296: F1194– F1201, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, Pop IL, Carlson NG, Kishore BK. Genetic deletion of the P2Y2 receptor offers significant resistance to development of lithium-induced polyuria accompanied by alterations in PGE2 signaling. Am J Physiol Renal Physiol 302: F70– F77, 2012. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y, Sands JM, Kohan DE, Nelson RD, Martin CF, Carlson NG, Kamerath CD, Ge Y, Klein JD, Kishore BK. Potential role of purinergic signaling in urinary concentration in inner medulla: insights from P2Y2 receptor gene knockout mice. Am J Physiol Renal Physiol 295: F1715– F1724, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]