

Abstract
The generation of NO by the various NO synthases in normal and malignant tissues is manifested by various biological effects that are involved in the regulation of cell survival, differentiation and cell death. The role of NO in the cytotoxic immune response was first revealed by demonstrating the induction of iNOS in target cells by immune cytokines (e.g. IFN-γ, IL-1, TNF-α, etc.) and resulting in the sensitization of resistant tumor cells to death ligands-induced apoptosis. Endogenous/exogenous NO mediated its immune sensitizing effect by inhibiting NF-κΒ activity and downstream, inactivating the repressor transcription factor YY1, which inhibited both Fas and DR5 expressions. In addition, NO-mediated inhibition of NF-κΒ activity and inhibition downstream of its anti-apoptotic gene targets sensitized the tumor cells to apoptosis by chemotherapeutic drugs. We have identified in tumor cells a dysregulated pro-survival/anti-apoptotic loop consisting of NF-κB/Snail/YY1/RKIP/PTEN and its modification by NO was responsible, in large, for the reversal of chemo and immune resistance and sensitization to apoptotic mechanisms by cytotoxic agents. Moreover, tumor cells treated with exogenous NO donors resulted in the inhibition of NF-κΒ activity via S-nitrosylation of p50 and p65, inhibition of Snail (NF-κΒ target gene), inhibition of transcription repression by S-nitrosylation of YY1 and subsequent inhibition of epithelial–mesenchymal transition (EMT), induction of RKIP (inhibition of the transcription repressor Snail), and induction of PTEN (inhibition of the repressors Snail and YY1). Further, each gene product modified by NO in the loop was involved in chemo-immunosensitization. These above findings demonstrated that NO donors interference in the regulatory circuitry result in chemo-immunosensitization and inhibition of EMT. Overall, these observations suggest the potential anti-tumor therapeutic effect of NO donors in combination with subtoxic chemo-immuno drugs. This combination acts on multiple facets including reversal of chemo-immune resistance, and inhibition of both EMT and metastasis.
Keywords: Apoptosis, Chemotherapeutic drugs, Nitric oxide, Nitrosylation, Sensitization, Trail DR5
Graphical abstract
Highlights
-
•
The induction of NO in tumor cells or exogenous NO donors reverse tumor cell resistance and chemo-immuno-sensitize the cells to apoptosis. The inhibition of the repressor YY1 by NO results in the upregulation of Fas and DR5 expressions on tumor cells and their sensitization to FasL, TRAIL and chemotherapeutic-induced apoptosis.
-
•
The mechanism by which NO mediates its sensitization is a result of its interference on the dysregulated pro-survival/anti-apoptotic NF-κΒ /Snail/YY1/RKIP/PTEN loop in tumor cells.
-
•
NO interference is the result of its direct inhibition of NF-κΒ, Snail, and YY1 and the induction of RKIP.
-
•
The inhibition of the repressor Snail and YY1 by NO results in the induction of RKIP and PTEN and inhibition of the pro-survival/anti-apoptotic NF-κΒ and AKT pathways, respectively.
-
•
NO donors are potential therapeutic agents to reverse resistance and to inhibit EMT and metastasis when used in combination with subtoxic chemo-immuno-cytotoxic agents.
1. Introduction
Since the early introduction of various nonspecific chemotherapeutic drugs and, subsequently, radiotherapeutics, hormonal drugs and immunotherapies, there have been significant clinical and objective responses in patients with various malignant diseases. While these therapeutic strategies continue to improve and are still currently in use in oncology, there remains a major problem in that a subset of cancer patients does not initially respond and another responding subset develops resistance to further treatments and both subsets succumb to the disease. Clearly, the non-responding patients underlie the rapid and successful development of new classes of targeted therapeutics based on the various molecular mechanisms that govern resistance. Noteworthy, it is recognized to date that many of the cytotoxic agents (drugs, hormones, immunotherapies) used in cancer therapy mediate their effects by inducing programmed cell death, or apoptosis, in the responding sensitive tumor cells. Hence, a major, but not the only mechanism of resistance, is the mechanism in acquisition of tumor cells of several molecular and genetic means to evade cell death by apoptosis.
The inhibition of pro-apoptotic pathways in cancer cells is the result of tumor cells exhibiting constitutively hyperactivated survival and proliferative signal pathways that counteract the pro-apoptotic pathways. A good example is the constitutively hyperactivated NF-κB pathway in the majority of cancers that was shown to play a pivotal role in tumorigenesis, angiogenesis, resistance, inflammation, immunomodulation, and metastasis. Further, other survival/proliferative/anti-apoptotic pathways are also upregulated in cancer cells such as the PI3K/AKT and MAPK pathways. These various pathways also crosstalk and synergize in their anti-apoptotic effects [1,2].
There have been several strategies to therapeutically inhibit anti-apoptotic pathways in cancer. Among these, many earlier and recent studies suggested the potential cytotoxic and sensitizing effects of NO in examined various cancer cell models, in vitro and in vivo [3–10]. Recently, Reynolds el al. [11] reviewed the use of NO in the inhibition of tumor cell proliferation and its consideration as a novel therapeutic.
2. Nitric oxide properties and applications
Nitric Oxide is a versatile and pleiotropic molecule. It has been reported to be involved in cell signaling and it regulates a variety of physiological functions in mammals [12,13]. NO is a lipophilic free radical gas that can readily diffuse through cell membranes and chemically reacts with metal containing proteins and other effector molecules. NO is synthesized from arginine by the nitric oxide synthases (NOS). Three types of NOS have been discovered, namely, eNOS (endothelial nitric oxide synthase or type III NOS), nNOS (neuronal nitric oxide synthase or type I NOS), and iNOS (inducible nitric oxide synthase or type II) [14]. Zinc finger proteins have been reported to be important targets of NO [15] and NO can also mediate nitrosation and nitrosylation of various proteins and affects their functions and activities [16–19].
The guanylyl cyclase (sGC) α-1 and α-2 heterodimers are found in many tissues and are the receptors for NO [20,21]. The generation of NO by nitroglycerine or nitroprusside activates soluble guanylate cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP) that, in turn, activates Protein kinase C (PKC) to phosphorylate proteins [22]. The recent findings by Bian and Murad [23] established that the NO/sGC/cGMP axis is involved in the regulation of proliferation and differentiation. They also reported that undifferentiated tumor cells and stem cells have low levels of sGC. Thus, by elevating endogenous cGMP it resulted in the inverse correlation with the proliferation and colony formation of glioma cells.
The various NOS isoforms' functions are interconnected and operate as a pivotal part of Red-OX-based molecular signaling systems that result in major impacts on the cell's physiology and behavior and include S-nitrothiols (SNOs), which are generally produced by S-nitrosation of cysteine thiols by NO. These result in the disruption or dysregulation of normal cell signaling and in the impairment of cellular functions [19,24]. Both eNOS and nNOS are calcium-dependent, but iNOs is calcium independent. Further, eNOS and nNOS release lower levels of NO whereas iNOS releases high levels of NO [25]. Of relevance, NO signaling has been reported to initiate apoptosis through its activity on the mitochondrial membrane permeability and the consequent release of cytochrome c oxidase [6, 26].
NO-mediated apoptosis consists also in the endogenous formation of reactive nitric oxide species (RNOS). These RNOS include superoxide (O−2) that reacts with NO to form peroxynitrite (ONOO−) that strongly induces apoptosis. Peroxynitrite acts as a DNA oxidant and induces single strand breaks in DNA [27,28]. Also, NO induces changes in the mitochondrial permeability transition pore (MPTP) and its opening results in the release of cytochrome c into the cytoplasm and induces apoptosis via binding Apaf-1 that activates caspase-9 leading to the activation of caspases 7 and 3 and downstream apoptosis [29]. The release of cytochrome c also results in the significant release of intracellular calcium resulting in cell death [30].
The consideration of the potential therapeutic application of NO donors has been the subject of many reports and reviews [7–9,11,13,31–33]. In this review, we will briefly discuss (1) the roles of NO-mediated regulation/interference of gene products that form a loop, namely, the NF-κB /Snail/YY1/RKIP/PTEN loop, that has been shown to be dysregulated in cancerous cells and that is also involved in drug/immune resistance (2) the roles of NO-mediated chemo- and immunosensitizations of resistant cancer cells to apoptosis by chemo-immuno cytotoxic agents (3) the role of NO donors in the inhibition in vivo in mice bearing tumor xenografts when used in combination with chemotherapy and (4) the clinical response in cancer patients treated with NO-mediated therapeutics.
