

Abstract
Cdc42 (cell division control protein 42) and Rac1 (Ras-related C3 botulinum toxin substrate 1) are attractive therapeutic targets in ovarian cancer based on established importance in tumor cell migration, adhesion and invasion. Despite a predicted benefit, targeting GTPases has not yet been translated to clinical practice. We previously established that Cdc42 and constitutively active Rac1b are overexpressed in primary ovarian tumor tissues. Through high throughput screening and computational shape homology approaches we identified R-ketorolac as a Cdc42 and Rac1 inhibitor; distinct from the anti-inflammatory, cyclooxygenase inhibitory activity of S-ketorolac. In the present study, we establish R-ketorolac as an allosteric inhibitor of Cdc42 and Rac1. Cell-based assays validate R-ketorolac activity against Cdc42 and Rac1. Studies on immortalized human ovarian adenocarcinoma cells (SKOV3ip), and primary, patient-derived ovarian cancer cells show R-ketorolac is a robust inhibitor of growth factor or serum dependent Cdc42 and Rac1 activation with a potency and cellular efficacy similar to small molecule inhibitors of Cdc42 (CID2950007/ML141) and Rac1 (NSC23766). Furthermore, GTPase inhibition by R-ketorolac reduces downstream p21-activated kinases (PAK1/PAK2) effector activation by >80%. Multiple assays of cell behavior using SKOV3ip and primary patient-derived ovarian cancer cells show that R-ketorolac significantly inhibits cell adhesion, migration and invasion. In sum, we provide evidence for R-ketorolac as direct inhibitor of Cdc42 and Rac1 that is capable of modulating downstream GTPase-dependent, physiological responses, which are critical to tumor metastasis. Our findings demonstrate the selective inhibition of Cdc42 and Rac1 GTPases by an FDA approved drug-racemic ketorolac that can be used in humans.
Keywords: enantiomer-selective, non-steroidal anti-inflammatory drug (NSAID), Rho GTPases, p21-activated kinases (PAK), effector-binding assay
Introduction
Ovarian cancer is the leading cause of gynecological cancer deaths in the United States with a 45% 5-year survival rate (1). Often associated with malignant ascites, which facilitates metastasis, most patients are diagnosed at advanced stages with dissemination throughout the intraperitoneal cavity (2). Recent understanding of the etiology of “ovarian cancer” suggests that the majority of serous ovarian cancers originate from dysplastic fallopian tubes (3). Metastatic spread of ovarian cancer beyond the ovary or fallopian tube is mediated by surface shedding of tumor cells and formation of free-floating multicellular aggregates in the ascites fluid. Subsequent mesothelial anchoring and invasion of the submesothelial matrix leads to secondary lesions on peritoneal organs and surfaces (4). Consequently, the first line of treatment involves major debulking surgery to remove as much tumor burden as possible, most often followed by intraperitoneal chemotherapeutic treatment in optimally debulked patients after recovery from surgery. Therefore, targeting tumor cell adhesion, migration and invasion in the perioperative period, during which tumor dissemination may occur and impact ovarian cancer recurrence, presents an important potential window of opportunity for treatment that has not yet been extensively explored (5, 6).
It is well established that Rho-family (Ras homolog) GTPases are central to dynamic actin cytoskeletal assembly and rearrangement that are the underpinnings of normal cell-cell adhesion, cell migration and even transformation (7). Among the Rho-family GTPases, Cdc42 (cell division control protein 42) and Rac1 (Ras-related C3 botulinum toxin substrate 1) are of particular interest on account of their frequent overexpression or hyperactivation in epithelial cancers including ovarian cancer (8-12). In response to extracellular stimuli, Cdc42 and Rac1 GTPase transition from an inactive GDP-bound status to an active GTP-bound status and interact with downstream effectors to propagate changes in cell behaviors (7). For example, activated Cdc42 and Rac1 phosphorylate p21-activated kinases (PAK1/PAK2) which in turn initiate actin reorganization and subsequently regulate cell adhesion, migration and invasion (13). Furthermore, the Rac1-PAK signaling axis lies immediately downstream of growth factor activated Ras signaling and in ovarian cancer has been shown to modulate epithelial to mesenchymal transition (EMT) and angiogenesis (14, 15). Therefore, it is of interest to investigate the utility of Cdc42 and Rac1 as points of intervention in the intraperitoneal spread of ovarian cancer.
While Cdc42 and Rac1 are potential high value, therapeutic targets in ovarian and other cancers, key gaps remain in translating known GTPase inhibitors into clinical application. Statins are HMG-CoA reductase inhibitors that indirectly act as broad spectrum GTPase inhibitors by interfering with the prenylation required for functionally essential membrane association (16). Cancer risk reductions associated with statin use are reported for breast, colorectal, and pancreatic cancers (17-19). The benefit in ovarian cancer is mixed and subtype specific (20), motivating the identification and testing of more selective GTPase inhibitors. Several GTPase targeted inhibitors have been identified with various modes of action. Yet, irrespective of whether they affect signaling (e.g. NSC23766, secramine) (21, 22), nucleotide binding ability (e.g. CID2950007 (PubChem compound identification for Cdc42 GTPase inhibitor)) (23), or effector binding activity (e.g. Y27632) (24), none have as yet been translated to human use.
Using a combination of high throughput screening and computational simulation, the R-enantiomer of ketorolac was identified as a selective Cdc42 and Rac1 inhibitor, without effect on RhoA (25). Ketorolac is clinically administrated as a racemic mixture for pain relief, based on the activity of S-ketorolac as a dual (COX-1/COX-2) inhibitor (26). The R-enantiomer was considered inert prior to our identification of its Cdc42 and Rac1 inhibitory activity (27, 28). Published literature on breast, lung and kidney cancer patients and our own data on ovarian cancer patients demonstrates that besides its analgesic function, ketorolac has significant benefit for patient survival (6, 12, 29, 30). The mechanism of this anti-cancer action is not clear, yet appears to be selectively ascribed to ketorolac and not a general property of cyclooxygenase inhibitors or other analgesics and anesthetics (6, 31, 32). This suggests that ketorolac possesses an additional property. In the present study, a human patient immortalized ovarian cancer cell line (SKOV3ip) and primary ascites-derived ovarian cancer cells were used to demonstrate that R-ketorolac acts as an allosteric inhibitor of Cdc42 and Rac1 nucleotide binding activities in vitro and blocks their activation and downstream activation of the PAK signaling axis. As a consequence of the inhibition there is a reduction in ovarian cancer cell adhesion, migration and invasion. Taken together the data demonstrate the potential for repurposing R-ketorolac, an FDA approved drug in the racemic form, for improved patient benefit in progression free and overall survival.
Materials and Methods
Cell and reagents
The human ovarian adenocarcinoma epithelial cell line SKOV3ip was derived from SKOV3 cell line by selecting for a peritoneal metastatic phenotype in the mice and was obtained under a Material Transfer Agreement with MD Anderson in June 24 2009. The ascites derived ovarian cancer cells were obtained from nine patients from 2012 to 2015. SKOV3ip cell line was authenticated using Short Tandem Repeat (STR) analysis (performed by Promega). SKOV3ip cells and primary ovarian cancer cells were cultured in RPMI 1640 media containing 5% FBS (Atlanta Biologicals). All cell culture media and reagents were purchased from Gibco® (Life Technologies). R-, and S-ketorolac were from Toronto Research Chemical Inc. BODIPY-GTP ((4,4-difluoro-4-bora-3a,4a-diaza-s-indacene or dipyrromethene boron difluoride) nucleotide analogue) was from Invitrogen Molecular Probes. Rat tail type I collagen was obtained from BD Biosciences. NSC23766 was from Santa Cruz Biotechnology and CID2950007 was from Sigma-Aldrich. GST (glutathione S-transferase)-tagged GTPases were purified as described previously (33). GST-PAK1 protein was from Millipore. A polyclonal antibody directed against Tks5 (Src tyrosine kinase substrate 5) was prepared as described (34). The following commercial antibodies were used: mouse mAb (monoclonal antibody) directed against Rac1 from BD Transduction Laboratories, mouse mAb directed against Cdc42 from Santa Cruz, FITC (fluorescein isothiocyanate)-conjugated mouse mAb directed against EpCAM (epithelial cell adhesion molecule) (clone Ber-EP4) from Dako; rabbit polyclonal Cy5-conjucated anti-CA125 (cancer antigen 125) from Bioss Inc., mouse mAb PE (Phycoerythrin)-conjugated anti-CD45 (lymphocyte common antigen 45) from eBioscience, rabbit polyclonal antibodies directed against phospho-PAK1 (Ser144)/PAK2(Ser141), phospho-PAK1(Ser199/204)/PAK2(Ser192/197), phospho-PAK1(Thr423)/PAK2(Thr402) and PAK1 from Cell Signaling Technology, Alexa 488 goat anti-mouse antibody and Alexa 647 goat anti-rabbit antibody from Life Technology, all used per manufacturers' instructions.
