

Abstract
Analysis of murine cerebrospinal fluid (CSF) by quantitative mass spectrometry is challenging because of low CSF volume, low total protein concentration, and the presence of highly abundant proteins such as albumin. We demonstrate that the CSF proteome of individual mice can be analyzed in a quantitative manner to a depth of several hundred proteins in a robust and simple workflow consisting of single ultra HPLC runs on a benchtop mass spectrometer. The workflow is validated by a comparative analysis of BACE1−/− and wild-type mice using label-free quantification. The protease BACE1 cleaves the amyloid precursor protein (APP) as well as several other substrates and is a major drug target in Alzheimer's disease. We identified a total of 715 proteins with at least 2 unique peptides and quantified 522 of those proteins in CSF from BACE1−/− and wild-type mice. Several proteins, including the known BACE1 substrates APP, APLP1, CHL1 and contactin-2 showed lower abundance in the CSF of BACE1−/− mice, demonstrating that BACE1 substrate identification is possible from CSF. Additionally, ectonucleotide pyrophosphatase 5 was identified as a novel BACE1 substrate and validated in cells using immunoblots and by an in vitro BACE1 protease assay. Likewise, receptor-type tyrosine-protein phosphatase N2 and plexin domain-containing 2 were confirmed as BACE1 substrates by in vitro assays. Taken together, our study shows the deepest characterization of the mouse CSF proteome to date and the first quantitative analysis of the CSF proteome of individual mice. The BACE1 substrates identified in CSF may serve as biomarkers to monitor BACE1 activity in Alzheimer patients treated with BACE inhibitors.
Cerebrospinal fluid (CSF)1 consists of interstitial fluid that is in continuous exchange with the central nervous system and the peripheral blood system. It represents the only body fluid in humans that is in direct contact with brain tissue and accessible in a routine clinical setting. Thus, the easy accessibility from the periphery renders CSF perfectly suited to study pathologic neurological processes (1). Human CSF has a relatively low protein content (∼ 0.4 mg/ml), but features a highly diverse proteome. It is thus increasingly studied by modern mass spectrometry based proteomics (2). The proteomic analysis of human CSF typically involves various protein concentration and fractionation steps as well as the depletion of highly abundant proteins, such as serum albumin. This allows the identification of several hundred up to 2600 proteins from several milliliters of human CSF (3).
Mice are the most popular animal model in preclinical research, because of their similarity to humans in genetics and physiology, their unlimited supply and their ease of genetic engineering. The study of their CSF can provide valuable insights into disease mechanisms and biomarker discovery and may allow the rapid translation of preclinical findings into human patients. However, the proteomic study of murine CSF has been limited because of several shortcomings. The low total CSF volume of ∼30 μl and an average yield of only ∼10 μl blood-free CSF pose a challenge for various protein concentration and depletion steps that are routinely applied to human CSF, where the sample volume is up to 1,000-fold more (4, 5). One study reported the identification of 289 proteins and the quantification of 103 proteins using pooled immunodepleted CSF from 10–12 mice per sample (6). A second study reported the identification of 566 proteins in murine CSF of individual mice, relying on time consuming fractionation by two dimensional liquid chromatography tandem MS (2D-LC-MS/MS) (7).
Here we show that label-free quantitative proteomics in murine CSF can be achieved in unprecedented depth in individual animals using single ultra HPLC runs on the benchtop Q Exactive mass spectrometer. We demonstrate the feasibility of our approach by comparing the CSF of BACE1 (β-site amyloid precursor protein (APP) cleaving enzyme 1) −/− mice with their wild-type littermates.
BACE1 is a membrane bound aspartyl protease that is essential in the pathogenesis of Alzheimer's disease. It is the rate-limiting enzyme in a proteolytic cascade leading to the liberation of the neurotoxic Aβ peptide from the much larger amyloid precursor protein (APP) into the extracellular space (8, 9). Inhibition of BACE1 abolishes Aβ generation, rendering BACE1 a prime drug target for the therapy of Alzheimer's disease (10). Besides APP, BACE1 processes numerous other substrates in vivo and in vitro, which raises concerns about mechanism based side effects on the therapeutic inhibition of this protease (11). Although BACE1 expression levels are the highest in the brain, it is currently unknown whether BACE1 substrate levels besides APP can be monitored in the CSF as a read-out of BACE1 activity. This would be desirable, as it would allow the longitudinal monitoring of BACE1 substrate levels on therapeutic inhibition of BACE1 in humans and thus an effective screening for possible adverse effects.
