

Abstract
The treatment options for cancer patients include surgery, chemotherapeutics, radiation therapy, antibody therapy and various combinations of these therapies. The challenge with each therapy is finding the balance between maximizing the anti-tumor efficacy while minimizing the dose limiting toxicities. Antibodies, unlike small molecule chemotherapeutics, selectively bind to cell surface tumor antigens and can be used to deliver radionucleotides or small molecule chemotherapeutic drugs directly to the tumor. Advances in antibody engineering, linker chemistry and the identification of potent cytotoxic drugs led to the recent approval of two antibody drug conjugates to treat breast cancer and lymphoma patients. We will discuss how the observations from the clinical development of antibody drug conjugates can guide the preclinical development of the next generation of antibody drug conjugates.
KEY WORDS: antibody drug conjugate, preclinical, site specific
Introduction
Small molecule chemotherapeutics and antibodies are commonly used to treat cancer patients. Small molecule chemotherapeutics, such as doxorubicin, paclitaxel and vinblastine, are delivered systemically, have relatively short serum half-lives and do not specifically target tumor cells. These molecules not only kill rapidly dividing tumor cells but they can also kill some normal cells, which results in various dose limiting toxicities (DLTs), such as neutropenia. DLTs reduce the amount of drug that can be administered to a patient which can reduce the effectiveness of a drug.
Antibodies are also delivered systemically but tend to have long serum half-lives and selectively bind to tumor cells. Antibody drug conjugates (ADCs) are a combination of the small molecule chemotherapeutic and the antibody thus allowing the selective delivery of the small molecule chemotherapeutic drugs to tumor cells and not normal cells.
Although average Drug/Antibody Ratios (DAR) are typically in the range from 2 to 4 drugs per antibody, most ADCs in clinical development today are a mix of antibodies carrying 0 to as many as 12 cytotoxic drugs covalently conjugated to specific amino acids on the antibody via a chemical linker. ADCs bind to cell surface antigens; they are typically internalized by the cells and processed by the lysosomes where the cytotoxic drugs are released into the cytotsol. Once the drugs are released from the lysosomes, they bind to their intracellular target, disrupt a critical cellular process and kill the cell.
The preclinical development of ADCs involves several steps which include the identification of the tumor antigen, the discovery and characterization of an antibody against the tumor antigen, the identification of the appropriate cytotoxic drug, the conjugation of the cytotoxic drug to the antibody and the characterization of the amount of aggregate and other physiochemical properties of the ADC. The preclinical evaluation of ADCs includes antibody/antigen binding studies, in vitro cytotoxic studies, in vivo anti-tumor efficacy studies, pharmacokinetic and the toxicology studies in rodent and non-human primates.
The observations from the clinical development of ADCs have been crucial in refining the preclinical development of ADCs. Improvements in antibody engineering, potency of cytotoxic drugs and improvements in the linker chemistry lead to the current generation of ADCs. We will discuss how data from the current clinical studies can be used to improve the preclinical development of the next generation of ADCs.
ADCs: A Historical Perspective
Paul Ehrlich, the German physician and scientist, described the concept of delivering a toxophore, a cytotoxic drug, selectively to tumors. ADCs are the embodiment of this concept. The first generation of ADCs used common chemotherapeutic drugs such as methotrexate, vinblastine and doxorubicin as cytotoxic drug payloads. KS1/4 and BR96 were the first antibodies to enter clinical development as ADCs.
KS1/4 was a murine IgG2a antibody against a 40 and 42 kD glycoprotein expressed by the human lung adenocarcinoma cell line, UCLA-P3 (1). The KS1/4 antigen is expressed by several cancers including ovarian, lung, pancreatic and colorectal cancers. KS1/4 was conjugated to methotrexate (KS1/4-methotrexate) or vinblastine (KS1/4-DAVLB) (2,3). There were 6 molecules of methotrexate and 4 to 6 molecules of vinblastine per antibody on lysines using hemisuccinate linkers. Preclinical in vivo anti-tumor efficacy was reported for the KS1/4-methotrexate and the KS1/4-DAVLB ADCs but no significant clinical responses were observed. Patients treated with the KS1/4 antibody or KS1/4 ADCs produced an antibody response against the mouse antibody, also known as a human anti mouse antibody (HAMA) response. Although the HAMA response has been reported to result in rapid systemic clearance of the antibody thus rendering the antibody or in this case ADC ineffective, high serum levels of the KS1/4 antibody were reported in patients treated with the higher doses of the KS1/4 antibody or ADCs. Subsequent ADCs used chimeric, humanized or fully human antibodies to reduce the patient’s immune response against the antibody.
BR96-Doxorubicin (SGN-15) was licensed by Seattle Genetics from Bristol-Meyer Squibb (BMS) (4). SGN-15 was a chimeric antibody against the Lewis Y (CD174) antigen that was conjugated to doxorubicin (adriamycin) using an acid labile, 6-maleimidocaproyl hydrazone linker (5,6). In preclinical studies, SGN-15 was able to selectively kill Lewis Y expressing cells in both in-vitro cytotoxicity and in in-vivo tumor efficacy studies yet it was unable to show statistically significant clinical benefit and further development was discontinued. The lack of clinical benefit has been attributed to several factors including the insufficient cytotoxic potency of doxorubicin, the instability of the hydrazone linker and the expression of Lewis Y by several normal tissues. (7–9).
CMD-193, which was developed by Wyeth Pharmaceuticals, Inc, was a humanized antibody (hu3S193) against the Lewis Y antigen that was conjugated to the DNA synthesis inhibitor, N-acetyl gamma calicheamicin dimethyl hydrazide (Calicheamicin) using the acid labile 4-(4′-acetylphenoxy) butanoic acid) linker (10). In preclinical studies, CMD-193, like SGN-15, was able to kill Lewis Y expressing tumors in both in vitro cytotoxicity studies and in vivo tumor efficacy studies (10) . In a phase I clinical study, myelosuppression and prolonged liver uptake which affected liver function were the most significant adverse events (11). Further clinical development of CMD-193 was terminated.
Gemtuzumab Ozogamacin (Mylotarg), which was also developed by Wyeth Pharmaceuticals, Inc (now Pfizer) and Celltech (now a part of UCB Brussels), was a humanized anti-CD33 IgG4 antibody, conjugated to Calicheamicin via the acid labile 4-4′-acetylphenoxy butanoic acid linker. Mylotarg had an average of 3 molecules of Calicheamicin per antibody and approximately 50% of the antibody did not contain Calicheamicin (12). Despite the high amount of unconjugated antibody, which could reduce its effectiveness, Mylotarg was able to kill CD33 expressing cells in in-vitro cytotoxicity studies and in preclinical in-vivo tumor efficacy studies (13). Mylotarg showed promise in clinical studies and was the first ADC to gain approval from the food and drug administration (FDA) in May 2000. Despite the promising clinical data, Mylotarg was eventually withdrawn from the market, in 2010, due to a lack of efficacy and an increased number of deaths in acute myelogenous leukemia (AML) patients as compared to patients treated with the standard of care chemotherapeutics (14).