3. NO-mediated interference in the dysregulated NF-κB/Snail/YY1/RKIP/PTEN loop responsible for chemo- and immune-resistance in cancer cells
Based on reported studies by us and others, we have established the presence of a dysregulated NF-κB/Snail/YY1/RKIP/PTEN loop in cancer cell model systems that regulates resistance and metastasis [34–38]. Each gene product of the loop was pivotal in the regulation of tumor cell sensitivity to various cytotoxic apoptotic stimuli (e.g., chemotherapeutic drugs, cytotoxic cells, cytotoxic ligands including FasL, TRAIL, and TNF-α). Treatment of tumor cells with high levels of NO donors (e.g., greater than 500 mM DETA-NONOate) resulted in the inhibition of the upregulated gene products in the loop, namely, NF-κB, Snail, and YY1 and the de-repression of down regulated gene products, namely, RKIP and PTEN. These NO-mediated effects were both direct and indirect and resulting in tumor cells' inhibition of cell proliferation and their sensitization to apoptotic stimuli.
Briefly, we present below the findings of NO-mediated effects on each of the gene products of the above loop.
3.1. NO-mediated inhibition of NF-κB
The transcription factor NF-κB is constitutively hyper-activated in many cancers and regulates, in large part, survival, proliferation, anti-apoptotic activity, angiogenesis, and metastatic properties of cancer cells [39]. The role of NF-κB in resistance to apoptotic stimuli is the result of the activation downstream of anti-apoptotic gene products such as XIAP, Bcl-xl, Bcl-2, and YY1 [40–43]. Hence, inhibition of NF-κB may reverse the above tumorigenic properties and reverse drug resistance. The inhibition of NF-κB by various agents, including drugs, small molecule inhibitors, siRNA, etc., results in the reversal of tumor cell resistance to apoptotic stimuli and sensitization to apoptosis by chemo-immunotherapeutic drugs [44–48].
Nitric oxide, endogenous or exogenous, mediates contrasting effects on tumor cells, namely, it can either promote or inhibit tumor cell survival and growth. These contrasting effects were found to be dependent on the level of NO; low level of NO is pro-tumorigenic whereas high level is anti-tumorigenic [49]. High level of NO inhibits NF-κB activity by inhibition of both the phosphorylation and degradation of IκBα and, thus, preventing NF-κB nuclear translocation and gene transcription [50]. NO will react with reactive oxygen species such as superoxide resulting in an impaired activation of NF-κB [51]. In addition, NO mediates direct effects by S-nitrosylation of NF-κB p50 and p65 and reduces its DNA-binding activity [52,53].
3.2. NO-mediated inhibition of Snail
Snail is a member of the Snail superfamily of zinc finger transcription factors. It plays important roles in embryonic development, neural differentiation, cell division, survival, invasion, EMT and metastasis [54,55]. Snail transcription is regulated, in part, by both NF-κB [56] and YY1 [57]. Snail represses the transcription of the metastasis suppressor E-cadherin [58] and RKIP [59]. Treatment of tumor cells with the NO donor, DETA-NONOate, inhibited Snail expression via NO-mediated inhibition of NF-κB and its target genes Snail and YY1[37,47]. Also, inhibition of Snail resulted in the derepression of E-cadherin and the induction of RKIP.
3.3. NO-mediated inhibition of YY1
The transcription factor YY1 is a DNA-binding factor that is multifunctional; it activates, represses or initiates transcription through either direct binding to DNA or indirectly through complexing with other DNA-binding proteins [60]. Our earlier findings demonstrated that YY1 represses the transcription of both Fas and DR5 and, hence, its role in immune resistance [61,62]. We have also reported that the treatment of tumor cells with the NO donor, DETA-NONOate, resulted in the S-nitrosylation of YY1 and further inhibition of its DNA-binding activity [63].
3.4. NO-mediated induction of RKIP
The Raf kinase inhibitory protein (RKIP) is a member of the phosphatidylethanolamine-binding protein family (PEBP) and it has been reported to play a role in lipid metabolism and phospholipid membrane biogenesis [64]. Yeung and colleagues were the first to clone RKIP and demonstrate its inhibitory effects on both the Raf/MEK/ERK and NF-κB pathways [65,66]. It has also been reported that RKIP negatively regulates the activation of G-protein coupled receptors (GPCRs) [67]. The metastatic inhibitor RKIP is significantly depressed in many tumors and almost absent in metastatic tumors [68–72].
Treatment of tumor cells with the NO donor, DETA-NONOate, resulted in significant upregulation of RKIP expression. This upregulation was the result of NO-mediated inhibition of NF-κB and its target the RKIP repressor Snail. The upregulation of RKIP subsequently results in the inhibition of both the NF-κB and the MAPK pathways and, thus, amplifying the inhibition of NF-κB, Snail and YY1 and upregulation of PTEN and RKIP.
3.5. NO-mediated induction of PTEN
PTEN is involved in the inhibition of the PI3K/AKT pathway [73]. It has been reported that both Snail and YY1 negatively regulate PTEN expression. In tumor cells whereby the NF-κB, Snail and YY1 are activated, they result in in the inactivation of PTEN and, thus, promoting the activation of the survival and anti-apoptotic PI3K/AKT pathway. The inhibition of NF-κB, Snail and YY1 by NO results in the derepression of PTEN and its upregulation and, thus, resulting in PTEN-mediated inhibition of the PI3K/AKT pathway and downstream target genes involved in both proliferation and anti-apoptosis [38].
4. NO-mediated immune-sensitization to apoptosis
Cytotoxic lymphocytes, primarily CTL and NK cells, mediate their cytotoxic activities by both necrosis and apoptosis. The induction of apoptosis is the result of CTL or NK activation following recognition and binding to the corresponding receptors on the target cell. The activation of the cytotoxic cell results in the utilization of the perforin/granzyme mechanism as well as the extracellular activation of the Fas-L, TRAIL and TNF-α- and their interactions and activation of their corresponding receptors [74].
There have been many approaches undertaken to reverse tumor cell resistance to cytotoxic stimuli. Among these are the use of nonspecific as well as specific sensitizing agents that modify the anti-apoptotic threshold of resistance and, thus, rendering the modified cells more susceptible to respond and be killed by the conventional therapeutic agents [75]. We have reported that treatment of drug/immune resistant tumor cells with NO donors sensitizes the cells to apoptosis by chemo-immunotherapeutic agents.
The majority of our studies used the NO donor, DETA-NONOate, a member of the family of diazoniumdiolate (formerly NONOate). DETA-NONOate spontaneously dissociates in a pH-dependent first order process and has a half-life of 20 h and 50 h at 37 °C and 22–25 °C, pH 7.4, respectively. It liberates two moles of NO per mole of the parent compound DETA-NONOate [76,77].
4.1. NO-mediated sensitization to FasL apoptosis
Our initial original findings regarding the role of NO-mediated sensitization of immune resistant cancer cells to apoptosis was the result of investigating the underlying mechanism by which interferon-γ (IFN-γ) sensitized Fas-resistant tumor cell lines to FasL apoptosis. We observed that treatment of the tumor cells with IFN-γ induced the upregulation of Fas expression on the cell surface [78]. It was also known that IFN-γ induces iNOS and NO release and, thus, we hypothesized that NO may be involved in the sensitization to FasL-mediated apoptosis. Hence, when the tumor cells were treated with an NO donor, SNAP, it resulted in the upregulation of Fas and sensitization to FasL-mediated apoptosis, mimicking IFN-γ treatment. The direct role of NO-mediated sensitization by IFN-γ was corroborated by the use of the NO inhibitor, L-NAME, which abrogated IFN-γ-mediated sensitization to FasL [78]. The molecular mechanism of NO-mediated upregulation of Fas was likely the result of either the activation of a transcription factor on the Fas promoter or the inhibition of a Fas transcription repressor. Analysis of the Fas promoter identified a silencer region with consensus binding sites for the transcription factor YY1. YY1 has been reported to act as both an activator and as a repressor [60]. Hence, deletion of the silencer region of the Fas promoter resulted in a significant augmentation of the Fas reporter activity. Also, treatment of tumor cells with DETA-NONOate resulted in the inhibition of YY1-DNA binding activity as assessed by EMSA [61]. In addition, DETA-NONOate treatment of the tumor cells resulted in the S-nitrosylation of YY1 and inhibition of its DNA-binding activity [41]. Overall, the findings revealed that treatment of tumor cells with DETA-NONOate resulted in the significant release of NO and consequent inhibition of the Fas YY1 repressor, the induction of Fas and sensitization to FasL-mediated apoptosis. The sensitization was the result of activation of the apoptotic pathway triggered by Fas and by NO and involved both type I and type II apoptotic pathways.
4.2. NO-mediated sensitization to TNF-alpha apoptosis
Several lines of evidence supported the role of the constitutively activated NF- κB pathway in cancer cells as pivotal of tumor cells resistance to TNF-α apoptosis [79,80]. TNF-α interacts with two distinct cell surface receptors, TNF-R1 and TNF-R2, and most cytotoxic effects of TNF-α are mediated by interaction with TNF-R2 [81]. TNF-α triggers the induction of various reactive oxygen species (ROS) [82], hence, the use of antioxidants inhibited TNF-α-mediated effects [83].
The ROS generated by TNF-α results from enzymatic partial reduction of oxygen yielding superoxide anion (O−2), which is immediately reduced by superoxide dismutase to H2O2 or reacts with NO to generate ONOO− [84].