Patient information
A Phase 0 trial investigating the use of postoperative ketorolac was reviewed and approved by the University of New Mexico Health Sciences Center Human Research Review Committee (NCT01670799 clinicaltrials.gov) (35). Informed patient consent was obtained prior to surgery. Eligible patients having suspected advanced stage ovarian, fallopian tube or primary peritoneal cancer underwent planned optimal cytoreductive surgery. Upon surgical entry into the abdomen, ascites fluid was retrieved and residual material was recovered and sent fresh to the investigators for processing. Ascites material used for this study was from patients confirmed to have stage III or IV at final pathologic diagnosis. Nine patient samples were included in the study (Supplementary Table 1).
Isolation and cell culture of ascites-derived primary ovarian cancer cells
Peritoneal ascites were obtained at the time of debulking surgery with an average volume of 200 ml. Cells were collected by centrifugation at 300g for 5 min. The Ficoll-Paque (1.073 ± 0.001 g/ml) PREMIUM density gradient media (GE Healthcare) was used to pellet and remove erythrocytes and polymorphonuclear cells. The mononuclear white cells and tumor cells found at the top of the Ficoll interface were transferred to a sterile tube and washed with RPMI 1640 media with 5% FBS. To deplete leukocytes and further enrich tumor cells, samples were incubated with CD45-coated Dynabeads® (Life technologies) for 1.5 h at 4 °C, washed and collected by centrifugation according to the manufacturer's protocol. Isolated tumor cells were cultured as described by T. Shepherd et. al (36). Briefly, enriched ovarian cancer cells were cultured on collagen-coated (20 μg/cm2) tissue culture dishes. After 3-4 d, when most of the tumor cells had adhered, the media was replaced and continued to be changed every 2-3 d until cells grew to confluence. The expanded cells were used for filopodia formation, cell adhesion, migration and extracellular matrix degradation assays.
Synthesis of glutathione (GSH) beads for flow cytometry assay
High density glutathione (GSH) conjugated beads were synthesized for flow cytometry assays by loading Superdex peptide beads with GSH as previously reported (37).
Flow cytometric BODIPY-GTP binding assay
The assay was carried out according to the protocols described previously (37). Briefly, individual GST-GTPases were attached to their respective GSH beads and combined. In the equilibrium assay, 2 × 103 GSH beads loaded with GST-GTPase were pre-incubated with either DMSO (dimethyl sulfoxide) or a fixed compound concentration for 15 min followed by the incubation with varying concentrations of BODIPY-GTP for 30 min at room temperature. In the dose-response assay, 2 × 103 GSH beads loaded with GST-GTPase were pre-incubated with DMSO or increasing concentration of the compound for 15 min followed by the incubation with a fixed concentration of BODIPY-GTP for 30 min at room temperature. For measurement, samples were diluted at least 10-fold and delivered and analyzed by the BD FACScan flow cytometer.
Flow cytometric GTPase effector binding assay for quantification of active, cellular GTPase levels
The assay was carried out according to the protocols described previously (38). Ketorolac treated and vehicle-control cells were lysed in RIPA (high stringency cell lysis buffer) buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 0.25% (w/v) Na-deoxycholate, 1 mM Na3VO4, 1 mM NaF, 1% (v/v) NP-40 (nonyl phenoxypolyethoxylethanol), 1 mM PMSF (phenylmethylsulfonyl fluoride) and protease inhibitors consisting 10 μg/ml each of chymostatin, leupeptin, pepstatin and antipain). Insoluble debris was removed by centrifugation and the supernatants were incubated with GTPase effector coated beads (PAK1-PBD for Cdc42 and Rac1) for 1 h. Primary antibodies directed against Cdc42 or Rac1 and secondary antibody Alexa 488 were incubated with the beads for 1 h. Fluorescence intensity MCF (mean channel fluorescence) was used to measure the amount of active intracellular GTPase. MCF was measured by flow cytometry (Accuri C6, BD Biosciences). GTPase activity was calculated as (MCFsample group − MCFunstimulated negative control) / MCFstimulated positive control.
Quantification of filopodia formation
The assay was conducted as described previously (23). Cells were cultured on coverslips to 50% confluence and serum-starved for 2 h, treated with individual drugs and stimulated with 10 ng/ml EGF (epidermal growth factor) for 20 min. Cells were gently rinsed 3× with ice cold PBS and fixed with 3% paraformaldehyde. Rhodamine phalloidin was used to label the actin. Images were collected using a Zeiss LSM (laser scanning microscope) 510 confocal microscope equipped with a 63× oil immersion lens. For each experiment, a battlement pattern was used to select 30 images at random. Length and number of filopodia per cell were quantified using Slidebook 5.5 software.
Cell adhesion assay
The adhesion assay was modified from the method described by M.J. Humphrie (39). Cells were cultured at least 48 h and disassociated with 0.05% trypsin/EDTA. Suspended cells were rested in 0% FBS media for 1 h and treated with individual drugs. Cells stimulated with 5% serum were seeded on a 96-well plate (5 × 104 cells/well) coated with 0.5 μg/cm2 fibronectin or collagen and permitted to attach for 1 h. The 96-well plate was gently washed with PBS and cells were fixed with 3% paraformaldehyde. Samples were stained with crystal violet and lysed with 10% acetic acid and absorbance (λ=595nm) quantified using on a plate reader.
Cell migration assay
SKOV3ip cells were plated at 1 × 105 cells/well in 24-well Boyden chambers and allowed to attach for 4 h. Ketorolac enantiomers were added to growth media at final concentrations ranging from 1 to 300 μM. After 48 h, inserts were removed and stained with DAPI (4′,6-diamidino-2-phenylindole). Membrane filters were imaged on a Zeiss inverted microscope using a 20× objective. Three representative fields were counted from each treatment group.
Gelatin degradation assay
The commercial gelatin degradation kit (Millipore) was used per manufacturer's instructions and modified to incorporate drug treatment and EGF stimulation as follows. Fluorescent gelatin was coated on coverslips as described previously (25). Cells were seeded on fluorescent coverslip and allowed to adhere for 6 h and then treated with individual drugs for 24 h. Cells were fixed with 3% paraformaldehyde, permeabilized with 0.1% Triton X-100 and immunostained for Tks5. Actin was visualized with FITC-phalloidin. Fifteen fields per coverslip were imaged on a Zeiss LSM 510 META with a 63× oil immersion objective. Gelatin degradation by invadopodia was analyzed using Slidebook 5.5 software. Percent degradation was quantified as: (degradation area)/(total cell area) for each image.
Western blot
SKOV3ip cells were serum-starved for 2 h and treated with individual drugs for 1 h. After simulation with 10 ng/ml EGF for 20 min, cells were washed with ice cold PBS and lysed with RIPA buffer. Cell lysates were processed for SDS-PAGE, transferred to polyvinylidene fluoride membranes and individual proteins were detected with specific antibodies directed against PAK and phosphor-PAK.
Statistical analyses
Prism 5 software (GraphPad) was used to analyze all data to determine statistical significance. One-way ANOVA (Analysis of Variance) with Dunnett's test for multiple comparisons was performed to compare differences between the means of each group relative to the control group for all assays. P-values less than 0.05 were considered statistically significant. For some assays conducted on primary ovarian patient samples (GTPase activity measurements and gelatin degradation assays), standard z-scores of the values were calculated for each patient to minimize large ranges of values and to compare across groups on a uniform scale. ANOVA was performed on these standardized values.
