

Abstract
Background
Mutations in Surfactant Protein C (SFTPC) can lead to fibrotic interstitial lung disease (ILD) with variable phenotypes, especially in children. The sources of phenotype variability are incompletely understood. A common MUC5B promoter variant rs35705950 is associated with adult Idiopathic Pulmonary Fibrosis (IPF). We examined whether MUC5B is similarly linked to ILD secondary to SFTPC mutations.
Methods
MUC5B concentration in bronchoalveolar lavage fluid (BALF) was measured in six pediatric patients with SFTPC mutations and diseased controls. Immunohistochemical localization of MUC5B was studied in fixed lung tissues in patients with SFTPC mutations, ABCA3 mutations, and controls. Genotyping for the MUC5B promoter variant rs35705950 was attempted in all samples.
Results
MUC5B glycoprotein was increased in BALF of patients with SFTPC mutations compared to diseased controls (P = 0.04). MUC5B was unexpectedly present in cells morphologically consistent with alveolar epithelial type II cells in patients with SFTPC mutations in the BRICHOS domain. Genotyping for the MUC5B promoter variant was successful in 18/27 patients, and there was no significant relationship between the MUC5B promoter variant and the BALF or MUC5B localization.
Conclusion
MUC5B may play a role in the development of fibrosis in patients with SFTPC mutations, especially in patients with BRICHOS mutations. Understanding the role of MUC5B in adult and pediatric lung diseases may lead to a better understanding of the etiology of fibrotic lung disease as well as development of novel therapies.
Keywords: surfactant, interstitial lung disease, mucins, pulmonary fibrosis, pediatrics, single nucleotide polymorphism, rare diseases, bronchoalveolar lavage
Introduction
Surfactant Protein C (SFTPC) mutations produce a rare type of children’s interstitial and diffuse lung disease (chILD) with a range of pathological findings including interstitial fibrosis. Patients can present with neonatal respiratory distress or as older adults with fibrosis.1,2 Different mutations account for the variability of disease between families, with mutations in the BRICHOS domain (a highly conserved region involved in protein folding) leading to more severe lung disease.3 The heterogeneity of phenotypes within families suggests variable penetrance and/or environmental or other genetic modifiers.1,4 Previous studies suggested that the mutations in SFTPC lead to endoplasmic reticulum (ER) stress, epithelial cell dysfunction, and inflammation.1,5–9 In other fibrotic lung diseases such as idiopathic pulmonary fibrosis (IPF), ER stress is thought to lead to epithelial-mesenchymal transition and ultimately fibrosis.10
MUC5B, a respiratory tract mucin, is typically located in the submucosal glands in normal bronchioles, but histological evidence in patients with IPF shows dysregulated, increased, and non-patchy MUC5B expression in the distal airways and honeycomb cysts.11–13 A common polymorphism in the promoter region of MUC5B (rs35705950) is strongly associated with familial and sporadic IPF14–18 but the MUC5B histologic localization appear to be characteristic of IPF and independent of the polymorphism.12,19 MUC5B is also increased in both mild and fatal asthma, most notably in the mucus plugs, but MUC5B was not found in the alveolar epithelial cells of asthmatic patients.20
We hypothesized that MUC5B protein expression is increased in fibroproliferative forms of pediatric lung disease, such as SFTPC mutations. In this pilot study, we evaluated MUC5B concentrations in bronchoalveolar lavage fluid (BALF) and MUC5B expression in lung tissue from pediatric patients with SFTPC mutations.
Materials and Methods
Two groups of patients were included in this study: patients who underwent bronchoalveolar lavage (15 patients, 6 with SFTPC mutations and 9 diseased controls) and patients who underwent lung biopsies (15 patients, 6 with SFTPC mutations, 3 with ABCA3 mutations, and 6 age matched controls). Three patients with SFTPC mutations underwent both bronchoalveolar lavage and surgical lung biopsy.