Treatment of tumor cells with IFN-γ sensitized the cells to TNF-α apoptosis and induction of iNOS. Treatment of tumor cells with the NOS inhibitor, L-NMA, inhibited IFN-γ-mediated sensitization to TNF-α. In contrast, treatment with an NO donor (e.g., S-nitroso-N-acetylpencillamine) sensitized tumor cells to TNF-α apoptosis [51]. This finding shows that NO interferes with TNF-α-induced NF-κB activity by reacting with O−2 and reducing the TNF-α H2O2-mediated activation of NF-κB.
4.3. NO-mediated sensitization to TRAIL apoptosis
Reported studies using chemotherapeutic drugs and NF-κB inhibitors showed that their use with TRAIL-resistant cancer cells resulted in their sensitization to TRAIL apoptosis concurrently with upregulation of DR-4 or DR-5 expression [34,85–87]. The finding that NO inhibits NF-κB led us to hypothesize that NO can also sensitize TRAIL-resistant tumor cells to apoptosis by TRAIL. Treatment of tumor cells with DETA-NONOate sensitized the tumor cells to TRAIL apoptosis concomitantly with upregulation of DR-5 [34]. Similar to our findings with Fas-induced upregulation by NO, there were YY1 consensus binding sites on the DR-5 promoter. Treatment with DETA-NONOate augmented DR-5 reporter activity and transcription of cells with DR-5 constructs lacking YY1 binding sites or mutation of the YY1 sites, all of which resulted in enhancement of reporter activity. The direct role of YY1 on the suppression of DR-5 transcript was corroborated by the use of siRNA for YY1. The sensitization by DETA-NONOate of TRAIL-induced apoptosis was mediated by the activation of type-I and type-II apoptotic pathways. The combination treatment activated both caspases 9 and 3 and PARP cleavage [34].
Overall, the above findings demonstrated that NO modulates the dysregulated NF-κB/Snail/YY1/RKIP loop and results in the inhibition of NF-κB, Snail and YY1 and the induction of RKIP. These findings suggested that each of the gene products in the loop contributes to the resistance and that each gene product alone is directly involved in the sensitization of tumor cells to immunotherapeutic agents.
5. NO-mediated chemosensitization to apoptosis
Several mechanisms have been postulated regarding drug-resistance of tumor cells. Drug-resistance is also a consequence of the anti-apoptotic machinery in cancer cells since many chemotherapeutic drugs mediate their cytotoxic activity via apoptosis. Inhibitors of survival pathways have been reported to reverse drug resistance. For instance, inhibitors of the constitutively activated NF-κB pathway have reversed Cis-Diammine-Dichloro-Platinum (CDDP)-resistant cancer cell lines to respond to CDDP treatments [88]. The finding that NO treatment inhibits NF-κB activity led us to postulate it will also sensitize tumor cells to apoptosis by chemotherapeutic drugs. Indeed, treatment of human prostate cancer cell lines with DETA-NONOate and CDDP resulted in significant synergistic apoptosis. Treatment with DETA-NONOate inhibited anti-apoptotic gene products, particularly, Bclxl and XIAP in these lines. The combination treatment activated the type-II apoptotic mitochondrial pathway [89]. Similar findings have also been reported with CDDP-resistant metastatic colon cancer cell lines [90]. These findings corroborated an earlier and a first report by Wink's Group, whereby, and under conditions of high concentrations of NO, tumor cells were sensitized to various chemotherapeutic drugs [91].
The NO-mediated chemosensitization in prostate cancer cell lines revealed that both YY1 and Bclxl were inhibited and played pivotal roles in chemosensitization. Treatment of tumor cells with either siRNA YY1 or siRNA Bclxl sensitized tumor cells to CDDP apoptosis. The in vitro findings of the synergistic apoptotic activity by the combination treatment were corroborated in vivo in mice bearing PC3 tumor xenografts. Treatment with the combination of CDDP and DETA-NONOate in vivo, but not a single agent alone, resulted in synergistic inhibition of tumor growth [89].
6. NO-mediated interference in the NF-κB/Snail/YY1/RKIP loop and the role of each gene product in chemo-immunosensitization
6.1. Role of NO-mediated inhibition of NF-kB in chemo-immunosensitization
The NF-κB activity is inhibited by NO and resulting in the inhibition of the anti-apoptotic target genes as well as the derepression of pro-apoptotic gene products [34,92,93]. Various NO donors have been reported which inhibited NF-κB activity and sensitized drug-resistant tumor cells to cytotoxicity [94,95]. The implication of NF-κB -induced inhibition by NO in chemo-immunosensitization was corroborated by the use of NF-κB inhibitors, for example DHMEQ, which mimicked NO in both chemo and immunosensitizations of tumor cells to CDDP and TRAIL apoptosis, respectively [34,96].
6.2. Role of NO-mediated inhibition of YY1 in chemo-immunosensitization
The transcription factor YY1 is under the transcriptional control of NF-κB [42]. Hence, inhibition of NF-κB will also result in the inhibition of YY1. In addition, NO directly nitrosylates YY1 and inhibits its DNA-binding activity [63]. The direct role of YY1 in chemo-sensitization was corroborated by the use of siRNA YY1. Treatment of tumor cells with siRNA YY1 sensitized tumor cells to CDDP apoptosis [89].
6.3. Role of NO-mediated inhibition of Snail in chemo-immunosensitization
The transcription regulation of the zinc transcription factor Snail is under the control of both NF-κB [56] and YY1 [57]. It has also been reported that Snail represses E-cadherin [58] and RKIP [59].
Treatment of tumor cells with the NO donor DETA-NONOate inhibited Snail mRNA and protein expressions. In addition, Snail was S-nitrosylated that inhibited its DNA-binding activity. Further, Snail was also involved in the reversal of chemo-immune resistance. The direct role of Snail was demonstrated by treating the resistant cells with Snail siRNA and the tumor cells were sensitized to apoptosis by both chemotherapeutics and immunotherapeutics [37,97]. Treatment with Snail siRNA resulted in modification of the regulatory loop by inhibiting NF-κB, YY1 and Snail and the induction of RKIP.
6.4. Role of RKIP-mediated upregulation by NO in chemo-immunosensitization
The first demonstration that RKIP plays a role in the regulation of drug resistance was reported by Chatterjee et al. [98], whereby they reported that the overexpression of RKIP in prostate cancer cell lines sensitized the cells to apoptosis by Camptothecin (CPT-1). Also, we have reported that overexpression of RKIP sensitizes cells to TRAIL apoptosis [45].
Since treatment of tumor cells with DETA-NONOate resulted in the upregulation of RKIP, we postulated that the induction of RKIP concomitantly with inhibition of the resistant factors NF-κB, Snail and YY1 will be involved in chemo-immunosensitization of resistant tumor cells. Treatment of tumor cells with DETA-NONOate resulted in the upregulation of RKIP mRNA and protein expressions in tumor cell lines and the cells were sensitized to apoptosis by cytotoxic drugs. The direct role of RKIP in chemo-immuno-sensitization was corroborated by the use of cells treated with siRNA RKIP, which resulted in the reversal of tumor cell sensitivity to TRAIL apoptosis [45].
Clearly, all of the above findings point out that each gene product of the loop plays a direct role in chemo-immunosensitization to apoptosis by both chemo and immunotherapeutic drugs. However, the direct role of Snail, YY1 and RKIP in chemo-immunosensitization is not totally clear since they are all interregulated. To approach the direct roles, one has to investigate tumor cells that are, for example, inhibited for NF-κB activity and then were transduced to overexpress Snail, YY1 or RKIP and examine their sensitization to drugs.
7. Comparison between NO donors and other chemo-immunosensitizing agents
There have been many reports of using various agents that reverse tumor cell resistance and sensitized the cells to apoptosis by chemo-immunotherapeutics [99]. Various agents have targeted the NF-κB, Snail, YY1, RKIP loop and the results showed similar findings to those mediated by treatment with NO donors. For example, we have reported that treatment of tumor cells with subtoxic concentrations of chemotherapeutic drugs sensitized the cells to both FasL and TRAIL apoptosis [100–102]. These findings were subsequently corroborated by treatment with NO donors [103]. Also, treatment with subtoxic concentrations of drugs sensitized the tumor cells to TRAIL apoptosis [104].