Subsequent preclinical efforts to develop ADCs focused on the identification of potent cytotoxic drugs, linker chemistries that provide sufficient in vitro and in vivo stability and refinement of conjugation conditions to reduce the amount of unconjugated antibody.
In 2011 Adcetris (Brentuximab vedotin) was approved by the FDA to treat systemic anaplastic large cell lymphoma (sALCL) and Hodgkin’s lymphoma (HL). Adcetris is a chimeric IgG1 antibody against CD30 that is conjugated to an average of four molecules of the cytotoxic drug, monomethyl auristatin E (MMAE), via the thiols on cysteines using a valine-citrulline dipeptide linker with a p-aminobenzyl alcohol (PABA) spacer. MMAE is a potent inhibitor of cell growth in vitro. MMAE’s average in vitro IC50 values, against a panel of human tumor cell lines, were commonly below 1 nM as opposed to doxorubicin which had an average IC50 value of 631 nM (15).
In 2013, Kadcyla (T-DM1) was approved by the FDA to treat Her2 expressing metastatic breast cancer patients. Kadcyla is a humanized IgG1 antibody against Her2 that is conjugated to an average of 3.5 molecules of the cytotoxic maytansine drug, N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine (DM1), via the amines on lysines using the 4-[N-maleimidomethyl] cyclohexane-1-carboxylate (MCC) linker. DM1’s average in vitro cytotoxicity IC50 values against several tumor cell lines were less than 1 nM (16,17).
The clinical development of ADCs is expanding rapidly. Over 30 ADCs are in clinical development (Table I). The majority of ADCs are in either Phase I, Phase II or have active clinical efforts in both Phase I and Phase II (Fig. 1). Only one ADC, Inotuzumab Ozogamicin, has an active Phase III clinical effort. Over 15 ADCs have been withdrawn from clinical development for various reasons, which include a lack of anti-tumor response, unacceptable toxicity or a change in company strategy. The results from the clinical development of ADCs influence the preclinical development efforts, which lead to the creation of the next generation of ADCs.
Table I.
ADCs in Clinical Development
| ADC | Target | Phase of Development | Indication | Isotype | Payload |
|---|---|---|---|---|---|
| ABT-414 | Activated EGFR/EGFRvIII | Phase I | GBM, SCC | IgG1 | MMAF |
| AGS16M8F | ENPP3 | Phase I | RCC | IgG2 | MMAF |
| ASG22M6E | Nectin 4 | Phase I | Solid tumors | IgG1 | MMAE |
| AGS67E | CD37 | Phase I | NHL, CLL, AML | IgG2 | MMAE |
| AMG 172 | CD27L | Phase I | RCC | IgG1 | DM1 |
| AMG-595 | EGFRvIII | Phase I | GBM | ND | DM1 |
| ASG15E | SLITRK6 | Phase I | Solid tumors | IgG2 | MMAE |
| Bay 94-9343 | Mesothelin | Phase I | Solid tumors | IgG1 | DM4 |
| BT-062 | CD138 | Phase I/II | Multiple myeloma | IgG4 | DM4 |
| Glembatumumab Vedotin (CDX-011) | GPNMB | Phase II | Breast cancer | IgG2 | MMAE |
| IMGN-853 | FOLR1 | Phase I | Solid tumors | IgG1 | DM4 |
| IMMU-130 | CEACAM5 | Phase I | Colorectal | IgG1 | SN-38 |
| IMMU-132 | TACSTD2 (TROP2) | Phase I | Epithelial cancer | IgG1 | SN-38 |
| IMGN529 | CD37 | Phase I | NHL, CLL | IgG1 | DM1 |
| Inotuzumab Ozogamicin (CMC-544) | CD22 | Phase III | ALL | IgG4 | CalichDMH |
| MDX-1203 | CD70 (MDX-1115) | Phase I | Clear cell RCC, B-NHL | ND | CC-1065 |
| Milatuzumab | CD74 | Phase I/II | Multiple myeloma | IgG1 | Doxorubicin |
| MLN0264 | Guanylyl cyclase C | Phase I | Colorectal | ND | MMAE |
| PF-0626350 | 5T4 | Phase I | Solid tumor | ND | MMAF |
| PSMA (BrUOG 263) | PSMA | Phase I/II | Prostate | IgG1 | MMAE |
| RG7450 (DSTP3086S) | STEAP1 | Phase I | Prostate | ND | MMAE |
| RG7458 (DMUC5754A) | MUC16 | Phase I | Ovarian | IgG1 | MMAE |
| RG7593 (DCDT2980S) | CD22 | Phase I | NHL | IgG1 | MMAE |
| RG7596 (DCDS4501A) | CD79b | Phase I/II | NHL | IgG1 | MMAE |
| RG7598 | ND | Phase I | ND | ND | Undisclosed |
| RG7599 (DNIB0600A) | NaPi2b | Phase I | Ovarian, NSCLC | IgG1 | MMAE |
| RG7600 | ND | Phase I | ND | ND | ND |
| RG7636 | Endothelin B receptor | Phase I | Melanoma | ND | MMAE |
| SAR3419 | CD19 | Phase I | DLBCL, ALL | IgG1 | DM4 |
| SAR566658 | CA6 | Phase I | Solid tumor | IgG1 | DM4 |
| SC16LD6.5 | Fyn3 | Phase I/II | SCLC | ND | D6.5 |
| SGN-CD19A | CD19 | Phase I | ALL, NHL | ND | MMAF |
| SGN-CD33A | CD33 | Phase I | AML | IgG1 | PBD (SGD-1882) |
| SGN-LIV1A | LIV-1 | Phase I | Breast cancer | ND | MMAE |
ND not disclosed, SN-30 7-ethyl-10-hydroxycamptothecin, PBD Pyrrolobenzodiazepine, MMAE Monomethylauristatin E, MMAF Monomethylauristatin F, GBM Glioblastoma multiforme, RCC Renal cell cancer, SCLC Small cell lung cancer, NSCLC Non-small cell lung cancer, AML Acute Lymphoblastic Leukemia, NHL Non-hodgkin lymphoma, ALL Acute lymphoblastic leukemia, DLBCL Diffuse large B-cell lymphoma, SCC Squamous cell carcinoma
Fig. 1.

Clinical development of ADCs.
There are several key areas of focus for the preclinical development of ADCs. These areas include the selection of an ADC target, the selection of an appropriate antibody and the selection of appropriate cytotoxic drugs and linkers. Furthermore, attention has to be given to characterizing the physiochemical properties of the ADCs, evaluation of the in vitro cytotoxicity, the in vivo anti- tumor efficacy of the ADCs, the pharmacokinetic properties of the ADCs and their toxicology/safety profiles (Fig. 2).
Fig. 2.