Bronochoalveolar lavage
Bronchoalveolar lavage fluid samples were obtained from children undergoing clinically indicated bronchoscopy (Table 1). These samples were processed and banked in the Colorado Pediatric Lung Bank under an IRB approved protocol with parental consent. The BALF was spun down and the supernatant was frozen.21 BALF from all patients with SFTPC mutations with banked BALF as well as diseased controls (patients with Neuroendocrine Cell Hyperplasia of Infancy (NEHI) and patients with chronic cough) were studied. A slot blot assay was used to determine MUC5B concentrations.22 A standard curve was used to control for variability between runs. Samples were pre-tested to determine the appropriate concentrations for the standard curve. Samples were dissolved in denaturing and reducing loading buffer, loaded into a 48-well slot blot manifold (Hoefer, Holliston, MA), and applied to a nitrocellulose membrane by vacuum (15 mm Hg). Membranes were then washed, blocked, and immunolabeled with anti-MUC5B antisera (Santa Cruz Biotechnology, Santa Cruz, CA) as described previously.23 Labels were detected using horseradish peroxidase chemiluminescence detection and were detected using Chemidoc gel imaging system image acquisition and Image One 1-D analysis software (BIO-RAD, Hercules, CA). Data are presented as quantities in arbitrary units.
Table 1.
Demographics, MUC5B density, and genotyping of patients included in BALF (bronchoalveolar lavage) analysis. Genotype is for the MUC5B promoter variant. GG = wildtype genotype, GT = heterozygous for the variant, and ? was unable to be obtained. (Note: patients 11, 12, and 15 correspond to patients 2, 6, and 4, respectively in Table 2)
| Patient # | Sex | Disease | Age at BALF | Culture | MUC5B Density | Genotype |
|---|---|---|---|---|---|---|
| 1 | F | control | 7 yr | no growth | 52402 | GG |
| 2 | F | control | 1 yr | Haemophilus | 120194 | GG |
| 3 | M | control | 5 yr | no growth | 37021 | GG |
| 4 | M | control | 1 yr | no growth | 78313 | ? |
| 5 | M | control | 11 yr | Gram Positive Cocci, no growth Eikenella, Streptococcus, | 64851 | ? |
| 6 | F | control | 1 yr | Moraxella | 153022 | ? |
| 7 | M | NEHI | 2 yr | H flu | 61227 | ? |
| 8 | F | NEHI | 1 yr | no growth | 73453 | ? |
| 9 | F | NEHI | 8 mo | no growth | 59842 | ? |
| 10 | F | SPC | 10 yr | no growth | 297059 | GG |
| 11 | M | SPC | 3 yr | Streptococcus | 90183 | GG |
| 12 | F | SPC | 1 yr | Moraxella | 474358 | GT |
| 13 | F | SPC | 2 yr | no growth | 213461 | GG |
| 14 | M | SPC | 3 yr | no growth | 268073 | GT |
| 15 | F | SPC | 3 yr | no growth | 170516 | GG |
Immunohistochemistry
15 patients were included in the evaluation of MUC5B localization: 6 patients with SFTPC mutations, 3 patients with ABCA3 mutations, and 6 age matched controls. Controls consisted of children who underwent surgical lung biopsy for a focal lesion with surrounding normal lung tissue. These specimens were reviewed under a retrospective IRB for which consent was waived. Five micron thick sections were cut from paraffin embedded blocks with surgical lung biopsy specimens. Immunohistochemistry was performed per standard protocols using a polyclonal rabbit-anti-human MUC5B primary antibody diluted to 1:500 (Santa Cruz Biotechnology). Tissues were stained with DAB and counterstained with Mayer’s hematoxilyn. An expert lung pathologist who was blinded to the underlying disease reviewed the slides for abnormal location of MUC5B (i.e. MUC5B not in the submucosal glands of the airways) and ultimately categorized them by positive MUC5B staining in cells morphologically consistent with alveolar epithelial type II cells or no MUC5B staining in these cells.
MUC5B genotype
We performed genotyping for rs35705950 using a Taqman assay, as previously described12,19 on the BALF and on small scrapings of tissue slides.
Statistics
A one-way ANOVA with median scores for the nonparametric linear rank statistic was employed to test the hypothesis that the median values of MUC5B density levels from each group are equal; due to small samples, exact p-values are reported. Secondary analyses were performed with a one-way ANOVA to investigate differences between BALF findings in patients with SFTPC mutations and with diseased controls.