Treatment with chemotherapeutic drugs resulted in the upregulation of DR4 or DR5, concomitantly with sensitization to TRAIL apoptosis [105,106]. However, the molecular underlying mechanisms were not known. In our prior findings [96] we reported that treatment with an NO donor upregulated DR5 concomitantly with sensitization to TRAIL apoptosis. The mechanism of DR5 upregulation was the result of NO-mediated inhibition of the DR5 repressor YY1. We have also reported that treatment of B-NHL tumor cells with rituximab sensitized the cells to both chemo and immunotherapeutics. Sensitization was the result of inhibition of the NF-κB, Raf-1, and PI3K pathways [36,107–109]. These findings were reproduced with NO donors and showed that rituximab and the NO donor behaved similarly. Inhibitors for NF-κB also mimicked NO donors in tumor cell sensitization. Hence, treatment of tumor cells with NF-κB inhibitors sensitized the tumor cells to apoptosis by cytotoxic drugs. One example is the use of the NF-κB inhibitor, DHMEQ, which resulted in sensitization to FasL and TRAIL and chemotherapeutic drugs [44]. Likewise, we have shown that the NO donor, which inhibited NF-κB, also resulted in the reversal of resistance and, thus, mimicking the NF-κB inhibitor. In addition, treatment with proteasome inhibitors resulted in tumor cell sensitization mimicking NO donors. For example, treatment with bortezomib was shown to sensitize tumor cells to TRAIL apoptosis. Bortezomib inhibits NF-κB activity like NO donors [45]. Like NO donors, treatment with another proteasome inhibitor, NPI0052, resulted in the inhibition of NF-κB, Snail, and YY1 and induction of RKIP.
The above findings demonstrated that treatment with NO donors mimicked many sensitizing agents, some of which are clinically used (Bortezomib) and suggest the use of NO donors in vivo in patients in combination with subtoxic cytotoxic therapies. Further, it is not clear whether the development of tumor sensitizing agents can be also applied for novel sensitizing NO donors.
8. Efficacy of pre-clinical and clinical applications of NO donors
8.1. Preclinical
An NO donor synthesized by Keefer's Lab at NIH, JS-K, is a diazonium diolate and releases NO enzymatically activated by glutathione and glutathione-S-transferase (GST) [110]. Findings by Weyerbroch et al. [111] reported the inhibition of proliferation and induction of apoptosis in vivo of U87 glioma tumors xenografts treated with JS-K. In a colo-rectal carcinoma model, Huerta et al. [112] used mice bearing the human metastatic CRC line, SW620, and treated the mice with DETA-NONOate and chemotherapy. This combination treatment resulted in inhibition of tumor cell growth. Pervin et al. [113] reported the treatment of tumor-bearing mice with the combination of DETA-NONOate and a farnesyl transferase inhibitor showed minimal toxicity and slowed the growth of the tumor. Also, the combination of DETA-NONOate and CDDP enhanced the cytotoxic activity of S29 lung fibroblasts [91], and similar results were achieved with head and neck cancers [114]. Huerta-Yepez et al. [89] reported tumor inhibition by treatment with the combination of DETA-NONOate and CDDP in mice bearing human prostate cancer xenografts.
In addition to NO donors, reports utilized gene therapy via inducible NOSII for cancer therapy [115]. Another NO donor, JS-K, was studied in castrated-resistant prostate cancer cells and resulted in the attenuation of functional androgen receptor signaling with inhibition of cell growth [116]. Also, NO-NSAID donors were examined in a variety of tumor cell lines and exhibited inhibition of cell proliferation and induction of apoptosis [117–119].
The anticancer effect of iNOS was first reported by Xie et al. [120]. The in vivo model used mice bearing tumors overexpressing iNOS and resulted in inhibition of tumor growth and additional studies were also reported by Xie and Fiddler [121]. Chung et al. [122] reported significant apoptosis in cells transfected with the iNOS gene and other studies have been reviewed elsewhere [33,115].
The antitumoral properties of NOS-3 overexpression by gene therapy was reported [123,124].
8.2. Clinical
Several reports have delineated the important role of NO in cancer therapy. It may act directly as a cytotoxic agent against cancer cells or sensitizes resistant tumor cells to chemo-immuno-radio-hormonal therapies [125,126].
The initial application of the NO donor, nitroglycerine (GTN), in patients was reported by Yasuda et al. [127,128] whereby they treated patients with lung cancer with the combination of GTN and vinorelbine and CDDP and showed an improved response of 72% and median time to progression compared to patients without GTN. Siemens et al. [129] reported a phase II study using patches releasing GTN in prostate cancer patients who failed primary treatment. This treatment reduced hypoxia-induced progression as measured by a decrease in the PSA doubling time. Siemens et al. [130] used a GTN patch that slowly releases GTN in combination with anti-cancer treatment with drugs that resulted in significant inhibition of PSA in patients with prostate cancer. Arrieta et al. [32] reported the first clinical study that the addition of GTN to induction chemotherapy and concurrent chemo-immunotherapy in patients with local advanced NSCCL resulted in acceptable toxicity profile.
9. Concluding remarks and future directions
It is clear that high levels of NO, either biologically generated by iNOS or by NO donors, exert several anti-tumor activities, namely, inhibition of cell survival and growth, sensitizing the tumor cells to both chemo, immune- and radio-toxicities, and also inhibit EMT and metastasis. The mechanisms mediating these various NO-mediated effects are, in part, under the regulation of the constitutively activated and dysregulated pro-survival/anti-apoptotic of the NF-κB/Snail/RKIP/PTEN loop in cancer cells. NO interferes directly in the loop by inhibiting the activities NF-κB, Snail and YY1 and derepressing the expressions of the anti-survival/pro-apoptotic gene products RKIP and PTEN (see schematic diagrams Figs. 1 and 2). These NO-mediated manifestations, both in vitro and in vivo in mouse models, support the potential therapeutic application of NO donors in combination with subtoxic therapeutic agents on the reversal of tumor resistance and the inhibition of both EMT and metastasis. Phase I/II clinical studies are under current investigation to support the contention of the anti-tumor-mediated effects of NO [see reviews in this volume]. Clearly, phase III clinical trials will be hopefully investigated in the future and we are eagerly awaiting the findings for the novel therapeutic application of NO in cancer. It must be noted also that while NO donors are not highly tumor specific, their pleiotropic activity in the reversal of resistance and sensitizing activity are paralleled with inhibition of EMT and metastasis. Various laboratories are developing novel NO donors with better efficacies as single agents or complexed with other targeting agents for specificity.
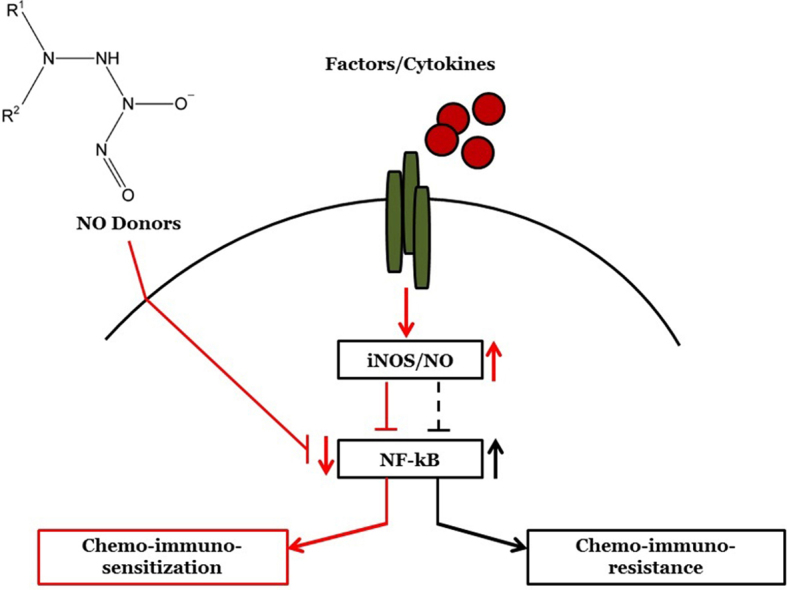
Fig. 1.

NO donors convert the anti-apoptotic NF-κB/Snail/YY1/RKIP/PTEN resistant loop into a sensitive pro-apoptotic loop. The schematic diagram represents tumor cells that have a dysregulated NF-κB/Snail/ YY1/RKIP/PTEN resistant loop involved in the maintenance of the tumor cell viability, proliferation, resistance, EMT, and metastasis. Hence, in tumor cells, the expression and the activities of NF-κB, Snail, YY1 are upregulated while those of RKIP and PTEN are downregulated. The loop is the result of NF-κB-mediated activation of its target Snail and YY1 and, in turn, Snail represses RKIP and YY1 represses PTEN. In addition, YY1 activates Snail. In the presence of NO donors, the activities of NF-κB, Snail and YY1 are inhibited and resulting in the de-repression of RKIP and PTEN. In turn, RKIP potentiates its inhibitory activity on NF-κB and its targets and, likewise, PTEN inhibits the PI3K/Akt pathway which controls NF-κB. Overall, the NO treatment inhibits tumor cell viability, proliferation and sensitizes the cells to chemo-immunotherapies-induced apoptosis.
Fig. 2.