Results
R-ketorolac acts as an allosteric inhibitor of guanine nucleotide binding to Cdc42 and Rac1
The enantiomeric selectivity of ketorolac (Fig. 1A) against Cdc42 and Rac1 was predicted by virtual screening and modeled in docking studies (25). To verify ketorolac inhibition of nucleotide binding activity and determine the mechanism of inhibition, a bead based flow cytometry assay – which measures the in vitro GTPase nucleotide binding activity – was used. First, equilibrium nucleotide binding assays were performed under conditions where the tested compound concentration was held fixed against increasing concentrations of fluorescent nucleotide. In the presence of R-ketorolac, both the maximum fluorescence (Bmax) and the apparent dissociation constant (Kd) of BODIPY-GTP for Cdc42 and Rac1 changed, which suggests a noncompetitive mechanism action (Fig. 1B, 1D, Supplementary Table 2). S-ketorolac did not change the Bmax and the Kd for Cdc42 (Fig. 1C, Supplementary Table 2) and exhibited a minor inhibition of Rac1 only at the maximum concentration of 100 μM (Fig. 1E). Single-plex dose-response measurements were used to verify the enantiomer selectivity of ketorolac for inhibiting nucleotide binding by Cdc42 and Rac1. The nucleotide concentrations in the assay were fixed at the determined dissociation constants (Kd≈ 300 nM). Increasing R-ketorolac concentrations reduced BODIPY-GTP binding, with respective EC50 (half-maximal effective concentration) values of 1.887 × 10-5 for Cdc42 and 2.308 × 10-5 for Rac1 (Fig. 1F, 1G). The maximal inhibitory response was ∼20% for both GTPases. S-ketorolac did not exhibit an inhibitory activity against either GTPase under the tested conditions. Together, these findings suggest an enantiomer-selective, inhibitory activity of R-ketorolac against the nucleotide binding activity of Cdc42 and Rac1. Mechanistically, R-ketorolac action is consistant with an allosteric inhibition of GTPase nucleotide binding.
Figure 1. R-ketorolac selectively inhibits Cdc42 and Rac1 GTPase BODIPY-GTP binding as an allosteric inhibitor.
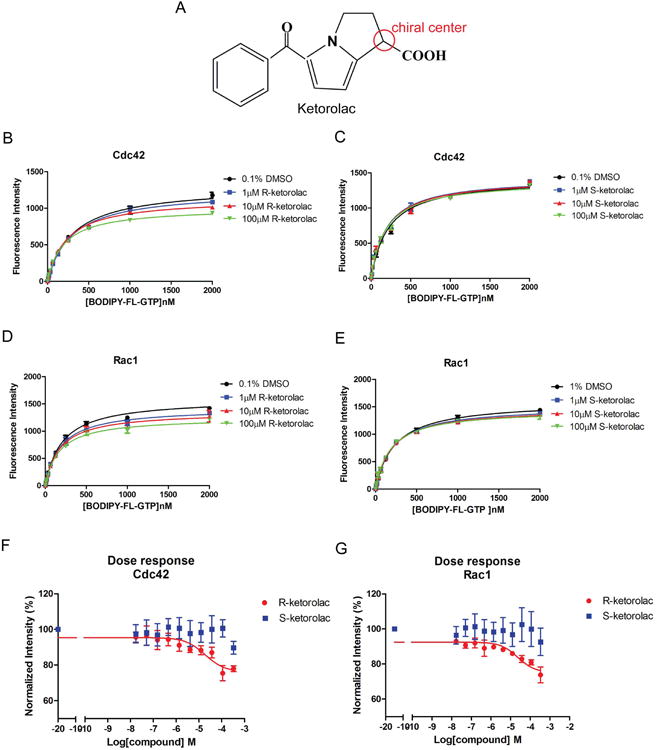
(A) Chemical structure of R- and S-ketorolac with chiral center noted. (B-C) The presence of R-ketorolac changed both the Bmax and the Kd (values are tabulated in supplementary table 2) of BODIPY-GTP binding to Cdc42 measured under equilibrium binding conditions, while S-ketorolac did not affect BODIPY-GTP binding. (D-E) R-ketorolac changed the Bmax and the Kd of BODIPY-GTP binding to Rac1 measured under equilibrium binding conditions, while S-ketorolac did not affect BODIPY-GTP binding except at 100 μM. The equilibrium binding of BODIPY-GTP to the GTPases was measured in the presence of 0.1% DMSO or various concentrations of R- or S-ketorolac (1∼100 μM). BODIPY-GTP binding curves were fitted to a hyperbolic one-site binding equation from GraphPad Prism 5. (F-G) R-ketorolac in contrast to S-ketorolac inhibited BODIPY-GTP binding to Cdc42 and Rac1 GTPases in a dose-dependent manner. BODIPY-GTP concentration was fixed at 300 nM for these experiments. The inhibition curves were fitted to the sigmoidal dose-response equation from GraphPad Prism5. In all experiments, GST-Cdc42 or Rac1 were immobilized on GSH beads and used to measure BODIPY-GTP binding. Bead-associated fluorescence intensity was quantified by flow cytometry and used to monitor drug treatment induced changes in nucleotide binding. All experiments were repeated three times independently.
R-ketorolac inhibits Cdc42 and Rac1 activation in immortalized and primary human ovarian cancer cells
We have developed a flow cytometry based assay for quantitatively assessing the levels of active GTPases in cell lysates (38). The assay was used to test the cellular impact of ketorolac isoforms on GTPase activation. Maximal Cdc42 and Rac1 activation in SKOV3ip cells treated with 10 ng/ml EGF occurred at different times; 20 min for Cdc42 and 5 min for Rac1 (Supplementary Fig. S1). Growth factor stimulated activation of Cdc42 and Rac1 was differentially inhibited in an enantiomer-selective manner (Fig. 2A, 2B). R-ketorolac substantially inhibited growth factor dependent activation of Cdc42 and Rac1 in a dose-dependent manner with an EC50 value of 2.577 μM for Cdc42 and 0.587 μM for Rac1 (Fig. 2A, 2B, circles). In contrast, even at the highest dose of 100 μM, S-ketorolac exhibited limited inhibition (less than 30% of maximum by R-ketorolac) of Cdc42 and Rac1(Fig. 2A, 2B, squares). Enantiomer-selective inhibition was also found in HeLa cells treated with R-ketorolac (25). Control cytotoxicity measurements in three different cell lines (SKOV3ip, OVCA 429, and OVCA 433) showed <10% inhibition of cell proliferation induced by either enantiomer of ketorolac at the highest tested dose and treatment time (300 μM for 96 h).
Figure 2. R-ketorolac preferentially inhibits Cdc42 and Rac1 activities in SKOV3ip cells and primary human ovarian cancer cells.
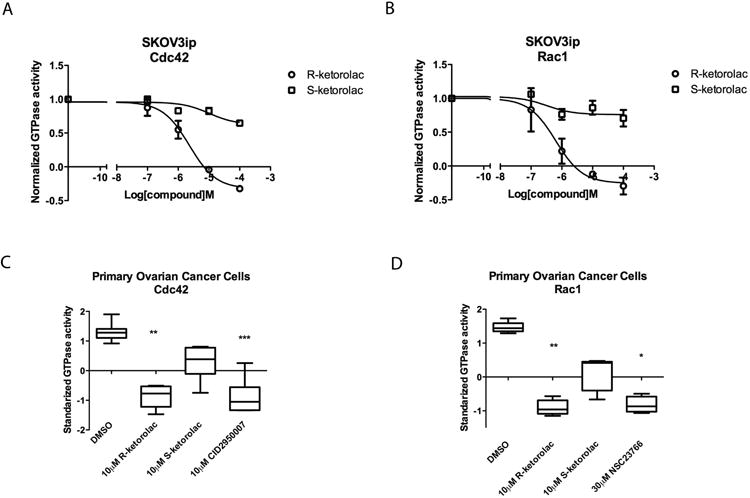
(A-B) R-ketorolac causes a dose-dependent, enantiomer selective inhibition of growth factor stimulated Cdc42 and Rac1 activation in SKOV3ip cells. SKOV3ip cells were starved in 0% FBS media for 2 h and pretreated with individual drugs for 2 h prior to EGF (10 ng/ml) stimulation. Activated Cdc42 and Rac1 levels in cell lysates were measured using a flow cytometry based GTPase effector binding assay. Flow intensities were normalized to untreated controls. All experiments were repeated three times independently. (C-D) R-ketorolac causes a dose-dependent, enantiomer-selective inhibition of endogenous Cdc42 and Rac1 activities in primary ovarian cancer cells. Isolated primary ovarian cancer cells from three independent patients were treated with drugs for 1 h and processed for flow cytometry GTPase effector binding assays to measure the GTPase activity. Six patient samples were tested for Cdc42 activity and five patient samples were tested for Rac1 activity in response (Supplementary Table 1). CID2950007 and NSC23766 served as Cdc42 and Rac1 specific inhibitors respectively. Plotted are z-scores. Quantification of three independent measurements are plotted ±SEM. Statistically significant differences are indicated: *p<0.05, **p<0.01, ***p<0.001.