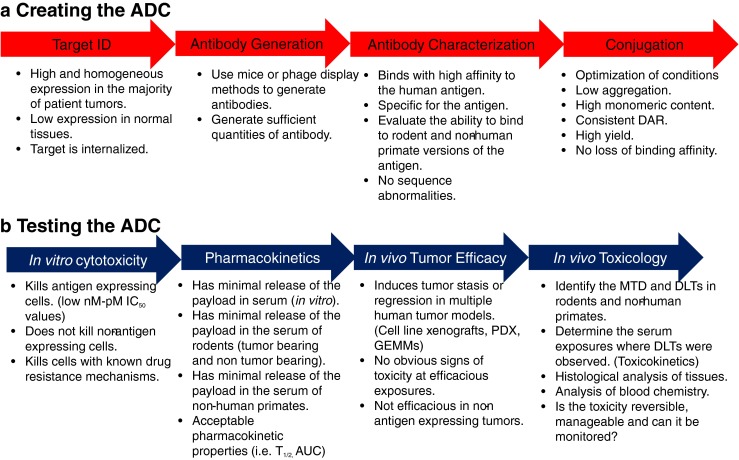
Preclinical development and evaluation of ADCs.
Target Selection
The optimal selection criteria for cell surface tumor antigens are that the antigen should be expressed selectively and at higher levels by tumors than normal tissues and the antigens are internalized by the tumor cell. Furthermore it would be preferred if the tumor antigens are expressed homogeneously to increase the number of cells killed by the ADC. It is unclear what the threshold receptor density or the internalization rate should be for a tumor antigen because the receptor density and the internalization rates may vary depending on the tumor antigen and tumor type. Thus far three ADCs have been approved to treat cancer patients. Those ADCs are Gemtuzumab Ozogamicin (Mylotarg), which targets CD33 expressing tumors; Adcetris, which targets CD30 expressing tumors and Kadcyla, which targets Her2 expressing tumors (Table II).
Table II.
List of Approved ADCs
| ADC | Target | Isotype | Payload | Indication | Date of Approval |
|---|---|---|---|---|---|
| Mylotarg | CD33 | IgG4 | Calicheamicin | AML | 2000 (US) a |
| Kadcyla (T-DM1) | Her2 | IgG1 | DM1 | Her2 positive late stage breast | 2013 (US) |
| Adcetris (SGN-35) | CD30 | IgG1 | MMAE | sALCL, relapsed/refractory Hodgkin lymphoma | 2011 (US) |
AML Acute Lymphoblastic Leukemia, ACLC Anaplastic large cell lymphoma
a Withdrawn in 2010
CD33
CD33 (Siglec-3) is expressed by the precursor myeloid and monocytic cells. CD33 is expressed in approximately 85–90% of adult and child AML patients and in 100% of acute promyelocytic leukemia (APL), which is a subset of AML (18,19). CD33 expression correlates with poor prognosis for children with ALL (20). High levels of CD33 expression have been reported in the bone marrow of AML patients while substantially lower levels of expression have been observed in normal bone marrow (21). The restricted expression of CD33 in normal tissues and the high levels of expression in AML made CD33 an attractive therapeutic target.
Three ADCs, Mylotarg, AVE9633, and SGN-CD33A, have been developed to treat patients with CD33 expressing tumors.
AVE9633 was a humanized IgG1 antibody conjugated to an average of 3–5 molecules of N2′-deacetyl-N2′-(4-mercapto-4-methyl-1-oxopentyl)-6-methylmaytansine (DM4) via a hindered disulfide linker on lysines. In preclinical studies, AVE9633 selectively killed AML cell lines and AML patient samples in vitro (22,23). Three phase I studies were performed and a total of 54 AML patients were treated with AVE9633. Patients were treated on day 1 of a 21 day cycle (Day 1), days 1 and 8 (Day 1/8) and days 1, 4 and 7 (Day1/4/7) of a 28 day cycle. The day 1 and day1/4/7 schedules were terminated early due lack of cytotoxic activity at doses higher than the doses that saturated binding to CD33 (22).
Mylotarg, as was previously mentioned, was removed from the market in 2010, which leaves SGN-CD33A as the only ADC currently in clinical development against CD33. SGN-CD33A is composed of humanized IgG1 antibody (h2H12), which contains two engineered cysteines at position 239 (S239C) on the antibody heavy chains. The DNA synthesis inhibitor, made by the former Spirogen LTD (now Astra-Zeneca/MedImmune), pyrrolobenzodiazepine (PBD), is covalently conjugated to each site specific cysteine using the maleimidocaproyl-valine-alanine linker. SGN-CD33A is the first publically disclosed site specific ADC in clinical development.
CD30
CD30 (TNFRSF8) is expressed on activated T and B cells and a small population of eosinophils. It is also expressed in sALCL, HL, mature T cell lymphomas and B cell derived non-Hodgkins lymphoma (NHL). sALCL is a rare type of NHL, comprising approximately 3% of all NHL but is one of the more common T cell lymphomas (24). CD30 is expressed uniformly on sALCL (25). A soluble form of CD30 has been reported in the sera of cancer patients but this does not seem to have a deleterious effect on Adcetris (26). Limited expression of CD30 has been reported in normal tissues and high expression has been reported in sALCL, HL and other malignancies (27).
Adcetris had objective response rates of 75% for Hodgkin’s lymphoma (HL) patients and 86% for sALCL patients. Tumor reductions were reported in 94% of the HL patients and 97% of the sALCL patients (28). Interestingly CD30 expression in diffuse large B cell lymphoma (DLBCL) patients did not correlate with patient response to Adcetris. Tumor responses were observed in DLBCL patients that had undetectable expression of CD30 via immunohistochemical evaluation (29). These data suggest that CD30 expression alone in DLBCL patients may not predict response to Adcetris. It also suggests that low CD30 expression is sufficient for responses to Adcetris.
Her2 (ERBB2)
Unlike CD33 and CD30, which are predominately expressed by hematological malignancies, Her2 is predominantly expressed by solid tumors. In particular, it is expressed in breast, gastric and ovarian cancer amongst others. Her2 is over expressed by approximately 25% of patients with invasive breast cancer. It is uniformly expressed in breast cancer and high expression of Her2 correlates with poor prognosis (30–33). Her 2 is also expressed by several normal tissues including, the skin, respiratory tract, heart, gastrointestinal epithelial cells and in the urinary and reproductive tracts (34). Kadcyla, also known as T-DM1, was approved in 2013 for Her2 expressing late stage breast cancer patients. The objective response rate in the pivotal EMILIA trial was 43.6% for patients treated with Kadcyla and 30.8% for patients treated with lapatinib and capecitabine (35).
Solid Tumor Antigens
Developing antibody therapeutics for targets expressed in solid tumors may offer some additional challenges that may not exist for hematological cancer targets. For hematological cancers, it is possible to determine the dose that achieves receptor saturation. Once that dose is achieved, the addition of higher amounts of ADC would not be beneficial. Antibody delivery to solid tumors, however, is very complex. Solid tumors have high intratumoral pressures, due to the lack of lymphatics (36). Solid tumors have leaky blood vessels and chaotic blood flow, which can result in heterogeneous distribution of the antibody within the tumor thus rendering portions of the tumor inaccessible to the antibody or ADC (36–38). Another factor to consider is the large size of antibodies, which have a molecular weight of ~150 kD. The large size of the antibodies may limit their ability to efficiently penetrate deep within a solid tumor mass thus leaving portions of the tumor untreated and free to continue to grow (39). In addition to the physical properties of the antibody, the expression of the antigen must also be taken into consideration. If the tumor antigen is heterogeneously expressed within the tumor, portions of the tumor could be devoid of ADC thus allowing portions of the tumor to be untreated, which enables tumor growth. Lastly antibody affinities may impact anti-tumor efficacy of an ADC. Antibodies with high affinities are tightly bound to the antigen and may be unable to penetrate the tumor as well as antibodies with lower affinities, which will reduce the effectiveness of the ADC (40,41).