Results
Bronchoalveolar Lavage
We analyzed BALF samples from 9 females and 6 males (Table 1) via a slot blot assay (Figure 1). The age at time of BALF ranged from 8 months to 11 years. Microrganisms grew in 5 BALF samples, including Haemophilus, Eikenella, Streptococcus, and Moraxella. An additional sample had Gram positive cocci, but no growth.
Figure 1. Slot Blot results for MUC5B density by disease.
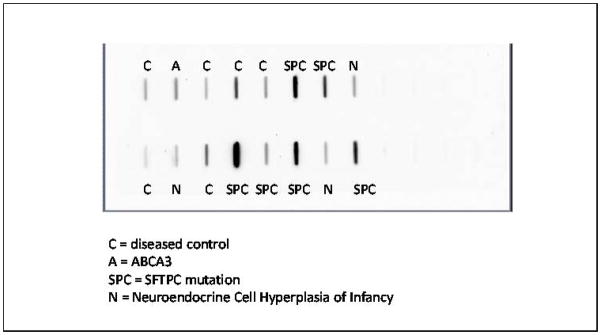
Darkness of line indicates increased density of MUC5B levels.
In patients with SFTPC mutations, the MUC5B concentration in BALF was significantly higher than in either patients with NEHI or other diseased controls (p = 0.04; Figure 2A and 2B).
Figure 2.

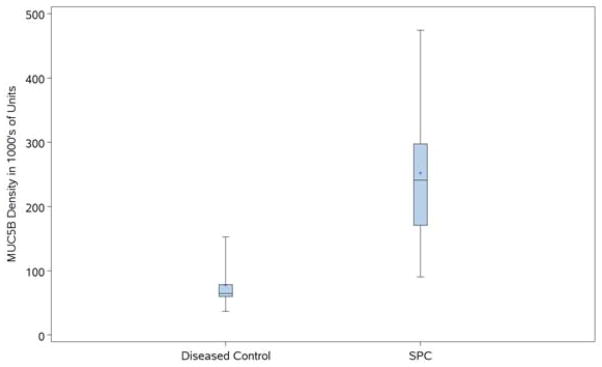
Figure 2A. MUC5B density in diseased controls and SFTPC mutations by culture. MUC5B density by disease group (controls with chronic cough, SFTPC with SFTPC mutations, and NEHI). Closed squares indicate positive cultures; open diamonds indicate negative cultures.
Figure 2B. MUC5B density in diseased controls (chronic cough and NEHI) compared to SFTPC mutations. MUC5B density was significantly higher in SFTPC mutation patients compared to diseased controls (p=0.0406).
Genotyping for the MUC5B promoter variant was successful in 9/15 samples and in only 3/6 patient with SFTPC mutations. Two patients were heterozygous for the MUC5B promoter variant and the remaining patients were either wildtype or unable to be obtained.
Correspondingly, white blood cell counts were also significantly higher in BALF from patients with SFTPC mutations (Supplemental Table 1). There were statistically significant increased percentages of neutrophils and eosinophils in BALF of patients with SFTPC mutations (Supplement Table 1). In all BALF, white blood cells correlated with log (MUC5B density) with a correlation of 0.63 (p=0.012)
Lung Tissue
Tissue was evaluated in 15 patients (Table 2). Each patient with a SFTPC mutation had a unique mutation. Four had mutations in the BRICHOS domain and two did not. MUC5B was located in typical regions of the tissue, such as submucosal glands, columnar airway epithelium, and in mucus plugs (Supplement Figure 1). However, in patients with SFTPC mutations, there was MUC5B staining in cells morphologically consistent with alveolar epithelial type II cells in 4/6 patients and 4/4 patients with BRICHOS mutations (Table 2). The positive staining was heterogeneous in the patients with BRICHOS mutations, with some foci of positively stained areas of cells morphologically consistent with alveolar epithelial type II cells (Figure 3). Supplement Figure 2 shows mildly positive MUC5B staining in the some of the alveolar macrophages for patients 7–15. These patients had no abnormally located MUC5B.
Table 2.