Mechanism of NO-mediated chemo-immunosensitization of resistant tumor cells to apoptosis. The schematic diagram represents the NO-mediated effects that results in the sensitization of resistant tumor cells to drug-immune-induced apoptosis. Both NO donors and iNOS induction will result in the upregulation of NO, resulting in the upregulation of death receptors and sensitization to death ligands (FasL, TRAIL, TNF-α) and activation of the effector caspase 3 and downstream apoptosis. Also, NO will inhibit anti-apoptotic gene products such as Bcl-2 and allowing cytotoxic drugs to act on the mitochondrial membrane and resulting on the activation of caspase 3 or induction of AIF, all of which result in apoptosis.
Acknowledgements
The authors acknowledge the various students, fellows and collaborators whose published reports were the subject of this review. They are Drs. S. Baritaki, F. Hongo, S. Huerta-Yepez, H. Liu, M. Vega, S. Huerta, and K. Yeung. The authors also acknowledge the Jonsson Comprehensive Cancer Center at UCLA for its assistance. The following funding agencies that supported the various publications that have been referred to in this review are listed below: the Bodossaki Foundation, the Department of Defense (DOD)/US Army, DAMD. 17-02-1-0023, the Fogarty Fellowships (D43DW0001 13-14), UC MEXUS, the UCLA AIDS Institute, the UCLA Gene Therapy Program, the UCLA SPORE in Prostate Cancer. The authors also acknowledge the assistance of Kathy Nguyen, Leah Moyal and Ailina Lao for their assistance in the preparation of this manuscript.
Footnotes
This article belongs to a special issue on Nitric Oxide and Cancer, edited by Jordi Muntané and Benjamin Bonavida.
References
- 1.Aggarwal B.B. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal B.B. Nuclear factor-kappa-B: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Fariaseisner R., Sherman M.P., Aeberhard E., Chaudhuri G. Nitric-oxide is an important mediator for tumoricidal activity in vivo. Proc. Natl. Acad. Sci. USA. 1994;91:9407–9411. doi: 10.1073/pnas.91.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binder C., Schulz M., Hiddemann W., Oellerich M. Induction of inducible nitric oxide synthase is an essential part of tumor necrosis factor-alpha-induced apoptosis in MCF-7 and other epithelial tumor cells. Lab. Investig. 1999;79:1703–1712. [PubMed] [Google Scholar]
- 5.Bredt D.S. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic. Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 6.Boyd C.S., Cadenas E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol. Chem. 2002;383:411–423. doi: 10.1515/BC.2002.045. [DOI] [PubMed] [Google Scholar]
- 7.Burgaud J.L., Ongini E., Del Soldato P. Nitric oxide-releasing drugs – a novel class of effective and safe therapeutic agents. In: Chiueh C.C., Hong J.S., Leong S.K., editors. Nitric Oxide: Novel Actions, Deleterious Effects and Clinical Potential. 2002. pp. 360–371. [DOI] [PubMed] [Google Scholar]
- 8.Huerta S., Chilka S., Bonavida B. Nitric oxide donors: novel cancer therapeutics (Review) Int. J. Oncol. 2008;33:909–927. [PubMed] [Google Scholar]
- 9.Switzer C.H., Flores-Santana W., Mancardi D., Donzelli S., Basudhar D., Ridnour L.A., Miranda K.M., Fukuto J.M., Paolocci N., Wink D.A. The emergence of nitroxyl (HNO) as a pharmacological agent. Biochim. Biophys. Acta. 2009;1787 doi: 10.1016/j.bbabio.2009.04.015. 835-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wink D.A., Hines H.B., Cheng R.Y., Switzer C.H., Flores-Santana W., Vitek M.P., Ridnour L.A., Colton C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011;89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds M.M., Witzeling S.D., Damodaran V.B., Medeiros T.N., Knodle R.D., Edwards M.A., Lookian P.P., Brown M.A. Applications for nitric oxide in halting proliferation of tumor cells. Biochem. Biophys. Res. Commun. 2013;431:647–651. doi: 10.1016/j.bbrc.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 12.Ignarro L.J. Elsevier Science; Burlington, MA: 2009. Nitric Oxide: Biology and Pathobiology. [Google Scholar]
- 13.Bonavida B. Springer; New York: 2010. Nitric Oxide (NO) and Cancer: Prognosis, Prevention, and Therapy. [Google Scholar]
- 14.Bredt D.S., Snyder S.H. Nitric-oxide: a physiological messenger molecule. Ann. Rev. Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 15.Kroncke K.D. Zinc finger proteins as molecular targets for nitric oxide-mediated gene regulation. Antioxid. Redox Signal. 2001;3:565–575. doi: 10.1089/15230860152542934. [DOI] [PubMed] [Google Scholar]
- 16.Stamler J.S., Jia L., Eu J.P., McMahon T.J., Demchenko I.T., Bonaventura J., Gernert K., Piantadosi C.A. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 17.Hess D.T., Matsumoto A., Nudelman R., Stamler J.S. S-nitrosylation: spectrum and specificity. Nat. Cell Biol. 2001;3:E46–E49. doi: 10.1038/35055152. [DOI] [PubMed] [Google Scholar]
- 18.Stamler J.S., Lamas S., Fang F.C. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 19.Garban H.J., Marquez-Garban D.C., Pietras R.J., Ignarro L.J. Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc. Natl. Acad. Sci. USA. 2005;102:2632–2636. doi: 10.1073/pnas.0409854102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braughler J.M., Mittal C.K., Murad F. Effects of thiols, sugars, and proteins on nitric-oxide activation of guanylate-cyclase. J. Biol. Chem. 1979;254:2450–2454. [PubMed] [Google Scholar]
- 21.Lewicki J.A., Brandwein H.J., Mittal C.K., Arnold W.P., Murad F. Proteins of purified soluble guanylate-cyclase activated by nitric-oxide and sodium-nitroprusside. J. Cycl. Nucleotide Res. 1982;8:17–25. [PubMed] [Google Scholar]
- 22.Bian K., Murad F. sGC-cGMP signaling: target for anticancer therapy. Adv. Exp. Med. Biol. 2014;814:5–13. doi: 10.1007/978-1-4939-1031-1_2. [DOI] [PubMed] [Google Scholar]
- 23.Bian K., Murad F. What is next in nitric oxide research? From cardiovascular system to cancer biology. Nitric Oxide. 2014;43:3–7. doi: 10.1016/j.niox.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Foster M.W., McMahon T.J., Stamler J.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 25.Andrew P.J., Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999;43:521–531. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 26.Sarti P., Forte E., Mastronicola D., Giuffre A., Arese M. Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochim. Biophys. Acta. 2012;1817 doi: 10.1016/j.bbabio.2011.09.002. 610-9. [DOI] [PubMed] [Google Scholar]
- 27.Kim P.K., Zamora R., Petrosko P., Billiar T.R. The regulatory role of nitric oxide in apoptosis. Int. Immunopharmacol. 2001;1:1421–1441. doi: 10.1016/s1567-5769(01)00088-1. [DOI] [PubMed] [Google Scholar]
- 28.Porasuphatana S., Tsai P., Rosen G.M. The generation of free radicals by nitric oxide synthase. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2003;134:281–289. doi: 10.1016/s1532-0456(02)00271-5. [DOI] [PubMed] [Google Scholar]
- 29.Zou H., Li Y., Liu X., Wang X. An APAF-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 30.Sedlak T.W., Snyder S.H. Messenger molecules and cell death: therapeutic implications. J. Am. Med. Assoc. 2006;295:81–89. doi: 10.1001/jama.295.1.81. [DOI] [PubMed] [Google Scholar]
- 31.Serafim R.A., Primi M.C., Trossini G.H., Ferreira E.I. Nitric oxide: state of the art in drug design. Curr. Med. Chem. 2012;19:386–405. doi: 10.2174/092986712803414321. [DOI] [PubMed] [Google Scholar]
- 32.Arrieta O., Blake M., de la Mata-Moya M.D., Corona F., Turcott J., Orta D., Alexander-Alatorre J., Gallardo-Rincon D. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother. Oncol. 2014;111:311–315. doi: 10.1016/j.radonc.2014.01.021. [DOI] [PubMed] [Google Scholar]
- 33.Bonavida B. Springer International Publishing; 2015. Nitric Oxide and Cancer: Pathogenesis and Therapy. [Google Scholar]
- 34.Huerta-Yepez S., Vega M., Jazirehi A., Garban H., Hongo F., Cheng G.H., Bonavida B. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-kappa B and inhibition of Bcl-(xL) expression. Oncogene. 2004;23:4993–5003. doi: 10.1038/sj.onc.1207655. [DOI] [PubMed] [Google Scholar]