The current operating hypothesis is an ADC must be internalized in order for the drug to be released from the antibody and subsequently kill the antigen expressing cell. This has resulted in linkers that are designed to release their drug in a low pH environment or upon proteolysis using lysosomal enzymes. Although this is one way ADCs function, it is possible that some ADC drugs could be released without being internalized. The anti-CEACAM5 ADC (Epratuzumab-SN-38), which uses the CL2A linker combined with the SN-38 drug, has been reported to deliver the ADC to the antigen expressing tumors in vivo but releases the SN-38 drug without being efficiently internalized (42). The authors state that SN-38 is slowly released from the anti-CEACAM5 antibody (@50%/day) and the in vitro internalization rate for the anti-CEACAM5 antibody is slow. However, it is not clear that in vitro rates of internalization necessarily predict in vivo rates. Thus, the use of pH sensitive dyes (i.e. pHRodoTM) or far red dyes to label ADCs to evaluate the internalization and intracellular trafficking of ADCs in vivo, could be a useful tool to determine the importance of internalization to in vivo efficacy.
Selecting the Antibody
Antibody Isotype
The first ADCs were developed using murine antibodies. Patients developed HAMA responses against the murine antibody, which may have limited the effectiveness of these ADCs (3). The majority of the ADCs currently in clinical development are chimeric, fully human or humanized IgG1 antibodies. IgG2 and IgG4 isotypes are also being developed as ADCs (Table I).
There are structural differences between the various antibody isoforms (Fig. 3). The IgG2 isotype has four interchain disulfide bonds in the hinge region while the IgG1 and IgG4 isoforms have two interchain disulfide bonds (Fig. 3). The conjugation of the cytotoxic drug to the antibody, on cysteines, requires a partial reduction of the disulfides in the hinge region of the antibody. Tris (2-carboxyethyl)-phosphine (TCEP) is commonly used to reduce the antibody thus increasing the number of free thiols available for conjugation. IgG2 antibodies require higher concentrations of TCEP and longer reaction times than is required for IgG1 antibodies (43). Furthermore the IgG2 isoform exists as an A form, a B form and a hybrid A/B form (44). The significance of how these different IgG2 isoforms affect the production or conjugation of a drug to an ADC is unclear.
Fig. 3.

IgG isotypes used as ADCs.
Some IgG1 antibodies have antibody-dependent cellular cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC), which is achieved through binding of the antibody’s Fc domain to the Fcγ receptors on natural killer cells, monocytes and macrophages (45). IgG2 and IgG4 antibodies have little to no CDC activity (46). Although IgG1 antibodies, such as Trastuzumab, can have ADCC and CDC activities, it is unclear whether these activities are important for an ADC (47). To address the potential differences between IgG1, IgG2 and IgG4 ADCs, an anti-CD70 ADC, which was conjugated to valine-citrulline monomethyl auristatin F (MMAF) (43). The authors also mutated the Fc domains of the anti-CD70 IgG1 (IgG1v1) and IgG4 (IgG4v1) ADCs to reduce binding affinity to the Fcγ receptors. The IgG1v1-vc4 ADC had better in vivo tumor efficacy than the native IgG1-vcF4 ADC, which was attributed to increased serum exposure of the Fc mutated IgG1 ADC. This provided only a partial explanation for the enhanced efficacy of the Fc mutated IgG1 ADC because the wild type IgG2-vcF4 ADC had comparable serum exposure as the Fc mutated IgG1 ADC yet it was not as efficacious as the mutated IgG1 ADC.
Cytotoxic Drugs and Linkers
Cytotoxic Drugs
The drugs used for ADCs inhibit cellular processes that are vital for cell proliferation and/or survival, such as tubulin polymerization or DNA replication (Table III). Inhibition of tubulin function or DNA synthesis induces apoptotic cell death. The current cytotoxic drugs, used as ADC payloads, are typically 100–2,000 fold more potent than doxorubicin, vinca alkaloids or taxanes (48–51). Although the majority of drugs in clinical development inhibit tubulin function or DNA synthesis, new drugs which inhibit critical cellular processes, such as RNA synthesis or membrane disruption are being developed as ADC payloads and may enter into clinical development. The cytotoxic drugs or their metabolites have varying degrees of membrane permeability. MMAE, for example, is membrane permeable and when released from a cell it may kill neighboring cells by a process known as bystander effect (52). MMAF has reduced membrane permeability and the metabolite, Cys-mcMMAF, does not exhibit bystander activity. Bystander activity may be useful in a solid tumor setting when the target antigen is heterogeneous expressed in the tumor. An anti-mesothelin ADC, Anetumab ravtansine, which is a fully human anti-mesothelin antibody conjugated to an average of 3.2 molecules of the cytotoxic maytansine drug DM4, was reported to exhibit bystander activity in tumor xenograft models (53). Tumor regressions were observed in tumor models where only 20% of the cells expressed mesothelin, which suggests that the non-mesothelin cells were killed by the release of the DM4 metabolite from the mesothelin expressing cells.
Table III.
ADC Payloads
| Drug/Payload (Payload name) | Mechanism of action | Stage of development |
|---|---|---|
| Auristatin (MMAE/MMMAF) | Tubulin assembly inhibitor | Approved/Clinical |
| Maytansinoid (DM1/DM4) | Tubulin assembly inhibitor | Approved/Clinical |
| Tubulysin | Tubulin assembly Inhibitor | Preclinical |
| SN-38 | Topoisomerase inhibitor | Clinical |
| Calicheamicin | DNA synthesis inhibitor | Clinical |
| Doxorubicin | DNA synthesis inhibitor | Clinical |
| Pyrrolobenzodiazepine (SGD-1882) | DNA synthesis inhibitor | Clinical |
| Duocarmycin (CC1065) | DNA synthesis inhibitor | Clinical |
| D6.5 | DNA synthesis inhibitor | Clinical |
| α- Amanitin | RNA polymerase II inhibitor | Preclinical |
Linkers
Linkers are grouped into two classes (Table IV). They are either cleavable or non-cleavable. Cleavable linkers can be dipeptide linkers, such as valine-citrulline, and are cleaved by proteases (i.e. Cathepsin B), which results in the release of the cytotoxic drug. Cleavable linkers can also be acid labile, such as hydrazones and hindered disulfides, where the lysosomal environment results in the release of the cytotoxic drug. ADCs with non-cleavable linkers are degraded in the lysosome, which results in the formation of an amino acid-drug combination (i.e. Cys-mcMMAF). Although the linkers can be processed by the enzymes or environment of the lysosome, some linkers such as maleimide linkers, can transfer the cytotoxic drug to free sulfhydryls such as the cysteine on human serum albumin (HSA) or glutathione via a retro-michael reaction (54,55). This can cause a 25% or greater decrease in the tumor exposure of the ADC (54). This decrease in tumor exposure may reduce the effectiveness of the ADC.
Table IV.