Demographics, immunohistochemistry for MUC5B, and MUC5B genotype. “AEII” represents cells morphologically consistent with alveolar epithelial type II cells. Genotype is for the MUC5B promoter variant. GG = wildtype genotype and GT = heterozygous for the variant. (Note: patients 2, 4, and 6 correspond to patients 11, 15, and 12, respectively in Table 1)
| Patient # | Age at biopsy | Disease state | Mutation | Interstitial Fibrosis | Brichos | MUC5B in “AEII” | Genotype |
|---|---|---|---|---|---|---|---|
| 1 | 2 months | SPC | G182R | no | yes | yes | GG |
| 2 | 2 months | SPC | c460+1G>A | no | yes | yes | GG |
| 3 | 3 months | SPC | 100Glu->Val and 103Leu->Val | no | no | no | GT |
| 4 | 5 months | SPC | 140delA | no | yes | yes | GG |
| 5 | 16 months | SPC | I73T | no | no | no | GG |
| 6 | 22 months | SPC | L188Q L->A | yes | yes | yes | GT |
| 7 | 21 days | ABCA3 | no | no | GG | ||
| 8 | 2 months | ABCA3 | no | no | GG | ||
| 9 | 16 days | ABCA3 | no | no | GG | ||
| 10 | 15 months | control | no | no | GG | ||
| 11 | 5 months | control | no | no | GG | ||
| 12 | 11 months | control | no | no | GG | ||
| 13 | 23 months | control | no | no | GG | ||
| 14 | 3 months | control | no | no | GG | ||
| 15 | 7 months | control | no | no | GG |
SPC = patients with SFTPC mutations, ABCA3 = patients with ABCA3 mutations
Figure 3. Immunohistochemical staining of MUC5B in lung tissue from four patients with SFTPC mutations.

Immunohistochemical staining reveal positive MUC5B staining in alveolar epithelial type II cells (black arrow). Alveolar epithelial cells without MUC5B staining, demonstrating patchiness (gray arrow). MUC5B staining positive in macrophages in airspaces (white arrow).
Genotyping for the MUC5B promoter variant was successful in all samples. Two of the cases with SFTPC mutations were heterozygotes for the MUC5B promoter variant and the remaining cases were wildtype.
Tissue was evaluated for fibrosis by both hematoxylin and eosin stain (Supplement Figure 3) and by Movat pentachrome stain. Patient 6 (SFTPC with mutation in the BRICHOS domain) was the only patient with interstitial fibrosis. Other patients had remodeling or reactive changes, but no significant interstitial fibrotic changes.
Discussion
Pulmonary fibrosis develops in patients with SFTPC mutations. In IPF, increased MUC5B expression is thought to impair mucus clearance, alveolar repair, and interfere with host defense.12 MUC5B expression may be increased because of a polymorphism in its promoter region or because of the underlying disease process itself. We suggest that increased MUC5B expression may be associated with an acceleration of the fibroproliferative pathway in patients with SFTPC mutations.
Our data demonstrated that the concentration of MUC5B in BALF is increased in patients with SFTPC mutations compared to diseased controls. As shown in Figure 2A, the patients with SFTPC mutations did not have higher MUC5B density because of bacterial infections leading to more mucus. However, the two controls with the highest MUC5B density did have bacterial growth, and therefore may have had a higher MUC5B density secondary to infection.
Immunohistochemistry for MUC5B in lung tissue demonstrated MUC5B located in cells morphologically consistent with alveolar epithelial type II cells in patients with SFTPC mutations, specifically in patients with protein trafficking issues (i.e. mutations in the BRICHOS domain). This abnormally localized MUC5B is focal; each slide only had 1–5 positive areas. Because the staining for MUC5B was patchy and our biopsy specimens were small, it is possible that abnormally located MUC5B was present in other patient samples. We propose that the MUC5B staining is patchy because of the early stage of disease in our patients with SFTPC mutations, especially as only one patient—the oldest biopsied patient with a surfactant dysfunction mutation—had significant interstitial fibrosis. The patient with fibrosis had positive MUC5B staining in cells morphologically consistent with alveolar epithelial type II cells and the highest MUC5B concentration in BALF. The additional patients with both tissue and BALF had abnormally located MUC5B, and surprisingly had the two lowest MUC5B densities in BALF of the patients with SFTPC mutations (Tables 1 and 2). However, these MUC5B density values were still higher than the average MUC5B density of the controls and than all non-infected controls.