- 35.Jazirehi A.R., Vega M.I., Chatterjee D., Goodglick L., Bonavida B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, BCL-xL down-regulation, and chemosensitization of non-Hodgkin’s lymphoma B cells by rituximab. Cancer Res. 2004;64:7117–7126. doi: 10.1158/0008-5472.CAN-03-3500. [DOI] [PubMed] [Google Scholar]
- 36.Vega M.I., Jazirehi A.R., Huerta-Yepez S., Bonavida B. Rituximab-induced inhibition of YY1 and Bcl-xL expression in Ramos non-Hodgkin’s lymphoma cell line via inhibition of NF-kappa B activity: role of YY1 and Bcl-xL in Fas resistance and chemoresistance, respectively. J. Immunol. 2005;175:2174–2183. doi: 10.4049/jimmunol.175.4.2174. [DOI] [PubMed] [Google Scholar]
- 37.Baritaki S., Yeung K., Palladino M., Berenson J., Bonavida B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009;69:8376–8385. doi: 10.1158/0008-5472.CAN-09-1069. [DOI] [PubMed] [Google Scholar]
- 38.Bonavida B., Baritaki S. The novel role of yin yang 1 in the regulation of epithelial to mesenchymal transition in cancer via the dysregulated NF-κB/Snail/YY1/RKIP/PTEN Circuitry. Crit. Rev. Oncog. 2011;16(3–4):211–226. doi: 10.1615/critrevoncog.v16.i3-4.50. [DOI] [PubMed] [Google Scholar]
- 39.DiDonato J.A., Mercurio F., Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 40.Karin M., Lin A. NF-kappa B at the crossroads of life and death. Nat. Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 41.Hongo F., Garban H., Huerta-Yepez S., Vega M., Jazirehi A.R., Mizutani Y., Miki T., Bonavida B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005;336:692–701. doi: 10.1016/j.bbrc.2005.08.150. [DOI] [PubMed] [Google Scholar]
- 42.Wang H., Hertlein E., Bakkar N., Sun H., Acharyya S., Wang J., Carathers M., Davuluri R., Guttridge D.C. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005;336:692–701. doi: 10.1016/j.bbrc.2005.08.150. ; Mol. Cell Biol 27 (2007) 4374–4387. [DOI] [PubMed] [Google Scholar]
- 43.Kleinberg L., Davidson B. Cell survival and apoptosis-related molecules in cancer cells in effusions: a comprehensive review. Diagn. Cytopathol. 2009;37:613–624. doi: 10.1002/dc.21095. [DOI] [PubMed] [Google Scholar]
- 44.Katsman A., Umezawa K., Bonavida B. Reversal of resistance to cytotoxic cancer therapies: DHMEQ as a chemo-sensitizing and immuno-sensitizing agent. Drug Resist. Updates. 2007;10:1–12. doi: 10.1016/j.drup.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Baritaki S., Suzuki E., Umezawa K., Spandidos D.A., Berenson J., Daniels T.R., Penichet M.L., Jazirehi A.R., Palladino M., Bonavida B. Inhibition of Yin Yang 1-dependent repressor activity of DR5 transcription and expression by the novel proteasome inhibitor NPI-0052 contributes to its TRAIL-enhanced apoptosis in cancer cells. J. Immunol. 2008;180:6199–6210. doi: 10.4049/jimmunol.180.9.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown M., Cohen J., Arun P., Chen Z., Van Waes C. NF-κB in carcinoma therapy and prevention. Expert Opin. Therap. Targets. 2008;12:1109–1122. doi: 10.1517/14728222.12.9.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baritaki S., Chapman A., Yeung K., Spandidos D.A., Palladino M., Bonavida B. Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: pivotal roles of Snail repression and RKIP induction. Oncogene. 2009;28:3573–3585. doi: 10.1038/onc.2009.214. [DOI] [PubMed] [Google Scholar]
- 48.Mauro C., Zazzeroni F., Papa S., Bubici C., Franzoso G. The NF-kappaB transcription factor pathway as a therapeutic target in cancer: methods for detection of NF-kappaB activity. Methods Mol. Biol. (Clifton, N.J.) 2009;512:169–207. doi: 10.1007/978-1-60327-530-9_10. [DOI] [PubMed] [Google Scholar]
- 49.Wink D.A., Ridnour L.A., Hussain S.P., Harris C.C. The reemergence of nitric oxide and cancer. Nitric Oxide. 2008;19:65–67. doi: 10.1016/j.niox.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katsuyama K., Shichiri M., Marumo F., Hirata Y. NO inhibits cytokine-induced iNOS expression and NF-kappa B activation by interfering with phosphorylation and degradation of I kappa B-alpha. Arterioscler. Thromb. Vasc. Biol. 1998;18:1796–1802. doi: 10.1161/01.atv.18.11.1796. [DOI] [PubMed] [Google Scholar]
- 51.Garban H.J., Bonavida B. Nitric oxide disrupts H2O2-dependent activation of nuclear factor kappa B. Role in sensitization of human tumor cells to tumor necrosis factor-alpha-induced cytotoxicity. J. Biol. Chem. 2001;276:8918–8923. doi: 10.1074/jbc.M008471200. [DOI] [PubMed] [Google Scholar]
- 52.Connelly L., Palacios-Callender M., Ameixa C., Moncada S., Hobbs A.J. Biphasic regulation of NF-kappa B activity underlies the pro- and anti-inflammatory actions of nitric oxide. J. Immunol. 2001;166:3873–3881. doi: 10.4049/jimmunol.166.6.3873. [DOI] [PubMed] [Google Scholar]
- 53.Marshall H.E., Stamler J.S. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 54.Nieto M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 55.Wu Y., Zhou B.P. Snail: More than EMT. Cell Adhes. Migr. 2010;4:199–203. doi: 10.4161/cam.4.2.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Julien S., Puig I., Caretti E., Bonaventure J., Nelles L., van Roy F., Dargemont C., de Herreros A.G., Bellacosa A., Larue L. Activation of NF-kappaB by Akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- 57.Palmer M.B., Majumder P., Cooper J.C., Yoon H., Wade P.A., Boss J.M. Yin yang 1 regulates the expression of snail through a distal enhancer. Mol. Cancer Res. 2009;7:221–229. doi: 10.1158/1541-7786.MCR-08-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cano A., Pérez-Moreno M.A., Rodrigo I., Locascio A., Blanco M.J., del Barrio M.G., Portillo F., Nieto M.A. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 59.Beach S., Tang H., Park S., Dhillon A.S., Keller E.T., Kolch W., Yeung K.C. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008;27:2243–2248. doi: 10.1038/sj.onc.1210860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gordon S., Akopyan G., Garban H., Bonavida B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 61.Garban H.J., Bonavida B. Nitric oxide inhibits the transcription repressor Yin-Yang 1 binding activity at the silencer region of the Fas promoter: a pivotal role for nitric oxide in the up-regulation of Fas gene expression in human tumor cells. J. Immunol. 2001;167:75–81. doi: 10.4049/jimmunol.167.1.75. [DOI] [PubMed] [Google Scholar]
- 62.Baritaki S., Katsman A., Chatterjee D., Yeung K.C., Spandidos D.A., Bonavida B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J. Immunol. 2007;179:5441–5453. doi: 10.4049/jimmunol.179.8.5441. [DOI] [PubMed] [Google Scholar]
- 63.Hongo F., Garban H., Huerta-Yepez S., Vega M., Jazirehi A.R., Mizutani Y., Miki T., Bonavida B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005;336:692–701. doi: 10.1016/j.bbrc.2005.08.150. [DOI] [PubMed] [Google Scholar]
- 64.Granovsky A.E., Rosner M.R. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 2008;18:452–457. doi: 10.1038/cr.2008.43. [DOI] [PubMed] [Google Scholar]
- 65.Yeung K., Seitz T., Li S., Janosch P., McFerran B., Kaiser C., Fee F., Katsanakis K.D., Rose D.W., Mischak H., Sedivy J.M., Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 66.Tang H., Park S., Sun S.C., Trumbly R., Ren G., Tsung E., Yeung K.C., Rose R.K.I.P. inhibits N.F.- D.W., Mischak H., Sedivy J.M. W. Kolcgnaling components of the IκB kinase complex. FEBS Lett. 2010;584:662–668. doi: 10.1016/j.febslet.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lorenz K., Lohse M.J., Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–579. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- 68.Fu Z., Smith P.C., Zhang L., Rubin M.A., Dunn R.L., Yao Z., Keller E.T. Effects of Raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J. Natl. Cancer Inst. 2003;95:878–889. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 69.Schuierer M.M., Bataille F., Hagan S., Kolch W., Bosserhoff A.K. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004;64:5186–5192. doi: 10.1158/0008-5472.CAN-03-3861. [DOI] [PubMed] [Google Scholar]