Linkers Used for ADCs
| Linker name | Cleavable/Non-cleavable | Conditions for payload release | Payload |
|---|---|---|---|
| Valine-citrulline (vc) | Cleavable | Protease | MMAE |
| SPP | Cleavable | Reducing/Oxidizing environment | DM1 |
| SPDB | Cleavable | Reducing/Oxidizing environment | DM4 |
| 6-maleimidocaproyl hydrazide | Cleavable | Acid labile | Doxorubicin |
| 4-(4′-acetylphenoxy) butanoic acid) | Cleavable | Acid labile | Calicheamicin |
| CL2A | Cleavable | pH mediated | SN-38 |
| SMCC | Non-cleavable | Antibody degradation | DM1 |
| Maleimidocaproyl (mc) | Non-cleavable | Antibody degradation | MMAF |
SPP N-succinimidyl 4-(2-pyridyldithio)pentanoate, SMCC N-succinimidyl 4-(N-maleimidomethyl)cyclohexane-1 carboxylate, SPDB N-succinimidyl-4-(2-pyridyldithio)butyrate, CL2A maleimido-[short PEG]-Lys-PABOCO-20-O-SN-38
The linker/drug combination can influence the pharmacokinetics and the toxicology profile of the ADC. A humanized antibody (huC242) against CanAg/MUC1 was conjugated to the SPP-DM1 and SPDB-DM4 linker/drug combinations and evaluated in preclinical and clinical studies (Table V). The serum half-life in humans for the huC242-SPP-DM1 (Cantuzumab Mertansine), using a Q3W dosing schedule, ranged between 18 and 48.5 hrs while the serum half-life for huC242-SPDB-DM4 (IMGN242) was between 60 and 120 hrs (56). In addition to the apparent differences in the half-lives of the ADCs, the dose limiting toxicities (DLT) were also different. The DLT for the huC242-SPP-DM1 ADC was elevated hepatic transaminase and the DLT for the huC242-SPDB-DM4 ADC was ocular toxicity, which was only observed in patients with low serum levels of CanAg (56). These data suggest that the different drug linker combinations can have significant effects on the pharmacokinetics of the ADC and the toxicology profiles. Interestingly, several ADCs have been reported to have reversible ocular toxicity in patients (Table VI). Ocular toxicity has been observed primarily for ADCs conjugated to DM4 or MMAF but Kadcyla, which is conjugated to DM1, reported ocular toxicity in 31.3% of the patients in a phase II study (57). These observations have resulted in the inclusion of ophthalmologic evaluation in preclinical toxicology studies. A patent application, US 20120282282 A1, was filed by Immunogen where the claims suggest that the ocular toxicity for DM4 ADCs can be decreased by modifying the charge of the linker. The uncharged SPDB linker was reported to display decreased ocular toxicity in a rabbit model than the charged sulfo-SPDB linker.
Table V.
Clinical Summary of huC242 ADCs
| Antibody | Payload | Linker | Schedule | MTD (mg/m2) | T1/2 (hours) | DLT | Clinical Phase |
|---|---|---|---|---|---|---|---|
| huC242 | DM1 | SPP | Q1W | 115 | 13–41 | Elevated hepatic transaminase | I |
| huC242 | DM1 | SPP | Q3W | 235 | 18–48.5 | Elevated hepatic transaminase | I |
| huC242 | DM4 | SPDB | Q3W | 168 | 60–120 | Reversible ocular toxicity | I |
| huC242 | DM4 | SPDB | Q3W | 168 | N/A | Reversible ocular toxicity | II |
Table VI.
ADCs with Ocular Toxicity in the Clinic
| Antibody | Target | Linker | Payload |
|---|---|---|---|
| SAR3419 | CD19 | SPDB | DM4 |
| IMGN853 | Folate Receptor a | SPDB | DM4 |
| SGN-75 | CD70 | mc | MMAF |
| SAR566658 | CA6 | SPDB | DM4 |
| BT-062 | CD138 | SPDB | DM4 |
| huC242-DM4 | CanAg | SPDB | DM4 |
| SGN-CD19A | CD19 | mc | MMAF |
| Kadcyla | Her2 | SMCC | DM1 |
Drug Resistance
One of the reasons why cancer therapies fail is due to drug resistance (58). There are several mechanisms responsible for resistance to chemotherapies, which includes the expression of drug-efflux pumps (59). Drug-efflux pumps lower the intracellular concentration of the drug which renders the drug ineffective. The expression of several drug efflux pumps including multi-drug resistance 1(MDR1/ABCB1), multi-drug resistance protein 1(MRP1/ABCC1) and breast cancer resistance protein (BCRP/ABCG2) have been implicated in resistance to various chemotherapeutics. Several cytotoxic drugs, used for ADCs, such as MMAE, DM1 and Calicheamicin, are known substrates of the drug efflux pump MDR1 (ABCB1) (60,61). MDR1 substrates tend to be hydrophobic drugs therefore modifying the metabolites to be polar or negatively charged may reduce the drugs from being pumped out of the cells via MDR1 (62).To counteract the MDR1 drug efflux pump, the linker used to conjugate DM1 to an anti-EpCam antibody was changed from N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate (SMCC) to the hydrophobic linker PEG4Mal. The resulting metabolite, lysine-PEG4Mal-DM1 was able to kill MDR1 expressing cells while the lysine-SMCC-DM1 was unable to kill MDR1 expressing cells (63). These data suggest that selection of the appropriate linker may be important to avoid resistance mediated via drug efflux pumps. In addition to selecting the appropriate linker to avoid drug efflux pumps, cytotoxic drugs that are not substrates for efflux pumps, such as PBDs, may also be considered. SGN-CD33A is conjugated to the PBD,SGD-1882, which is not an MDR1 substrate. SGN-CD33A can kill MDR1 expressing CD33 positive AML patient samples and is currently in Phase I.
Several methods, using fluorescent substrates for the various drug efflux pumps, have been used to assess the activity of the pumps and may be useful in characterizing the contributions of the various pumps to resistance to the ADC metabolites (64). Rhodamine 123, for example, can be used to assess drug efflux pump activity in cell lines and specific inhibitors of various efflux pumps can be used to determine the specific drug efflux pump’s contribution to the efflux activity in a cell line or patient derived cells (65).
Antibody Conjugation Options
There are two methods used to covalently attach a cytotoxic drug to an antibody. Currently, the more conventional method, conjugates the cytotoxic drugs to an antibody’s solvent exposed cysteine or lysine residues. The site specific conjugation method conjugates the cytotoxic drugs to natural or non-natural amino acids that have been engineered at different locations on the antibody or to glycosyl moieties.
Conventional Conjugation
The majority of the ADCs in clinical development are in this category. The cytotoxic drugs are conjugated to antibodies via the thiols on solvent exposed cysteines or the amines on lysines. The conjugation of cytotoxic drugs to the antibodies results in a heterogeneous mixture of ADCs where the number of cytotoxic drugs conjugated to an antibody typically varies from 0 to 8 drugs per antibody. Antibodies with a DAR of 8 or more have a higher clearance rate than antibodies with a lower DAR (11). The majority of the ADCs have an average DAR of approximately 4 drugs per antibody, which offers an acceptable balance between pharmacokinetic properties, toxicities and anti-tumor efficacy.