The effect of ketorolac was also examined in primary ovarian cancer cells purified from patient ascites. Surgery samples were obtained and analyzed as part of an approved clinical study (see Materials and Methods and Supplementary Table 1). Primary human ovarian cancer cells were enriched via Ficoll gradient centrifugation and depletion of lymphocytes using CD45 Dynabeads. Purified primary human ovarian cancer cells were positive for CA125 and EpCAM, which are commonly overexpressed in epithelial ovarian carcinomas (Supplementary Fig. S2)(40, 41). Isolated tumor cells were treated with individual ketorolac isoforms or specific Cdc42 and Rac1 inhibitors and the endogenous Cdc42 and Rac1 activities were tested. Due to the heterogeneity in activity levels of the GTPases for individual patient samples, z-scores were used to comparatively evaluate the impact of ketorolac on tumor cell GTPase activities from different patients. Treatment with 10 μM R-ketorolac reproducibly inhibited both Cdc42 and Rac1 in a statistically significant manner. The magnitude of R-ketorolac inhibition of Cdc42 or Rac1 was similar to the inhibition observed in positive controls treated with CID2950007, an allosteric Cdc42 inhibitor, or NSC23766, a Rac1 inhibitor that interferes with GEF (guanine nucleotide exchange factor)-mediated GTPase activation (21, 23). S-ketorolac exhibited limited inhibition and was not statistically significant (Fig. 2C-D). Taken together, the results demonstrate that R-ketorolac is a potent, enantiomer-selective inhibitor of Cdc42 and Rac1 in primary human ovarian cancer cells.
Inhibition of Cdc42 and Rac1 by R-ketorolac decreases activation of downstream p21 activated kinases (PAK1/PAK2)
The impact of R-ketorolac on PAK1/PAK2 effector signaling directly downstream of the Cdc42 and Rac1 GTPases was examined (Supplementary Fig. S3A). Cells stimulated with 10 ng/ml EGF exhibited robust phosphorylation of the Ser144/141 residues compared to unstimulated control cells. In contrast, there was a dose-dependent suppression of p-PAK1/PAK2 following R-ketorolac treatment that was similar to positive controls treated with Cdc42 or Rac1 specific inhibitors. S-ketorolac on the other hand had limited effect on p-PAK1/PAK2 levels at either 1 μM or 10 μM (Fig. 3A-C). The expression of total PAK1 was not affected by either R- or S-ketorolac treatment (Fig. 3A). These results demonstrate the suppression of p-PAK1/PAK2 paralleled the inhibition of Cdc42 and Rac1 in a dose-dependent manner following R-ketorolac treatment; and S-ketorolac exhibited no significant inhibition of p-PAK1/PAK2.
Figure 3. R-ketorolac decreases the phosphorylation of PAK1(Ser144)/PAK2(Ser141) without affecting total PAK levels.
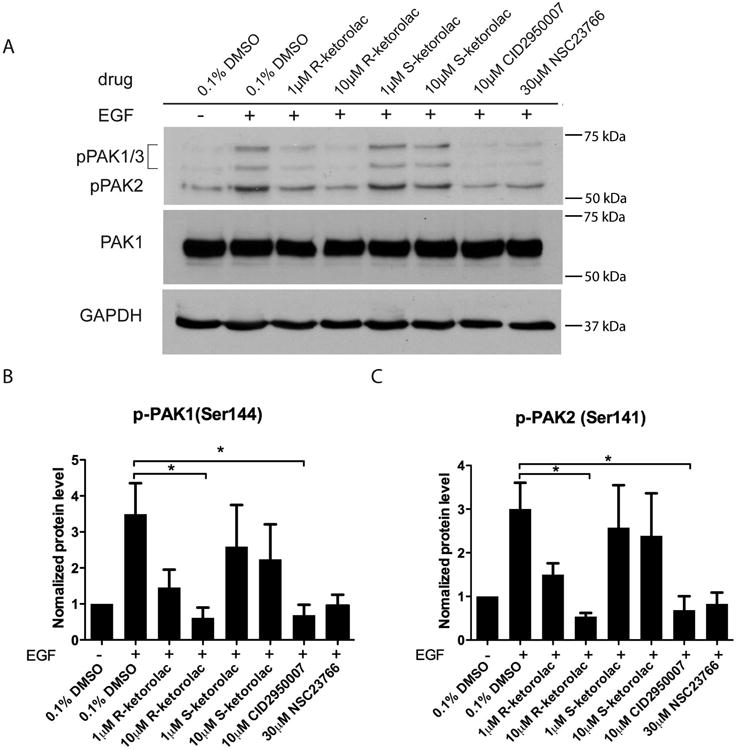
(A) SKOV3ip cells were cultured overnight, then starved in 0% FBS media for 2 h and pretreated with individual drugs for 2 h followed by EGF stimulation (10 ng/ml) for 20 min. Representative blots from one of three independent experiments are shown. Equal amounts of cell lysate protein were resolved by SDS-PAGE and immunoblotted for p-PAK1(Ser144)/PAK2(Ser141). GAPDH served as the loading control. Cdc42 inhibitor, CID2950007 and Rac1 inhibitor, NSC232766 were used as positive controls. (B-C) Films from three independent experiments were quantified by ImageJ analysis. p-PAK1/PAK2 levels were normalized to unstimulated controls. Quantification of three independent measurements are plotted ±SEM. Statistically significant differences are indicated: *p<0.05, **p<0.01, ***p<0.001.
Phosphorylation of PAK1/PAK2 on two further residues (Thr423/402 or Ser199/204/ Ser192/197) that are downstream of other signaling pathways were also examined. Interestingly, p-PAK1(Thr423)/PAK2(Thr402) and p-PAK1(Ser199/204)/PAK2(Ser192/197) were reduced by R and S-ketorolac with greater variability and less pronounced enantiomeric selectivity (Supplementary Fig. S3A-E). For example, R-ketorolac consistently inhibited the phosphorylation at pPAK1/PAK2 (Thr423) with statistical significance at 1 μM though more variably at 10 μM. S-ketorolac exhibited statistically significant inhibitory effects at 10 μM against Thr432/402, but not against Ser199/204 / Ser192/197. Inhibition of phosphorylation by the Rac1 inhibitor NSC23766 and the Cdc42 inhibitor CID2950007 was also more modest and failed to reach statistical significance in some instances. The explanation for the differences in responsiveness of individual phosphorylation sites is likely due to the fact that Ser144/141 is localized in an N-terminal regulatory domain and directly interacts with Cdc42 and Rac1 (Supplementary Fig. S3A). While Thr423/402 residues in the catalytic kinase domain and Ser199/204/ Ser192/197 adjacent to auto-inhibitory domain (AID) are targeted by other regulatory molecules in a GTPase-independent manner (42, 43). For example, S-ketorolac may inhibit phosphorylation on these sites by affecting lipid-dependent stimulation of PAK1/PAK2 (e.g. sphingosine) or through crosstalk between COX-1/COX-2 and PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase)-GTPase-PAK1/PAK2 (44, 45).