Alveolar epithelial cells normally do not produce mucins, thus MUC5B in cells morphologically consistent with alveolar epithelial type II cells of patients with SFTPC mutations in the BRICHOS domain represents a novel pathological finding.
Because a MUC5B promoter variant is a risk factor for developing IPF,12,14–19 MUC5B genotyping was attempted in all samples to determine if this promoter variant might explain abnormal MUC5B staining. Genotyping was successful in 18/27 individuals. BALF supernatant contains minimal DNA and therefore it was not surprising that it was difficult to genotype the BALF. While the sample size was small, there was no evidence of a relationship between MUC5B promoter variant and positive MUC5B staining in cells morphologically consistent with alveolar epithelial type II cells. The patient with fibrosis and the highest concentration of MUC5B in BALF and notably was heterozygous for the MUC5B promoter variant.
MUC5B expression is increased in the lungs of patients with IPF and a single nucleotide polymorphism in the MUC5B promoter variant is associated with increased risk of disease and improved outcome.12,19,24 Efforts are underway to better understand how this polymorphism in MUC5B contributes to lung fibrosis in IPF and to develop agents to target MUC5B. If MUC5B is confirmed as a disease modifier/contributor in chILD, then these novel agents could be tested in pediatric fibrotic lung disease. This is the first time that MUC5B expression has been evaluated in pediatric lung disease generally and SFTPC mutations specifically, and due to our small sample size, we are currently unable to directly link rare or even common variants in MUC5B to pediatric ILD. Indeed, studying a rare disease has obvious limitations, and this pilot study was limited by the number of patients with disease. Given the limited samples, we were unable to control for age, BALF culture results, MUC5B promoter variant, or sex. An additional limitation is that alveolar epithelial type II cells were identified morphologically and no staining for alveolar epithelial type II cell markers was completed.
Nevertheless, our data do demonstrate that MUC5B levels in BALF are increased in patients with SFTPC mutations compared to diseased controls and that MUC5B is abnormally expressed in lung tissue with SFTPC mutations in the BRICHOS domain. These initial findings suggest a role for MUC5B in pediatric fibrotic lung disease that may inform pathogenesis or function as a diagnostic biomarker as well as provide future, novel therapeutics for these rare lung diseases.
Supplementary Material
Supplement Figure 1: Normal MUC5B staining in airway submucosal glands and goblet cells.
Supplement Figure 2: Immunohistochemical staining of MUC5B in lung tissue from patients with ABCA3 mutations (Patients 7–9) and controls (Patients 10–15). Mildly positive staining of macrophages in airspaces.
Supplement Figure 3: Hematoxylin and eosin staining of patients 1–9 (patients with surfactant dysfunction mutations.
WBC = white blood cells. PMNs = neutrophils. Lymphs = lymphocytes. Eos = eosinophils. Monos = monocytes. Mphages = macrophages.
Acknowledgments
Supported by Academic Training Program in Pediatric Pulmonary Disease 5T32HL007670 (D.R.L.) and NIH-NHLBI: P01-HL092870 (D.A.S), NIH-NHLBI: R01-HL097163 (D.A.S.), and VAMC: 1I01BX001534 (D.A.S.).