Site Specific Conjugation
Cytotoxic drugs are conjugated to amino acids, typically cysteines or non-natural amino acids, which have been incorporated at distinct locations on the antibody. The resulting ADC is homogeneous composition where all of the antibodies contain a defined number of drugs per antibody. Typically a DAR of 2 is used although higher DARs could be achieved. The incorporation of a cytotoxic drug on specific positions of the antibody can influence the pharmacokinetic properties of the ADC. Several examples of site specifically conjugated ADCs, using antibodies containing engineered cysteines or the non-natural amino acid, p-acetyl phenyalanine (pAF), have reported improved in vitro and in vivo serum stability, improved pharmacokinetic properties, comparable anti-tumor efficacy and improved toxicology profiles as compared to the conventional ADCs (55,66,67). Cytotoxic drugs conjugated to pAF via an oxime bond are reported to have improved serum stability when compared to cytotoxic drugs conjugated to thiols via maleimide (10,55).
One cysteine engineered site specifically conjugated ADC, SGN-CD33A, is currently in phase I clinical development. This is probably the first of many site specific ADCs that will be evaluated in the clinic.
A couple of novel approaches for site-specific conjugation not requiring molecular engineering of the antibody are cysteine bridging approaches, which can specifically conjugate all of the cysteines with a defined DAR or ½ the number of cysteines (4 for a typical IgG1), and conjugation of glycosyl moieties.
There are several companies developing site specific conjugation technologies but most are currently in preclinical development (Table VII). This is an exciting area of development for ADCs.
Table VII.
Companies Developing Site Specific ADC Technologies
| Company | Type of Technology | Stage of Development |
|---|---|---|
| Ambrx Inc | Non-natural amino acid (NNAA) | Preclinical |
| Genentech/Roche, Novartis | Engineered cysteine (Thiomab) | Preclinical |
| Igenica Inc | (SNAP ADC) no mutation or NNAA | Preclinical |
| Meditope Bioscience Inc | Cetuximab derived residue grafts into Fab | Preclinical |
| Pfizer | FXIII substrate peptide - acceptor glutamine | Preclinical |
| Poly Therics Ltd | ThioBridge | Preclinical |
| Redwood Bioscience Inc (acquired by Catalent Pharma Solutions) | Aldehyde tag | Preclinical |
| Seattle Genetics | Engineered Cysteine | Clinical |
| Sutro Biopharma Inc | Non-natural amino acid | Preclinical |
| SynAffix | Glycosyl conjugation | Preclinical |
Challenges of Preclinical Development of ADCs
The data from clinical evaluation of ADCs has led to significant changes in the preclinical development of ADCs. The current cytotoxic drugs used as ADC payloads are more potent than methotrexate, doxorubicin and vinblastine, which were previously used as ADC payloads. The current sets of linkers, used to conjugate the drugs to the antibodies, are more stable than the previous linkers and the amount of unconjugated antibody has been dramatically reduced. These changes were incorporated into the preclinical development of the two FDA approved ADCs, Kadcyla and Adcetris.
The clinical development of more than 15 ADCs has been discontinued because the ADCs did not provide sufficient anti-tumor responses, had unacceptable toxicology profiles or the company decided to support other products in their pipeline. These ADCs apparently had an acceptable preclinical data package that supported the decision to progress them into clinical development. Inotuzumab Ozogamicin (CMC-544) has an active Phase III clinical effort and is currently the most advanced ADC in clinical development. CMC-544 showed significant inhibition of tumor growth in various preclinical human B-cell lymphoma cell line models (68). Pfizer, which is developing CMC-544, recently announced that it is discontinuing a Phase III trial in NHL but will continue the Phase III INO-VATE ALL study (B1931022) in ALL. This suggests that preclinical in vivo anti-tumor efficacy studies may not predict patient responses to the ADCs and additional preclinical studies may be warranted to increase the correlation between the data from the preclinical studies and clinical responses.
An area of focus in the preclinical development of ADCs could be in improving the tumor models used to evaluate the ADCs. Preclinical studies have suggested that human tumor xenografts, derived from cell lines, do not accurately reflect the original patient tumors they were derived from and may not predict clinical anti-tumor activity (69,70). Patient derived tumor xenografts (PDX) are derived from freshly isolated patient tumors that are implanted, grown and propagated in immunodeficient mice. A large panel of patient derived colon tumors was reported to accurately represent the histology and the range of molecular heterogeneity observed in colon cancer patients (71). The authors conducted an evaluation of cetuximab in the colorectal cancer PDX tumors and confirmed the key role of KRAS mutation in cetuximab resistance. The increased use of PDX models may provide better predictive value than the use of human tumor cell lines. ADCs against 5T4 and tissue factor have reported preclinical in vivo anti-tumor activity in PDX models (72,73).
Perhaps attention to the pharmacokinetics and pharmacodynamics of the ADC may add value to the preclinical effort. An evaluation of a PK/PD modeling approach reported that tumor growth rate and the ratio between the exposures and concentrations that induced tumor stasis were important in predicting tumor responses to Kadcyla (74). This PK/PD model was modified using the clinical PK data for Kadcyla and was used to predict the clinically efficacious exposures for an ADC against the 5T4 tumor antigen (75). The utility of this modeling approach is currently being evaluated.
The use of site specific ADCs, improvements in the pharmacokinetic properties of the ADCs through the development of better linkers and the use of potent cytotoxic drugs as ADC payloads combined with improvements in tumor model selection and target identification may result in higher success rates for ADCs in the clinic.