R-ketorolac inhibits Cdc42-dependent filopodia formation in SKOV3ip cells and primary ovarian cancer cells
Filopodia are membrane protrusions that are constructed of bundled actin filaments and directly regulated by Cdc42 (46). Therefore, we tested filopodia formation, including numbers and length of filopodia to evaluate the effect of ketorolac on cell behaviors. When resting cells were stimulated with 10 ng/ml EGF, both numbers and length of filopodia increased with the most striking activation seen 20 min post-stimulation. Two concentrations of R-, and S-ketorolac were tested to take into consideration differential sensitivities of Cdc42 to the two enantiomers. In comparison to the vehicle treatment group, R-ketorolac (at both 1 μM and 10 μM concentrations) decreased numbers length of filopodia to basal levels and similar to CID2950007; while S-ketorolac only showed a significant inhibition on filopodia length at 10 μM and did not significantly inhibit filopodia numbers at either 1 μM or 10 μM. (Fig. 4A-C). These results demonstrate that R-ketorolac impedes filopodia formation, which depends on the upstream activation of Cdc42.
Figure 4. R-ketorolac decreases Cdc42 dependent filopodia formation in SKOV3ip cells and primary human ovarian cancer cells following EGF stimulation.
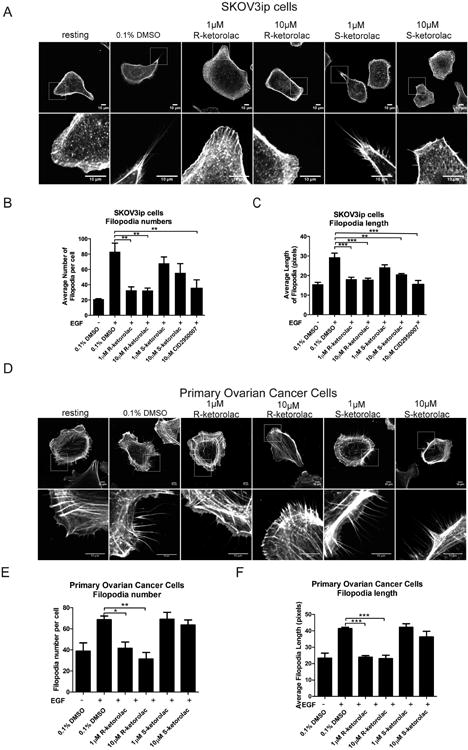
(A) Representative confocal images of filopodia formation for each treatment group in SKOV3ip cells are shown. Lower panels are magnified images of the boxed regions. (B-C) Quantification of numbers and length of filopodia on each cell. (D) Representative confocal images of filopodia formation for each treatment group in primary human ovarian cancer cells are shown. Lower panels are magnified images of the boxed regions. (E-F) Quantification of numbers and length of filopodia per cell in primary human ovarian cancer cells. For all experiments, cells were plated on coverslips and cultured overnight, then starved in 0% FBS media for 2 h and pretreated with drug or 0.1% DMSO vehicle for 2 h, followed by EGF (10 ng/ml) stimulation for 20 min. Cells were permeabilized and stained with rhodamine-phalloidin. Confocal images were taken on a Zeiss LSM 510 META confocal microscope. All quantification is based on three independent trials with 30 cells imaged in a battlement pattern and analyzed using Slidebook 5.5 software. Mean lengths or numbers of filopodia are plotted ±SEM. Statistically significant differences are indicated: *p<0.05, **p<0.01, ***p<0.001.
Filopodia formation assays performed using primary ovarian cancer cells yielded similar results and reinforced the finding of the enantiomer-selective effect. R-ketorolac treated primary ovarian cancer cells exhibited significantly decreased filopodia numbers and length; while S-ketorolac exerted no inhibitory effect (Fig. 4D-F).
R-ketorolac inhibits Cdc42- and Rac1-mediated cell adhesion, migration and invasion
Cell adhesion
Cdc42 and Rac1 are involved in cellular signaling pathways that transduce extracellular signals to the assembly of integrin-mediated focal adhesion complexes (47). The process of cell-substrate adhesion plays critical roles during cancer metastasis which is required for the anchoring of malignant spheroids to the extracellular matrix (ECM) of target abdominal organs (4). A cell attachment assay was used to test the effect of ketorolac on cell adhesion. Stimulated cells were allowed to adhere to matrix-coated substrates. R-ketorolac reduced SKOV3ip cells adherence to both fibronectin and collagen, and S-ketorolac had no significant effect (Fig. 5A-B). R-ketorolac also inhibited human primary ovarian cancer cell adhesion to collagen; while S-ketorolac had no significant effect (Fig. 5C). This observation suggests that the R-enantiomer of ketorolac inhibits cell-substrate adhesion likely as a biological consequence of its inhibitory effect on upstream Cdc42 and Rac1 activation.
Figure 5. R-ketorolac inhibits cell-substrate adhesion and migration in SKOV3ip cells and primary human ovarian cancer cells.
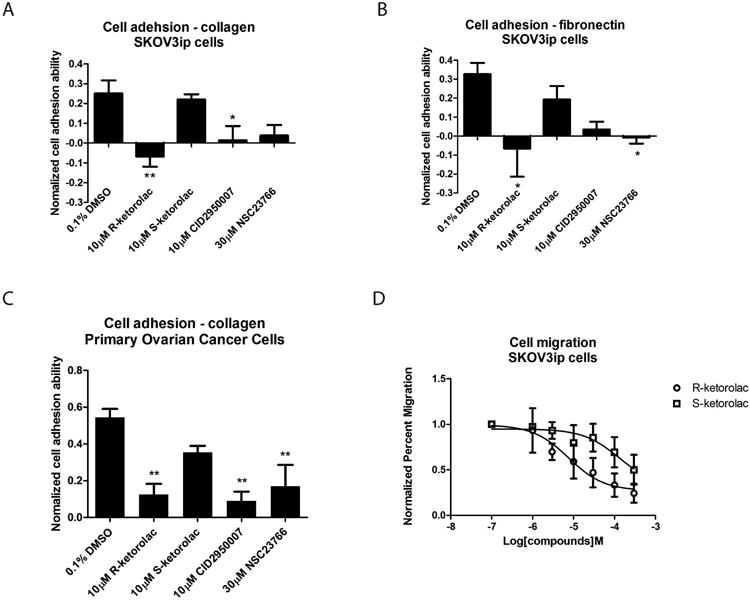
(A-B) R-ketorolac significantly inhibits cell adhesion in SKOV3ip cells compared to S-ketorolac. SKOV3ip cells were trypsinized, starved in 0% FBS media for 1 h, pretreated with drug for 1 h, followed by serum stimulation (5% (v/v) final concentration) and seeded on matrix pre-coated 96-well plate for 1 h. Cells were fixed and stained with crystal violet and finally lysed with acetic acid. Cell substrate adhesion was quantified based on absorbance of cell lysates at 595 nm and normalized to unstimulated controls. (C) Similar to SKOV3ip cells, R-ketorolac significantly inhibits cell substrate adhesion in primary human ovarian cancer cells as well. Primary ovarian cancer cells were purified, cultured and used for the cell attachment assays to test the effect of drugs on cell substrate adhesion. CID2950007 and NSC23766 served as positive controls. Each group was normalized to unstimulated controls. (D) R-ketorolac inhibits cell migration in a dose-dependent manner with maximal inhibition of 80% in SKOV3ip cells, whereas S-ketorolac is 50-fold less potent. SKOV3ip cells were seeded in Boyden chambers in the presence of drug for 48 h. Cell migration was normalized to no treatment controls. Three fields of view were counted for each treatment group. Data represented three independent trials. Each data point represents the mean of measured values ±SEM. Statistically significant differences are indicated: *p<0.05, **p<0.01, ***p<0.001.
Cell migration
Activated by extracellular stimuli and dependent on GTPase activation, cell migration is initiated by the formation of membrane protrusions, including Cdc42-dependent filopodia and Rac1-mediated lamellipodia (7). SKOV3ip cell migration in the presence of R- or S-ketorolac was tested in a Boyden chamber assay. Migration was inhibited in a dose dependent manner with R-ketorolac beginning to impinge on cell migration at 1 μM. The inhibitory effect increased in a dose-dependent manner with maximum inhibition of ∼80% at 300 μM R-ketorolac (Fig. 5D). Modest inhibition by S-ketorolac was detected at about 50 μM with a maximum inhibition less than 50% at 300 μM. The results demonstrate that cell migration was inhibited as a direct biological consequence mediated by Cdc42 and Rac1 inhibition by R-ketorolac, whereas S-ketorolac was 50-fold less potent.