Footnotes
This research was presented at the American Thoracic Society Annual Meeting, San Diego, CA. 5/18/2014 Word Count: 1899
References
- 1.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–8. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 2.Abou Taam R, Jaubert F, Emond S, Le Bourgeois M, Epaud R, Karila C, Feldmann D, Scheinmann P, de Blic J. Familial interstitial disease with I73T mutation: A mid- and long-term study. Pediatr Pulmonol. 2009;44:167–75. doi: 10.1002/ppul.20970. [DOI] [PubMed] [Google Scholar]
- 3.Thouvenin G, Abou Taam R, Flamein F, Guillot L, Le Bourgeois M, Reix P, Fayon M, Counil F, Depontbriand U, Feldmann D, et al. Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch Dis Child. 2010;95:449–54. doi: 10.1136/adc.2009.171553. [DOI] [PubMed] [Google Scholar]
- 4.Hallik M, Annilo T, Ilmoja ML. Different course of lung disease in two siblings with novel ABCA3 mutations. Eur J Pediatr. 2013:1–4. doi: 10.1007/s00431-013-2087-3. [DOI] [PubMed] [Google Scholar]
- 5.Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol. 2005;67:663–96. doi: 10.1146/annurev.physiol.67.040403.101937. [DOI] [PubMed] [Google Scholar]
- 6.Thurm T, Kaltenborn E, Kern S, Griese M, Zarbock R. SFTPC mutations cause SP-C degradation and aggregate formation without increasing ER stress. Eur J Clin Invest. 2013;43:791–800. doi: 10.1111/eci.12107. [DOI] [PubMed] [Google Scholar]
- 7.Bridges JP, Wert SE, Nogee LM, Weaver TE. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem. 2003;278:52739–46. doi: 10.1074/jbc.M309599200. [DOI] [PubMed] [Google Scholar]
- 8.Maguire JA, Mulugeta S, Beers MF. Endoplasmic reticulum stress induced by surfactant protein C BRICHOS mutants promotes proinflammatory signaling by epithelial cells. Am J Respir Cell Mol Biol. 2011;44:404–14. doi: 10.1165/rcmb.2009-0382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci. 2003;116:683–92. doi: 10.1242/jcs.00267. [DOI] [PubMed] [Google Scholar]
- 10.Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech. 2013;6:9–17. doi: 10.1242/dmm.010736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, Schwarz MI, Schwartz DA, Reynolds SD. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8:e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–12. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma P, Dudus L, Nielsen PA, Clausen H, Yankaskas JR, Hollingsworth MA, Engelhardt JF. MUC5B and MUC7 are differentially expressed in mucous and serous cells of submucosal glands in human bronchial airways. Am J Respir Cell Mol Biol. 1998;19:30–7. doi: 10.1165/ajrcmb.19.1.3054. [DOI] [PubMed] [Google Scholar]
- 14.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, Russell AM, Denton CP, Abraham DJ, Hansell DM, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68:436–41. doi: 10.1136/thoraxjnl-2012-201786. [DOI] [PubMed] [Google Scholar]
- 16.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–17. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, Airo P, Matucci-Cerinic M, Wallaert B, Israel-Biet D, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8:e70621. doi: 10.1371/journal.pone.0070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei R, Li C, Zhang M, Jones-Hall YL, Myers JL, Noth I, Liu W. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. 2014;163:494–502. doi: 10.1016/j.trsl.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–7. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groneberg DA, Eynott PR, Lim S, Oates T, Wu R, Carlstedt I, Roberts P, McCann B, Nicholson AG, Harrison BD, et al. Expression of respiratory mucins in fatal status asthmaticus and mild asthma. Histopathology. 2002;40:367–73. doi: 10.1046/j.1365-2559.2002.01378.x. [DOI] [PubMed] [Google Scholar]
- 21.Sagel SD, Sontag MK, Accurso FJ. Relationship between antimicrobial proteins and airway inflammation and infection in cystic fibrosis. Pediatr Pulmonol. 2009;44:402–9. doi: 10.1002/ppul.21028. [DOI] [PubMed] [Google Scholar]
- 22.AMW, Brown KK, Thornton DJ, Rousseau K, Evans CM, Schwartz DA. American Thoracic Society. San Diego, CA: American Journal of Respiratory and Critical Care Medicine; 2014. Serum Muc5b, A Potential Novel Diagnostic Biomarker for Idiopathic Pulmonary Fibrosis. [Google Scholar]
- 23.Piccotti L, Dickey BF, Evans CM. Assessment of intracellular mucin content in vivo. Methods Mol Biol. 2012;842:279–95. doi: 10.1007/978-1-61779-513-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, Steele MP, Loyd JE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–9. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1: Normal MUC5B staining in airway submucosal glands and goblet cells.
Supplement Figure 2: Immunohistochemical staining of MUC5B in lung tissue from patients with ABCA3 mutations (Patients 7–9) and controls (Patients 10–15). Mildly positive staining of macrophages in airspaces.
Supplement Figure 3: Hematoxylin and eosin staining of patients 1–9 (patients with surfactant dysfunction mutations.
WBC = white blood cells. PMNs = neutrophils. Lymphs = lymphocytes. Eos = eosinophils. Monos = monocytes. Mphages = macrophages.