Abbreviations
- ALCL
Anaplastic large cell lymphoma
- ADC
Antibody drug conjugate
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloid leukemia
- CL2A
maleimido-[short PEG]-Lys-PABOCO-20-O-SN-38
- DAR
Drug to antibody ratio
- DLBCL
Diffuse large B-cell lymphoma
- GBM
Glioblastoma multiforme
- kD
Kilodalton
- MMAE
Monomethylauristatin E
- MMAF
Monomethylauristatin F
- ND
Not disclosed
- NHL
Non-hodgkin lymphoma
- NNAA
Non-natural amino acid
- NSCLC
Non-small cell lung cancer
- PBD
Pyrrolobenzodiazepine
- PDX
Patient derived xenograft
- PK/PD
Pharmacokinetic/Pharmacodynamic
- Q3W
Every 3 weeks
- RCC
Renal cell cancer
- SCC
Squamous cell carcinoma
- SCLC
Small cell lung cancer
- SMCC
N-succinimidyl 4-(N-maleimidomethyl)cyclohexane-1 carboxylate
- SN-30
7-ethyl-10-hydroxycamptothecin
- SPDB
N-succinimidyl-4-(2-pyridyldithio)butyrate
- SPP
N-succinimidyl 4-(2-pyridyldithio)pentanoate
References
- 1.Varki NM, Reisfeld RA, Walker LE. Antigens associated with a human lung adenocarcinoma defined by monoclonal antibodies. Cancer Res. 1984;44:681–7. [PubMed] [Google Scholar]
- 2.Schneck D, Butler F, Dugan W, Littrell D, Petersen B, Bowsher R, et al. Disposition of a murine monoclonal antibody vinca conjugate (KS1/4-DAVLB) in patients with adenocarcinomas. Clin Pharmacol Ther. 1990;47:36–41. doi: 10.1038/clpt.1990.5. [DOI] [PubMed] [Google Scholar]
- 3.Elias DJ, Hirschowitz L, Kline LE, Kroener JF, Dillman RO, Walker LE, et al. Phase I clinical comparative study of monoclonal antibody KS1/4 and KS1/4-methotrexate immunconjugate in patients with non-small cell lung carcinoma. Cancer Res. 1990;50:4154–9. [PubMed] [Google Scholar]
- 4.Goldmacher VS, Kovtun YV. Antibody-drug conjugates: using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther Deliv. 2011;2:397–416. doi: 10.4155/tde.10.98. [DOI] [PubMed] [Google Scholar]
- 5.Trail PA, Willner D, Lasch SJ, Henderson AJ, Hofstead S, Casazza AM, et al. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science. 1993;261:212–5. doi: 10.1126/science.8327892. [DOI] [PubMed] [Google Scholar]
- 6.Willner D, Trail PA, Hofstead SJ, King HD, Lasch SJ, Braslawsky GR, et al. (6-Maleimidocaproyl)hydrazone of doxorubicin--a new derivative for the preparation of immunoconjugates of doxorubicin. Bioconjug Chem. 1993;4:521–7. doi: 10.1021/bc00024a015. [DOI] [PubMed] [Google Scholar]
- 7.Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest. 1970;49:673–80. doi: 10.1172/JCI106279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol. 2012;30:631–7. doi: 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- 9.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–44. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 10.Tian F, Lu Y, Manibusan A, Sellers A, Tran H, Sun Y, et al. A general approach to site-specific antibody drug conjugates. Proc Natl Acad Sci U S A. 2014;111:1766–71. doi: 10.1073/pnas.1321237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 12.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs. 2011;3:161–72. doi: 10.4161/mabs.3.2.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golay J, Di Gaetano N, Amico D, Cittera E, Barbui AM, Giavazzi R, et al. Gemtuzumab ozogamicin (Mylotarg) has therapeutic activity against CD33 acute lymphoblastic leukaemias in vitro and in vivo. Br J Haematol. 2005;128:310–7. doi: 10.1111/j.1365-2141.2004.05322.x. [DOI] [PubMed] [Google Scholar]
- 14.Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res. 2011;17:6417–27. doi: 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- 15.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–84. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 16.Widdison WC, Wilhelm SD, Cavanagh EE, Whiteman KR, Leece BA, Kovtun Y, et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J Med Chem. 2006;49:4392–408. doi: 10.1021/jm060319f. [DOI] [PubMed] [Google Scholar]
- 17.Oroudjev E, Lopus M, Wilson L, Audette C, Provenzano C, Erickson H, et al. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol Cancer Ther. 2010;9:2700–13. doi: 10.1158/1535-7163.MCT-10-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8:521–34. doi: 10.1016/0145-2126(84)90001-8. [DOI] [PubMed] [Google Scholar]
- 19.Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67:1048–53. [PubMed] [Google Scholar]
- 20.Mejstrikova E, Kalina T, Trka J, Stary J, Hrusak O. Correlation of CD33 with poorer prognosis in childhood ALL implicates a potential of anti-CD33 frontline therapy. Leukemia. 2005;19:1092–4. doi: 10.1038/sj.leu.2403737. [DOI] [PubMed] [Google Scholar]
- 21.Jilani I, Estey E, Huh Y, Joe Y, Manshouri T, Yared M, et al. Differences in CD33 intensity between various myeloid neoplasms. Am J Clin Pathol. 2002;118:560–6. doi: 10.1309/1WMW-CMXX-4WN4-T55U. [DOI] [PubMed] [Google Scholar]
- 22.Lapusan S, Vidriales MB, Thomas X, de Botton S, Vekhoff A, Tang R, et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Invest New Drugs. 2012;30:1121–31. doi: 10.1007/s10637-011-9670-0. [DOI] [PubMed] [Google Scholar]
- 23.Tang R, Cohen S, Perrot JY, Faussat AM, Zuany-Amorim C, Marjanovic Z, et al. P-gp activity is a critical resistance factor against AVE9633 and DM4 cytotoxicity in leukaemia cell lines, but not a major mechanism of chemoresistance in cells from acute myeloid leukaemia patients. BMC Cancer. 2009;9:199. doi: 10.1186/1471-2407-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Younes A, Yasothan U, Kirkpatrick P. Brentuximab vedotin. Nat Rev Drug Discov. 2012;11:19–20. doi: 10.1038/nrd3629. [DOI] [PubMed] [Google Scholar]
- 25.Deng C, Pan B, O’Connor OA. Brentuximab vedotin. Clin Cancer Res. 2013;19:22–7. doi: 10.1158/1078-0432.CCR-12-0290. [DOI] [PubMed] [Google Scholar]
- 26.Vaklavas C, Forero-Torres A. Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Ther Adv Hematol. 2012;3:209–25. doi: 10.1177/2040620712443076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katz J, Janik JE, Younes A. Brentuximab Vedotin (SGN-35) Clin Cancer Res. 2011;17:6428–36. doi: 10.1158/1078-0432.CCR-11-0488. [DOI] [PubMed] [Google Scholar]
- 28.Garnock-Jones KP. Brentuximab vedotin: a review of its use in patients with hodgkin lymphoma and systemic anaplastic large cell lymphoma following previous treatment failure. Drugs. 2013;73:371–81. doi: 10.1007/s40265-013-0031-5. [DOI] [PubMed] [Google Scholar]
- 29.Bartlett NL, Sharman JP, Oki Y, Advani RH, Bello CM, Winter JN, et al. A phase 2 study of brentuximab vedotin in patients with relapsed or refractory CD30-positive non-hodgkin lymphomas: interim results in patients with DLBCL and other B-cell lymphomas. 2013. [PubMed]
- 30.Yonemura Y, Ninomiya I, Yamaguchi A, Fushida S, Kimura H, Ohoyama S, et al. Evaluation of immunoreactivity for erbB-2 protein as a marker of poor short term prognosis in gastric cancer. Cancer Res. 1991;51:1034–8. [PubMed] [Google Scholar]
- 31.Sasano H, Date F, Imatani A, Asaki S, Nagura H. Double immunostaining for c-erbB-2 and p53 in human stomach cancer cells. Hum Pathol. 1993;24:584–9. doi: 10.1016/0046-8177(93)90236-A. [DOI] [PubMed] [Google Scholar]