Invasion
Invadopodia are membrane protrusions that are dependent on activated Cdc42 and Src kinase. Invadopodia concentrate matrix metalloproteinases at their tips and based on their matrix degradative properties serve as surrogates for cell invasive behaviors (48). To study the effect of ketorolac on invadopodia formation, the invadopodia-dependent degradation of gelatin matrices was quantified. In Skov3ip cells, invadopodia were observed as puncta of F-actin and Tks5 positive structures, which colocalized with the degradation sites on the gelatin matrix (Fig. 6A). R-ketorolac treatment also resulted in reduced gelatin degradation in a dose-dependent manner with 70- 80% inhibition at 1 and 10 μM. However, S-ketorolac did not exert any obvious inhibition of gelatin degradation at 1 μM and the reduction of gelatin degradation with10 μM S-ketorolac treatment was ∼50% but failed to reach the statistical significance (Fig. 6B, D). Tks5 is an early marker of invadopodia formation that precedes matrix metalloprotease localization to the tips of mature structures. To distinguish if R-ketorolac had a direct inhibitory effect on metalloproteinase activity or if the observed effect on gelatin degradation was primarily due to inhibition of invadopodia formation, the impact on metalloproteinase activity secreted into the culture medium was also measured and found to be similar +/- ketorolac. Therefore, we conclude that the primary effect of R-ketorolac is to prevent Cdc42-dependent invadopodia formation. When the gelatin degradation assay was conducted in primary ovarian cancer cells, the standardized z-scores showed the degradation of matrix by tumor cells was also significantly decreased in a dose dependent manner by R-ketorolac treatment, while the inhibition by S-ketorolac was less and reached significance only at the highest dose (Fig. 6C, 6E). Z-scores were used to account for large differences in absolute GTPase activity levels observed for individual patient samples (Fig. 2C, 2D), likely related to known differences in GTPase overexpression seen in ovarian cancer (11, 12). The inhibitory effect of R-ketorolac in primary ovarian cancer cells is consistent with the observations in SKOV3ip cells.
Figure 6. R-ketorolac inhibits invadopodia formation and gelatin degradation in SKOV3ip and primary human ovarian cancer cells.
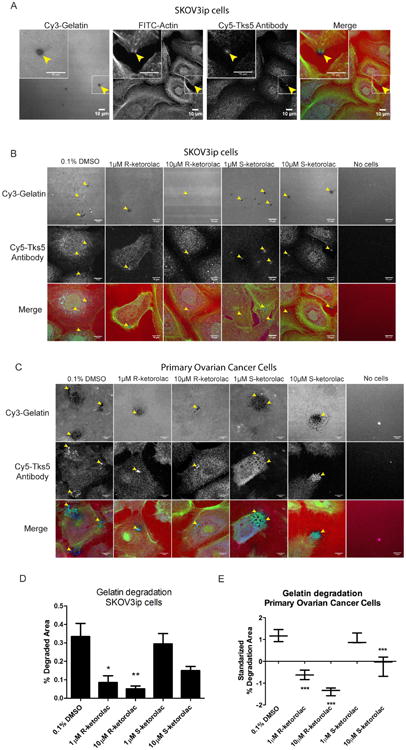
(A) Colocalization of gelatin degradation, actin and Tks5 antibody in SKOV3ip cells. Insets are magnified images of the boxed regions. Arrowheads denote points of invadopodia formation that are coincident with gelatin degradation. (B) Representative confocal images of gelatin degradation for each treatment group in SKOV3ip cells are shown. Lower panels are magnified images of the boxed regions. Arrowheads denote the gelatin degradation sites. (C) Representative confocal images of gelatin degradation for each treatment group in primary ovarian cancer cells are shown. Lower panels are magnified images of the boxed regions. Arrowheads denote the gelatin degradation sites. (D) Quantification of the gelatin degradation area in SKOV3ip cells. (E) Quantification of the gelatin degradation area in primary ovarian cancer cells. Due to the variability of the extent of gelatin degradation observed with primary human ovarian cancer cells, z-scores are plotted. For all experiments, SKOV3ip cells or primary ovarian cancer cells were seeded on Cy3-gelatin labeled coverslips for 24 h in the presence of different drugs, including two enantiomers of ketorolac, and Cdc42 and Rac1 specific inhibitors (CID2950007 and NSC23766, respectively) as positive controls. DMSO (0.1%) served as the vehicle control treatment. All quantification is based on three independent trials with 15 representative fields counted for each treatment. Statistically significant differences are indicated: *p<0.05, **p<0.01, ***p<0.001.
Discussion
In the present study, we establish R-ketorolac as an allosteric, non-competitive inhibitor of guanine nucleotide binding to Cdc42 and Rac1. In its clinical application, ketorolac is given for its potent analgesic activity. It is administered as a racemic mixture via diverse routes (intravenous, injectable, oral, intranasal) and is known as Toradol™. The pain relieving activity is ascribed primarily to the S-enantiomer, which is a known non-selective cyclooxygenase (COX-1/COX-2) inhibitor (27, 28). The R-enantiomer lacks significant activity against cyclooxygenases (e.g. IC50>100 μM against COX1 and COX2 and ∼3 orders of magnitude greater than the IC50 of S-ketorolac) and has until now been considered inert (27, 28). The previously unrecognized and novel mechanism of action for the R-enantiomer has important ramifications when considering the efficacy and novel activities of clinically administered ketorolac. For example, breast cancer patients treated with ketorolac perioperatively had a decreased rate of early breast cancer relapse (6). Our own studies demonstrate ovarian cancer patients receiving ketorolac exhibit an enrichment of R-ketorolac in the peritoneum, a reduction in GTPases activation of residual tumor cells, and improved 5-year survival (12). The cell based studies using human immortalized and primary human ovarian cancer cells presented here, demonstrate that R-ketorolac treatment alters tumor cell adhesion, migration and invasion—all behaviors that are central to ovarian cancer metastasis. The findings offer unprecedented mechanistic insights regarding the R-ketorolac activity against GTPases, which may explain the clinically observed survival benefit in patients treated perioperatively with racemic ketorolac.
The biochemical basis for GTPase inhibition by R-ketorolac is provided by computational and experimental approaches. A model of R-ketorolac docked near the GTPase nucleotide binding pocket was generated based on DOCK9 GEF facilitated nucleotide exchange on Cdc42 (25, 49) and is consistent with the observed allosteric, non-competitive inhibition mechanism in the present studies. We suggest that the activity of R-ketorolac against Cdc42 and Rac1 GTPases may result from a similar magnesium exclusion mechanism, except with magnesium exclusion being induced by the carboxyl moiety at the chiral center of R-ketorolac instead of by Val1951 on DOCK9 GEF. The consequent reduced stabilization of bound nucleotide by such magnesium exclusion could reduce nucleotide binding affinity and promote release when R-ketorolac is present. The model is enantiomer-selective, with S-ketorolac being poorly positioned to exert the same magnesium exclusion and therefore could explain the observed differences in EC50 values measured for R- and S-ketorolac in the in vitro nucleotide binding assay. The model is further supported by X-ray crystallography data, which identified the preferential binding of S-naproxen to COX-2 (50). R-naproxen, like R-ketorolac, exhibits Cdc42 and Rac1 inhibitory activity, albeit with less potency and naproxen, like ketorolac has a constrained alpha methyl carboxyl that may account for the stereo-selective activities of the R- and S-enantiomers (25).