- 70.Hagan S., Al-Mulla F., Mallon E., Oien K., Ferrier R., Gusterson B., Curto García J.J., Kolch W. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin. Cancer Res. 2005;11:7392–7397. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 71.Al-Mulla F., Hagan S., Behbehani A.I., Bitar M.S., George S.S., Going J.J., Curto García J.J., Scott L., Fyfe N., Murray G.I., Kolch W. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J. Clin. Oncol. 2006;24:5672–5679. doi: 10.1200/JCO.2006.07.5499. [DOI] [PubMed] [Google Scholar]
- 72.Fu Z., Kitagawa Y., Shen R., Shah R., Mehra R., Rhodes D., Keller P.J., Mizokami A., Dunn R., Chinnaiyan A.M., Yao Z., Keller E.T. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2006;66:248–256. doi: 10.1002/pros.20319. [DOI] [PubMed] [Google Scholar]
- 73.Carracedo A., Pandolfi P.P. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 74.Zamai L., Ahmad M., Bennett I.M., Azzoni L., Alnemri E.S., Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998;188:2375–2380. doi: 10.1084/jem.188.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonavida B. Human Press of Springer Science+Business Media; Totowa, NJ: 2008. Sensitization of Cancer Cells for Chemo/Immuno/Radio-therapy. [Google Scholar]
- 76.Keefer L.K., Nims R.W., Davies K.M., Wink D.A. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 77.Smith D.J., Chakravarthy D., Pulfer S., Simmons M.L., Hrabie J.A., Citro M.L., Saavedra J.E., Davies K.M., Hutsell T.C., Mooradian D.L., Hanson S.R., Keefer L.K. Nitric oxide-releasing polymers containing the [N(O)NO]- group. J. Med. Chem. 1996;39:1148–1156. doi: 10.1021/jm950652b. [DOI] [PubMed] [Google Scholar]
- 78.Garban H.J., Bonavida B. Nitric oxide sensitizes ovarian tumor cells to Fas-induced apoptosis. Gynecol. Oncol. 1999;73:257–264. doi: 10.1006/gyno.1999.5374. [DOI] [PubMed] [Google Scholar]
- 79.Beg A.A., Baltimore D. An essential role for NF-kappa B in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 80.Wang C.Y., Mayo M.W., Baldwin A.S., Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 81.Tartaglia L.A., Goeddel D.V. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 82.GarciaRuiz C., Colell A., Mari M., Morales A., FernandezCheca J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species – role of mitochondrial glutathione. J. Biol. Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 83.Suzuki Y.J., Forman H.J., Sevanian A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 84.Fukuto J.M. Chemistry of nitric oxide: biologically relevant aspects. Adv. Pharmacol. (San Diego, Calif.) 1995;34:1–15. doi: 10.1016/s1054-3589(08)61078-9. [DOI] [PubMed] [Google Scholar]
- 85.Leverkus M., Sprick M.R., Wachter T., Mengling T., Baumann B., Serfling E., Brocker E.B., Goebeler M., Neumann M., Walczak H. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol. Cell Biol. 2003;23:777–790. doi: 10.1128/MCB.23.3.777-790.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang S., El-Deiry W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 87.Baritaki S., Huerta-Yepez S., Sakai T., Spandidos D.A., Bonavida B. Chemotherapeutic drugs sensitize cancer cells to TRAIL-mediated apoptosis: up-regulation of DR5 and inhibition of Yin Yang 1. Mol. Cancer Therap. 2007;6:1387–1399. doi: 10.1158/1535-7163.MCT-06-0521. [DOI] [PubMed] [Google Scholar]
- 88.Jazirehi A.R., Bonavida B. Resveratrol modifies the expression of apoptotic regulatory proteins and sensitizes non-Hodgkin’s lymphoma and multiple myeloma cell lines to paclitaxel-induced apoptosis. Mol. Cancer Ther. 2004;3:71–84. [PubMed] [Google Scholar]
- 89.Huerta-Yepez S., Baritaki S., Baay-Guzman G., Hernandez-Luna M.A., Hernandez-Cueto A., Vega M.I., Bonavida B. Contribution of either YY1 or BclXL-induced inhibition by the NO-donor DETANONOate in the reversal of drug resistance, both in vitro and in vivo YY1 and BclXL are overexpressed in prostate cancer. Nitric oxide: Biol. Chem. / Off. J. Nitric Oxide Soc. 2013;29:17–24. doi: 10.1016/j.niox.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 90.Huerta S., Baay-Guzman G., Gonzalez-Bonilla C.R., Livingston E.H., Huerta-Yepez S., Bonavida B. In vitro and in vivo sensitization of SW620 metastatic colon cancer cells to CDDP-induced apoptosis by the nitric oxide donor DETANONOate: involvement of AIF. Nitric Oxide-Biol. Chem. 2009;20:182–194. doi: 10.1016/j.niox.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 91.Wink D.A., Cook J.A., Christodoulou D., Krishna M.C., Pacelli R., Kim S., DeGraff W., Gamson J., Vodovotz Y., Russo A., Mitchell J.B. Nitric oxide and some nitric oxide donor compounds enhance the cytotoxicity of cisplatin. Nitric Oxide. 1997;1:88–94. doi: 10.1006/niox.1996.0108. [DOI] [PubMed] [Google Scholar]
- 92.Kumar A., Takada Y., Boriek A.M., Aggarwal B.B. Nuclear factor-kappaB: its role in health and disease. J. Mol. Med. (Berl.) 2004;82:434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 93.Vega M.I., Jazirehi A.R., Huerta-Yepez S., Bonavida B. Rituximab-induced inhibition of YY1 and Bcl-xL expression in Ramos non-Hodgkin’s lymphoma cell line via inhibition of NF-ompounds enhance the cytotoxicity of cisplatin, Nitric Od chemoresistance, respectively. J. Immunol. 2005;175:2174–2183. doi: 10.4049/jimmunol.175.4.2174. [DOI] [PubMed] [Google Scholar]
- 94.Santos-Silva M.C., Sampaio de Freitas M., Assreuy J. Killing of lymphoblastic leukemia cells by nitric oxide and taxol: involvement of NF-kappaB activity. Cancer Lett. 2001;173:53–61. doi: 10.1016/s0304-3835(01)00664-4. [DOI] [PubMed] [Google Scholar]
- 95.Sun Y., Rigas B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: implications for the mechanism of action of anticancer agents. Cancer Res. 2008;68:8269–8277. doi: 10.1158/0008-5472.CAN-08-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huerta-Yepez S., Vega M., Escoto-Chavez S.E., Murdock B., Sakai T., Baritaki S., Bonavida B. Nitric oxide sensitizes tumor cells to TRAIL-induced apoptosis via inhibition of the DR5 transcription repressor Yin Yang 1. Nitric Oxide. 2009;20:39–52. doi: 10.1016/j.niox.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 97.Baritaki S., Huerta-Yepez S., Sahakyan A., Karagiannides I., Bakirtzi K., Jazirehi A., Bonavida B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: inhibition of the metastasis-inducer snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010;9:4931–4940. doi: 10.4161/cc.9.24.14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chatterjee D., Bai Y., Wang Z., Beach S., Mott S., Roy R., Braastad C., Sun Y., Mukhopadhyay A., Aggarwal B.B., Darnowski J., Pantazis P., Wyche J., Fu Z., Kitagwa Y., Keller E.T., Sedivy J.M., Yeung K.C. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J Biol Chem. 2004;279:17515–17523. doi: 10.1074/jbc.M313816200. [DOI] [PubMed] [Google Scholar]
- 99.Bonavida B. Springer; New York: 2013. Resistance to Immunotherapeutic Antibodies in Cancer 2: Strategies to Overcome Resistance. [Google Scholar]
- 100.Morimoto H., Yonehara S., Bonavida B. Overcoming tumor necrosis factor and drug resistance of human tumor cell lines by combination treatment with anti-Fas antibody and drugs or toxins. Cancer Res. 1993;53:2591–2596. [PubMed] [Google Scholar]
- 101.Uslu R., Borsellino N., Frost P., Garban H., Ng C.P., Mizutani Y., Belldegrun A., Bonavida B. Chemosensitization of human prostate carcinoma cell lines to anti-fas-mediated cytotoxicity and apoptosis. Clin. Cancer Res. 1997;3:963–972. [PubMed] [Google Scholar]
- 102.Mizutani Y., Nakao M., Ogawa O., Yoshida O., Bonavida B., Miki T. Enhanced sensitivity of bladder cancer cells to tumor necrosis factor related apoptosis inducing ligand mediated apoptosis by cisplatin and carboplatin. J. Urol. 2001;165:263–270. doi: 10.1097/00005392-200101000-00076. [DOI] [PubMed] [Google Scholar]
- 103.Garban H.J., Bonavida B. Nitric oxide inhibits the transcription repressor Yin-Yang 1 binding activity at the silencer region of the Fas promoter: a pivotal role for nitric oxide in the up-regulation of Fas gene expression in human tumor cells. J. Immunol. 2001;167:75–81. doi: 10.4049/jimmunol.167.1.75. [DOI] [PubMed] [Google Scholar]