- 32.Uchino S, Tsuda H, Maruyama K, Kinoshita T, Sasako M, Saito T, et al. Overexpression of c-erbB-2 protein in gastric cancer. Its correlation with long-term survival of patients. Cancer. 1993;72:3179–84. doi: 10.1002/1097-0142(19931201)72:11<3179::AID-CNCR2820721108>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 33.Nakajima M, Sawada H, Yamada Y, Watanabe A, Tatsumi M, Yamashita J, et al. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer. 1999;85:1894–902. doi: 10.1002/(SICI)1097-0142(19990501)85:9<1894::AID-CNCR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 34.Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5:953–62. [PubMed] [Google Scholar]
- 35.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milosevic MF, Fyles AW, Hill RP. The relationship between elevated interstitial fluid pressure and blood flow in tumors: a bioengineering analysis. Int J Radiat Oncol Biol Phys. 1999;43:1111–23. doi: 10.1016/S0360-3016(98)00512-4. [DOI] [PubMed] [Google Scholar]
- 37.Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2:a006536. doi: 10.1101/cshperspect.a006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi IK, Strauss R, Richter M, Yun CO, Lieber A. Strategies to increase drug penetration in solid tumors. Front Oncol. 2013;3:193. doi: 10.3389/fonc.2013.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–34. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31:1191–8. [PubMed] [Google Scholar]
- 41.Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61:4750–5. [PubMed] [Google Scholar]
- 42.Sharkey RM, Govindan SV, Cardillo TM, Goldenberg DM. Epratuzumab-SN-38: a new antibody-drug conjugate for the therapy of hematologic malignancies. Mol Cancer Ther. 2012;11:224–34. doi: 10.1158/1535-7163.MCT-11-0632. [DOI] [PubMed] [Google Scholar]
- 43.McDonagh CF, Kim KM, Turcott E, Brown LL, Westendorf L, Feist T, et al. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Ther. 2008;7:2913–23. doi: 10.1158/1535-7163.MCT-08-0295. [DOI] [PubMed] [Google Scholar]
- 44.Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem. 2008;283:16194–205. doi: 10.1074/jbc.M709987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. 2010;2:181–9. doi: 10.4161/mabs.2.2.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Presta LG. Engineering antibodies for therapy. Curr Pharm Biotechnol. 2002;3:237–56. doi: 10.2174/1389201023378256. [DOI] [PubMed] [Google Scholar]
- 47.Petricevic B, Laengle J, Singer J, Sachet M, Fazekas J, Steger G, et al. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Transl Med. 2013;11:307. doi: 10.1186/1479-5876-11-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 49.Pietersz GA, Rowland A, Smyth MJ, McKenzie IF. Chemoimmunoconjugates for the treatment of cancer. Adv Immunol. 1994;56:301–87. doi: 10.1016/S0065-2776(08)60455-1. [DOI] [PubMed] [Google Scholar]
- 50.Tolcher AW, Sugarman S, Gelmon KA, Cohen R, Saleh M, Isaacs C, et al. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J Clin Oncol. 1999;17:478–84. doi: 10.1200/JCO.1999.17.2.478. [DOI] [PubMed] [Google Scholar]
- 51.Tassone P, Gozzini A, Goldmacher V, Shammas MA, Whiteman KR, Carrasco DR, et al. In vitro and in vivo activity of the maytansinoid immunoconjugate huN901-N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res. 2004;64:4629–36. doi: 10.1158/0008-5472.CAN-04-0142. [DOI] [PubMed] [Google Scholar]
- 52.Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–97. doi: 10.1158/1078-0432.CCR-09-2069. [DOI] [PubMed] [Google Scholar]
- 53.Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13:1537–48. doi: 10.1158/1535-7163.MCT-13-0926. [DOI] [PubMed] [Google Scholar]
- 54.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, et al. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19:759–65. doi: 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- 55.Jackson D, Atkinson J, Guevara CI, Zhang C, Kery V, Moon SJ, et al. In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLoS One. 2014;9:e83865. doi: 10.1371/journal.pone.0083865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tolcher AW, Ochoa L, Hammond LA, Patnaik A, Edwards T, Takimoto C, et al. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the CanAg antigen: a phase I, pharmacokinetic, and biologic correlative study. J Clin Oncol. 2003;21:211–22. doi: 10.1200/JCO.2003.05.137. [DOI] [PubMed] [Google Scholar]
- 57.Burris HA, 3rd, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 58.Lippert TH, Ruoff HJ, Volm M. Intrinsic and acquired drug resistance in malignant tumors. The main reason for therapeutic failure. Arzneimittelforschung. 2008;58:261–4. doi: 10.1055/s-0031-1296504. [DOI] [PubMed] [Google Scholar]
- 59.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 60.Sammet B, Steinkuhler C, Sewald N. Antibody-drug conjugates in tumor therapy. Pharm Pat Anal. 2012;1:65–73. doi: 10.4155/ppa.12.4. [DOI] [PubMed] [Google Scholar]
- 61.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 62.Loo TW, Clarke DM. Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J Membr Biol. 2005;206:173–85. doi: 10.1007/s00232-005-0792-1. [DOI] [PubMed] [Google Scholar]
- 63.Kovtun YV, Audette CA, Mayo MF, Jones GE, Doherty H, Maloney EK, et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010;70:2528–37. doi: 10.1158/0008-5472.CAN-09-3546. [DOI] [PubMed] [Google Scholar]
- 64.Lebedeva IV, Pande P, Patton WF. Sensitive and specific fluorescent probes for functional analysis of the three major types of mammalian ABC transporters. PLoS One. 2011;6:e22429. doi: 10.1371/journal.pone.0022429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forster S, Thumser AE, Hood SR, Plant N. Characterization of rhodamine-123 as a tracer dye for use in in vitro drug transport assays. PLoS One. 2012;7:e33253. doi: 10.1371/journal.pone.0033253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 67.Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res. 2010;16:4769–78. doi: 10.1158/1078-0432.CCR-10-0987. [DOI] [PubMed] [Google Scholar]
- 68.DiJoseph JF, Armellino DC, Boghaert ER, Khandke K, Dougher MM, Sridharan L, et al. Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood. 2004;103:1807–14. doi: 10.1182/blood-2003-07-2466. [DOI] [PubMed] [Google Scholar]
- 69.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–9. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006;66:3351–4. doi: 10.1158/0008-5472.CAN-05-3627. [DOI] [PubMed] [Google Scholar]
- 71.Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goere D, Mariani P, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18:5314–28. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 72.Breij EC, de Goeij BE, Verploegen S, Schuurhuis DH, Amirkhosravi A, Francis J, et al. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014;74:1214–26. doi: 10.1158/0008-5472.CAN-13-2440. [DOI] [PubMed] [Google Scholar]
- 73.Sapra P, Damelin M, Dijoseph J, Marquette K, Geles KG, Golas J, et al. Long-term tumor regression induced by an antibody-drug conjugate that targets 5T4, an oncofetal antigen expressed on tumor-initiating cells. Mol Cancer Ther. 2013;12:38–47. doi: 10.1158/1535-7163.MCT-12-0603. [DOI] [PubMed] [Google Scholar]
- 74.Jumbe NL, Xin Y, Leipold DD, Crocker L, Dugger D, Mai E, et al. Modeling the efficacy of trastuzumab-DM1, an antibody drug conjugate, in mice. J Pharmacokinet Pharmacodyn. 2010;37:221–42. doi: 10.1007/s10928-010-9156-2. [DOI] [PubMed] [Google Scholar]
- 75.Haddish-Berhane N, Shah DK, Ma D, Leal M, Gerber HP, Sapra P, et al. On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: a PK/PD approach. J Pharmacokinet Pharmacodyn. 2013;40:557–71. doi: 10.1007/s10928-013-9329-x. [DOI] [PubMed] [Google Scholar]