In ovarian cancer, cyclooxygenases are upregulated and have been considered as potential targets, however, clinical trials have been mixed and do not support significant benefit in combinatorial treatment with chemotherapy and a cyclooxygenase inhibitor versus treatment with chemotherapy alone (31). Although the survival benefit of ketorolac use in breast cancer was postulated to be due to possible inhibitory roles in anti-angiogenesis, inhibition of prostaglandin synthesis and decreased immune suppression as compared to opioids and other analgesics (51), there was previously no precedence for other pharmacologic activities associated with R- or S-ketorolac. We speculate that the slight inhibitory effects of S-ketorolac on GTPase activation and actin dependent cell behaviors may arise from crosstalk between COX-2 and PI3K and integrin regulated pathways. COX-2 dependent crosstalk has been shown through knockout and pharmacologic means to regulate Cdc42 and Rac1 activation, and adhesion and migration of macrophages and endothelia (45, 52). Because R-ketorolac is a much less potent COX inhibitor than S-ketorolac, its more pronounced effect on GTPase activation and downstream cell behaviors indicates that it is primarily targeting Cdc42 and Rac1 through a direct mechanism. Furthermore, pharmacokinetic analyses show that S-ketorolac is more rapidly excreted, leading to an increased prevalence of R-ketorolac in vivo (12, 27, 28). Taken together, our identification of R-ketorolac as a GTPase inhibitor helps to explain the significant benefits of racemic ketorolac in human breast and ovarian cancer patient survival, and why other NSAIDs that preferentially target COX have not yielded similar benefit (6, 12, 32).
Ketorolac is an FDA (Food and Drug Administration)-approved drug for human use and is in active clinical use as the racemic mix. Therefore, R-ketorolac has significant potential for rapid repurposing. Implementation of R-ketorolac in human clinical trials would offer the first opportunity to directly test the predicted benefit of a Cdc42 and Rac1 selective inhibitor for cancer patients. Such an application could circumvent current renal and hematologic toxicities that limit racemic ketorolac use to a maximum of 5 days. Analyses of Rac1 inhibition in cancer cell lines and animal models with NSC23766 treatment suggests benefit for inhibiting metastasis and angiogenesis (53). Additionally, Rac1 inhibition may mitigate Trastuzumab resistance in breast cancer cell lines, thus suggesting that the availability of a clinically accessible selective Cdc42 and Rac1 inhibitors could offer new paradigms for combinatorial therapies (53). We envision that the use of R-ketorolac in the perioperative window and thereafter presents a unique opportunity to block tumor reseeding and spread, as well as angiogenesis, which may enhance the efficacy of combined chemotherapies or targeted therapies.
Supplementary Material
Acknowledgments
Financial support: NCI R21CA170375, DOD OC110514 W81XWH-11-OCRP-TEA, UNM Science and Technology Corporation Gap Fund (A. Wandinger-Ness and L.G. Hudson), Core Facility support (Flow Cytometry Shared Resource, Fluorescence Microscopy Shared Resource, Clinical Trials Office) from the University of New Mexico Cancer Research and Treatment Center (P30 CA118100) (C. Willman). Focus Interactive Group grants from UNM Cancer Center (0990MD; 0990Q8 to A. Wandinger-Ness and L.G. Hudson). NCI R25CA153825 (predoctoral fellowship) and the George D. Montoya Scholarship to Y. Guo; INBRE NCRR 5P20RR016480 (predoctoral fellowship to S.R. Kenney).
Footnotes
Conflict of Interest: Authors Angela Wandinger-Ness, Larry A. Sklar, and Laurie Hudson are listed as co-inventors on a US patent application (allowance pending) entitled “Modulators of GTPases and Their Use”. The patent is owned by the University of New Mexico and its technology transfer office, STC.unm, and is under an option to license.
References
- 1.Howlader N NA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, et al. National Cancer Institute; Bethesda, MD: SEER Cancer Statistics Review, 1975-2011. Based on November 2012 SEER data submission, posted to the SEER web site, April 2013. Available from: http://seer.cancer.gov/csr/1975_2010/ [Google Scholar]
- 2.Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nature reviews Clinical oncology. 2013;10:211–24. doi: 10.1038/nrclinonc.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erickson BK, Conner MG, Landen CN., Jr The role of the fallopian tube in the origin of ovarian cancer. American journal of obstetrics and gynecology. 2013;209:409–14. doi: 10.1016/j.ajog.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudson LG, Zeineldin R, Stack MS. Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clinical & experimental metastasis. 2008;25:643–55. doi: 10.1007/s10585-008-9171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horowitz M, Neeman E, Sharon E, Ben-Eliyahu S. Exploiting the critical perioperative period to improve long-term cancer outcomes. Nature reviews Clinical oncology. 2015;12:213–26. doi: 10.1038/nrclinonc.2014.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Retsky M, Rogers R, Demicheli R, Hrushesky WJ, Gukas I, Vaidya JS, et al. NSAID analgesic ketorolac used perioperatively may suppress early breast cancer relapse: particular relevance to triple negative subgroup. Breast cancer research and treatment. 2012;134:881–8. doi: 10.1007/s10549-012-2094-5. [DOI] [PubMed] [Google Scholar]
- 7.Sadok A, Marshall CJ. Rho GTPases: masters of cell migration. Small GTPases. 2014;5:e29710. doi: 10.4161/sgtp.29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leve F, Morgado-Diaz JA. Rho GTPase signaling in the development of colorectal cancer. Journal of cellular biochemistry. 2012;113:2549–59. doi: 10.1002/jcb.24153. [DOI] [PubMed] [Google Scholar]
- 9.Ma J, Xue Y, Liu W, Yue C, Bi F, Xu J, et al. Role of activated rac1/cdc42 in mediating endothelial cell proliferation and tumor angiogenesis in breast cancer. PloS one. 2013;8:e66275. doi: 10.1371/journal.pone.0066275. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Grise F, Bidaud A, Moreau V. Rho GTPases in hepatocellular carcinoma. Biochimica et biophysica acta. 2009;1795:137–51. doi: 10.1016/j.bbcan.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Leng R, Liao G, Wang H, Kuang J, Tang L. Rac1 expression in epithelial ovarian cancer: effect on cell EMT and clinical outcome. Medical oncology. 2015;32:329. doi: 10.1007/s12032-014-0329-5. [DOI] [PubMed] [Google Scholar]
- 12.Guo Y, Kenney SR, Cook L, Adams S, Rutledge T, Romero E, et al. A novel pharmacologic activity of ketorolac for therapeutic benefit in ovarian cancer patients. Clinical Cancer Research an official journal of the American Association for Cancer Research. 2015 Jun 12; doi: 10.1158/1078-0432.CCR-15-0461. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eswaran J, Li DQ, Shah A, Kumar R. Molecular pathways: targeting p21-activated kinase 1 signaling in cancer--opportunities, challenges, and limitations. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:3743–9. doi: 10.1158/1078-0432.CCR-11-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker NM, Yee Chow H, Chernoff J, Der CJ. Molecular pathways: targeting RAC-p21-activated serine-threonine kinase signaling in RAS-driven cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4740–6. doi: 10.1158/1078-0432.CCR-13-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Villasana V, Fuentes-Mattei E, Ivan C, Dalton HJ, Rodriguez-Aguayo C, Fernandez-de Thomas RJ, et al. Rac1/Pak1/p38/MMP-2 Axis Regulates Angiogenesis in Ovarian Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2127–37. doi: 10.1158/1078-0432.CCR-14-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adam O, Laufs U. Rac1-Mediated Effects of HMG-CoA Reductase Inhibitors (Statins) in Cardiovascular Disease. Antioxidants & redox signaling. 2014;20:1238–50. doi: 10.1089/ars.2013.5526. [DOI] [PubMed] [Google Scholar]
- 17.Sendur MA, Aksoy S, Yazici O, Ozdemir NY, Zengin N, Altundag K. Statin use may improve clinicopathological characteristics and recurrence risk of invasive breast cancer. Medical oncology. 2014;31:835. doi: 10.1007/s12032-013-0835-x. [DOI] [PubMed] [Google Scholar]
- 18.Cardwell CR, Hicks BM, Hughes C, Murray LJ. Statin use after colorectal cancer diagnosis and survival: a population-based cohort study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:3177–83. doi: 10.1200/JCO.2013.54.4569. [DOI] [PubMed] [Google Scholar]
- 19.Walker EJ, Ko AH, Holly EA, Bracci PM. Statin use and risk of pancreatic cancer: Results from a large, clinic-based case-control study. Cancer. 2015;121:1287–94. doi: 10.1002/cncr.29256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habis M, Wroblewski K, Bradaric M, Ismail N, Yamada SD, Litchfield L, et al. Statin therapy is associated with improved survival in patients with non-serous-papillary epithelial ovarian cancer: a retrospective cohort analysis. PloS one. 2014;9:e104521. doi: 10.1371/journal.pone.0104521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7618–23. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, et al. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nature chemical biology. 2006;2:39–46. doi: 10.1038/nchembio751. [DOI] [PubMed] [Google Scholar]
- 23.Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Noth J, et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. The Journal of biological chemistry. 2013;288:8531–43. doi: 10.1074/jbc.M112.435941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, et al. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Molecular pharmacology. 2000;57:976–83. [PubMed] [Google Scholar]
- 25.Guo Y, Kenney SR, Romero R, Oprea T, Adams S, Muller C, et al. Selected NSAIDs target GTPases for ovarian cancer therapy. Clin Cancer Res. 2013;19:19s. suppl; abstr B81. [Google Scholar]
- 26.Hoffmann-La Roche Limited. PrToradol® IM. Copyright 1990-2015 by Hoffmann-La Roche Ltd. Date of Revision Feb. 19, 2015. Available from: http://www.rochecanada.com/content/dam/internet/corporate/rochecanada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Toradol/Toradol_PM_CIE.pdf.