- 104.Zisman A., Ng C.P., Pantuck A.J., Bonavida B., Belldegrun A.S., Actinomycin D. and gemcitabine synergistically sensitize androgen-independent prostate cancer cells to Apo2L/TRAIL-mediated apoptosis. J. Immunother. 2001;24:459–471. doi: 10.1097/00002371-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 105.LaVallee T.M., Zhan X.G.H., Johnson M.S., Herbstritt C.J., Swartz G., Williams M.S., Hembrough W.A., Green S.J., Pribluda V.S. 2-methoxyestradiol up-regulates death receptor 5 and induces apoptosis through activation of the extrinsic pathway. Cancer Res. 2003;63:468–475. [PubMed] [Google Scholar]
- 106.Shigeno M., Nakao K., Ichikawa T., Suzuki K., Kawakami A., Abiru S., Miyazoe S., Nakagawa Y., Ishikawa H., Hamasaki K., Nakata K., Ishii N., Eguchi K. Interferon-alpha sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-kappa B inactivation. Oncogene. 2003;22:1653–1662. doi: 10.1038/sj.onc.1206139. [DOI] [PubMed] [Google Scholar]
- 107.Jazirehi A.R., Bonavida B. Cellular and molecular signal transduction pathways modulated by rituximab (rituxan, anti-CD20m Ab) in non-Hodgkin’s lymphoma: implications in chemosensitization and therapeutic intervention. Oncogene. 2005;24:2121–2143. doi: 10.1038/sj.onc.1208349. [DOI] [PubMed] [Google Scholar]
- 108.Jazirehi A.R., Huerta-Yepez S., Cheng G., Bonavida B. Rituximab (chimeric anti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-{kappa}B signaling pathway in non-Hodgkin’s lymphoma B-cell lines: role in sensitization to chemotherapeutic drug-induced apoptosis. Cancer Res. 2005;65:264–276. [PubMed] [Google Scholar]
- 109.Vega M.I., Huerta-Yepez S., Jazirehi A.R., Garban H., Bonavida B. Rituximab (chimeric anti-CD20) sensitizes B-NHL cell lines to Fas-induced apoptosis. Oncogene. 2005;24:8114–8127. doi: 10.1038/sj.onc.1208954. [DOI] [PubMed] [Google Scholar]
- 110.Shami P.J., Saavedra J.E., Wang L.Y., Bonifant C.L., Diwan B.A., Singh S.V., Gu Y., Fox S.D., Buzard G.S., Citro M.L., Waterhouse D.J., Davies K.M., Ji X., Keefer L.K. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol. Cancer Ther. 2003;2:409–417. [PubMed] [Google Scholar]
- 111.Weyerbrock A., Osterberg N., Psarras N., Baumer B., Kogias E., Werres A., Bette S., Saavedra J.E., Keefer L.K., Papazoglou A. JS-K, a glutathione S-transferase-activated nitric oxide donor with antineoplastic activity in malignant gliomas. Neurosurgery. 2012;70:497–510. doi: 10.1227/NEU.0b013e31823209cf. discussion 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huerta S., Gao X., Bonavida B. DETANONOate is a potent chemo\radio-sensitizing agent in colon and colorectal cancers as assessed in in vitro and in vivo established tumor xenografts. Forum Immunopathol. Dis. Ther. 2010;1:281–295. [Google Scholar]
- 113.Pervin S., Singh R., Gau C.L., Edamatsu H., Tamanoi F., Chaudhuri G. Potentiation of nitric oxide-induced apoptosis of MDA-MB-468 cells by farnesyltransferase inhibitor: implications in breast cancer. Cancer Res. 2001;61:4701–4706. [PubMed] [Google Scholar]
- 114.Azizzadeh B., Yip H.T., Blackwell K.E., Horvath S., Calcaterra T.C., Buga G.M., Ignarro L.J., Wang M.B. Nitric oxide improves cisplatin cytotoxicity in head and neck squamous cell carcinoma. Laryngoscope. 2001;111:1896–1900. doi: 10.1097/00005537-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 115.McCarthy H.O., Coulter J.A., Robson T., Hirst D.G. Gene therapy via inducible nitric oxide synthase: a tool for the treatment of a diverse range of pathological conditions. J. Pharm. Pharmacol. 2008;60:999–1017. doi: 10.1211/jpp.60.8.0007. [DOI] [PubMed] [Google Scholar]
- 116.Laschak M., Spindler K.-D., Schrader A.J., Hessenauer A., Streicher W., Schrader M., Cronauer M.V. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide releasing prodrug inhibits androgen receptor and WNT-signaling in prostate cancer cells. BMC Cancer. 2012;12 doi: 10.1186/1471-2407-12-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Williams J.L., Borgo S., Hasan I., Castillo E., Traganos F., Rigas B. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: Implications for colon cancer chemoprevention. Cancer Res. 2001;61:3285–3289. [PubMed] [Google Scholar]
- 118.Rigas B., Kashfi K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol. Med. 2004;10:324–330. doi: 10.1016/j.molmed.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 119.Kozoni V., Rosenberg T., Rigas B. Development of novel agents based on nitric oxide for the control of colon cancer. Acta Pharmacol. Sin. 2007;28:1429–1433. doi: 10.1111/j.1745-7254.2007.00696.x. [DOI] [PubMed] [Google Scholar]
- 120.Xie K., Huang S., Dong Z., Juang S.H., Gutman M., Xie Q.W., Nathan C., Fidler I.J. Transfection with the inducible nitric oxide synthase gene suppresses tumorigenicity and abrogates metastasis by K-1735 murine melanoma cells. J. Exp. Med. 1995;181:1333–1343. doi: 10.1084/jem.181.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xie K., Fidler I.J. Therapy of cancer metastasis by activation of the inducible nitric oxide synthase. Cancer Metastasis Rev. 1998;17:55–75. doi: 10.1023/a:1005956721457. [DOI] [PubMed] [Google Scholar]
- 122.Chung P., Cook T., Liu K.H., Vodovotz Y., Zamora R., Finkelstein S., Billiar T., Blumberg D. Overexpression of the human inducible nitric oxide synthase gene enhances radiation-induced apoptosis in colorectal cancer cells via a caspase-dependent mechanism. Nitric Oxide-Biol. Chem. 2003;8:119–126. doi: 10.1016/s1089-8603(02)00147-7. [DOI] [PubMed] [Google Scholar]
- 123.González R., Ferrín G., Aguilar-Melero P., Ranchal I., Linares C.I., Bello R.I., De la Mata M., Gogvadze V., Bárcena J.A., Alamo J.M., Orrenius S., Padillo F.J., Zhivotovsky B., Muntané J. Targeting hepatoma using nitric oxide donor strategies. Antioxid. Redox Signal. 2013;18:491–506. doi: 10.1089/ars.2011.4476. [DOI] [PubMed] [Google Scholar]
- 124.De la Rosa A., Rodríguez-Hernández A., González R., Romero-Brufau S., Navarro-Villarán E., Barrera-Pulido L., Pereira S., Marín L.M., López-Bernal F., Álamo J.M., Gómez-Bravo M.A., Padillo F.J., Muntané J. Antitumoral gene-based strategy involving nitric oxide synthase type III overexpression in hepatocellular carcinoma. Gene Therapy. 2015 doi: 10.1038/gt.2015.79. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 125.Aranda E., Lopez-Pedrera C., De La Haba-Rodriguez J.R., Rodriguez-Ariza A. Nitric oxide and cancer: the emerging role of s-nitrosylation. Curr. Mol. Med. 2012;12:50–67. doi: 10.2174/156652412798376099. [DOI] [PubMed] [Google Scholar]
- 126.Burke A.J., Sullivan F.J., Giles F.J., Glynn S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34:503–512. doi: 10.1093/carcin/bgt034. [DOI] [PubMed] [Google Scholar]
- 127.Yasuda H., Nakayama K., Watanabe M., Suzuki S., Fuji H., Okinaga S., Kanda A., Zayasu K., Sasaki T., Asada M., Suzuki T., Yoshida M., Yamanda S., Inoue D., Kaneta T., Kondo T., Takai Y., Sasaki H., Yanagihara K., Yamaya M. Nitroglycerin treatment may enhance chemosensitivity to docetaxel and carboplatin in patients with lung adenocarcinoma. Clin. Cancer Res. 2006;12:6748–6757. doi: 10.1158/1078-0432.CCR-06-1124. [DOI] [PubMed] [Google Scholar]
- 128.Yasuda H., Yamaya M., Nakayama K., Sasaki T., Ebihara S., Kanda A., Asada M., Inoue D., Suzuki T., Okazaki T., Takahashi H., Yoshida M., Kaneta T., Ishizawa K., Yamanda S., Tomita N., Yamasaki M., Kikuchi A., Kubo H., Sasaki H. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J. Clin. Oncol. 2006;24:688–694. doi: 10.1200/JCO.2005.04.0436. [DOI] [PubMed] [Google Scholar]
- 129.Siemens D.R., Heaton J.P.W., Adams M.A., Kawakami J., Graham C.H. Phase II Study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology. 2009;74:878–883. doi: 10.1016/j.urology.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 130.Siemens D.R., Heaton J., Adams M., Graham C. A phase I/II pilot trial of low-dose, sustained-release GTN for prostate cancer patients with recurrence after primary therapy. Nitric Oxide-Biol. Chem. 2007;17 S15–S15. [Google Scholar]