- 27.Handley DA, Cervoni P, McCray JE, McCullough JR. Preclinical enantioselective pharmacology of (R)- and (S)- ketorolac. Journal of clinical pharmacology. 1998;38:25S–35S. doi: 10.1002/j.1552-4604.1998.tb04414.x. [DOI] [PubMed] [Google Scholar]
- 28.Jett MF, Ramesha CS, Brown CD, Chiu S, Emmett C, Voronin T, et al. Characterization of the analgesic and anti-inflammatory activities of ketorolac and its enantiomers in the rat. The Journal of pharmacology and experimental therapeutics. 1999;288:1288–97. [PubMed] [Google Scholar]
- 29.Forget P, Machiels JP, Coulie PG, Berliere M, Poncelet AJ, Tombal B, et al. Neutrophil:lymphocyte ratio and intraoperative use of ketorolac or diclofenac are prognostic factors in different cohorts of patients undergoing breast, lung, and kidney cancer surgery. Annals of surgical oncology. 2013;20(Suppl 3):S650–60. doi: 10.1245/s10434-013-3136-x. [DOI] [PubMed] [Google Scholar]
- 30.Mahller Y, Walsh C, Cass I, Rimel B, Karlan B, Li A. Peri-operative ketorolac is associated with improved epithelial ovarian cancer survival. Gynecologic oncology. 2012;127:S25. [Google Scholar]
- 31.Reyners AK, de Munck L, Erdkamp FL, Smit WM, Hoekman K, Lalisang RI, et al. A randomized phase II study investigating the addition of the specific COX-2 inhibitor celecoxib to docetaxel plus carboplatin as first-line chemotherapy for stage IC to IV epithelial ovarian cancer, Fallopian tube or primary peritoneal carcinomas: the DoCaCel study. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2012;23:2896–902. doi: 10.1093/annonc/mds107. [DOI] [PubMed] [Google Scholar]
- 32.Forget P, Berliere M, van Maanen A, Duhoux FP, Machiels JP, Coulie PG, et al. Perioperative ketorolac in high risk breast cancer patients. Rationale, feasibility and methodology of a prospective randomized placebo-controlled trial. Medical hypotheses. 2013;81:707–12. doi: 10.1016/j.mehy.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz SL, Tessema M, Buranda T, Pylypenko O, Rak A, Simons PC, et al. Flow cytometry for real-time measurement of guanine nucleotide binding and exchange by Ras-like GTPases. Analytical biochemistry. 2008;381:258–66. doi: 10.1016/j.ab.2008.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacob A, Jing J, Lee J, Schedin P, Gilbert SM, Peden AA, et al. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. Journal of cell science. 2013;126:4647–58. doi: 10.1242/jcs.126573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller C. A Pilot Trial to Study the Availability and Effect of Post-OP IV Ketorolac on Ovarian, Fallopian Tube or Primary Peritoneal Cancer, Cells Retrieved From the Peritoneal Cavity. [cited 2015 May 18]; Available from: https://clinicaltrials.gov/ct2/show/NCT01670799?term=ketorolac+and+ovarian+cancer&rank=1.
- 36.Shepherd TG, Theriault BL, Campbell EJ, Nachtigal MW. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nature protocols. 2006;1:2643–9. doi: 10.1038/nprot.2006.328. [DOI] [PubMed] [Google Scholar]
- 37.Agola JO, Hong L, Surviladze Z, Ursu O, Waller A, Strouse JJ, et al. A competitive nucleotide binding inhibitor: in vitro characterization of Rab7 GTPase inhibition. ACS chemical biology. 2012;7:1095–108. doi: 10.1021/cb3001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buranda T, Basuray S, Swanson S, Agola J, Bondu V, Wandinger-Ness A. Rapid parallel flow cytometry assays of active GTPases using effector beads. Analytical biochemistry. 2013;442:149–57. doi: 10.1016/j.ab.2013.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphries MJ. Cell adhesion assays. Molecular biotechnology. 2001;18:57–61. doi: 10.1385/MB:18:1:57. [DOI] [PubMed] [Google Scholar]
- 40.Bellone S, Siegel ER, Cocco E, Cargnelutti M, Silasi DA, Azodi M, et al. Overexpression of epithelial cell adhesion molecule in primary, metastatic, and recurrent/chemotherapy-resistant epithelial ovarian cancer: implications for epithelial cell adhesion molecule-specific immunotherapy. International journal of gynecological cancer: official journal of the International Gynecological Cancer Society. 2009;19:860–6. doi: 10.1111/IGC.0b013e3181a8331f. [DOI] [PubMed] [Google Scholar]
- 41.Taylor DD, Gercel-Taylor C, Parker LP. Patient-derived tumor-reactive antibodies as diagnostic markers for ovarian cancer. Gynecologic oncology. 2009;115:112–20. doi: 10.1016/j.ygyno.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Zhao ZS, Manser E. PAK family kinases: Physiological roles and regulation. Cellular logistics. 2012;2:59–68. doi: 10.4161/cl.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer metastasis reviews. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bokoch GM, Reilly AM, Daniels RH, King CC, Olivera A, Spiegel S, et al. A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. The Journal of biological chemistry. 1998;273:8137–44. doi: 10.1074/jbc.273.14.8137. [DOI] [PubMed] [Google Scholar]
- 45.Diaz-Munoz MD, Osma-Garcia IC, Iniguez MA, Fresno M. Cyclooxygenase-2 deficiency in macrophages leads to defective p110gamma PI3K signaling and impairs cell adhesion and migration. Journal of immunology. 2013;191:395–406. doi: 10.4049/jimmunol.1202002. [DOI] [PubMed] [Google Scholar]
- 46.Arjonen A, Kaukonen R, Ivaska J. Filopodia and adhesion in cancer cell motility. Cell adhesion & migration. 2011;5:421–30. doi: 10.4161/cam.5.5.17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nature reviews Molecular cell biology. 2008;9:446–54. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 48.Courtneidge SA. Cell migration and invasion in human disease: the Tks adaptor proteins. Biochemical Society transactions. 2012;40:129–32. doi: 10.1042/BST20110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Zhang Z, Roe SM, Marshall CJ, Barford D. Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science. 2009;325:1398–402. doi: 10.1126/science.1174468. [DOI] [PubMed] [Google Scholar]
- 50.Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, et al. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. The Journal of biological chemistry. 2010;285:34950–9. doi: 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forget P, Vandenhende J, Berliere M, Machiels JP, Nussbaum B, Legrand C, et al. Do intraoperative analgesics influence breast cancer recurrence after mastectomy? A retrospective analysis. Anesthesia and analgesia. 2010;110:1630–5. doi: 10.1213/ANE.0b013e3181d2ad07. [DOI] [PubMed] [Google Scholar]
- 52.Dormond O, Foletti A, Paroz C, Ruegg C. NSAIDs inhibit alpha V beta 3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nature medicine. 2001;7:1041–7. doi: 10.1038/nm0901-1041. [DOI] [PubMed] [Google Scholar]
- 53.Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Molecular cancer therapeutics. 2013;12:1925–34. doi: 10.1158/1535-7163.MCT-13-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.