Our approach allows the accurate identification and quantification of several hundred proteins in as little as 2 μl of murine CSF in ∼4.5 h per sample, at a much greater speed and proteomic depth than in previous studies, despite using lower sample amounts (6, 7). Overall, 715 proteins were identified with at least two unique peptides and 522 proteins were quantified in at least three biological replicates of both BACE1−/− and wild-type mice. We provide evidence that BACE1 activity is reflected in the composition of the CSF, as the secreted ectodomains of well-known BACE1 substrates were reduced in BACE1−/− animals. In addition, we identified and validated a previously unknown BACE1 substrate candidate and confirmed two recently described novel BACE1 substrates. The three proteins may represent novel prognostic or diagnostic biomarkers and may aid in the development of APP-specific BACE1 inhibitors.
EXPERIMENTAL PROCEDURES
Materials
The following antibodies were used: pAb APLP1 antibody (Proteintech, Chicago, IL; 12305–2-AP), pAb APLP2 antibody (Calbiochem; 171617), mouse mAb HA.11 (Covance, Emeryville, CA), rat mAb HA 3F10 (Roche, Rotkreutz, Switzerland), mouse mAb FLAG M2 (Sigma, St. Louis, MO), mouse mAb 3D5 (specific for BACE1, kind gift of R. Vassar)(12), rabbit pAb calnexin (Stressgen, Enzo Life Sciences, Lörrach, Germany), HRP- coupled anti-rabbit and anti-mouse antibody (DAKO, Glostrup, Denmark), HRP coupled anti- rat antibody (Santa Cruz, Dallas, TX). The following reagents were used: Acetonitrile, water, formic acid (all LC-MS/MS grade), dithiothreitol and iodoacetamide were purchased from Sigma-Aldrich. BACE Inhibitor C3 (β-Secretase Inhibitor IV, Calbiochem)(13), Lipofectamine 2000 (Invitrogen).
Mouse Strains
BACE1−/− mice that are commercially available from the Jackson Laboratory (www.jax.org, strain B6.129-Bace1tm1Pcw/J) were maintained according to the European community council directive (86/609/ECC). Wild-type and BACE1−/− mice were both on a C57BL/6 background and obtained from BACE1 ± x BACE1 ± matings. Animals were kept under a 12/12 h light-dark cycle with food and water ad libitum. All animal procedures were carried out in accordance with the European Communities Council Directive (86/609/EEC) and with an animal protocol approved by the Ludwigs-Maximilians-University Munich and the government of Upper Bavaria.
Immunoblots
For the analysis of murine CSF, 7 μl of CSF plus 2.3 μl of 4x Laemmli buffer per sample were incubated at 95 °C for 5 min and loaded on 8% acrylamide gels for immunoblot analysis and detection of APLP1 and APLP2. PVDF membranes (Millipore) were incubated overnight at 4 °C. Blots were developed using horseradish peroxidase-conjugated secondary antibodies and the ECL chemiluminescence system (Millipore). Immunoblots were quantified using the LAS-4000 Fujifilm chemiluminescence camera and software (Fuji Film, Inc.) and quantification was based on three independent replicates. For analysis of HEK293T cell lysates and conditioned media, samples were boiled in reducing Laemmli buffer for 5 min at 95 °C and subsequently applied to 8% SDS-polyacrylamide-gel electrophoresis. Nitrocellulose membranes pore size 0.45 μm were used for transfer, emulsified nonfat dry milk for blocking. Primary antibodies were incubated overnight at 4 °C. The full-length proteins were detected with HA.11 antibody and FLAG M2 antibody. The soluble proteins were detected with HA 3F10 antibody. Blots were developed and quantified as described above for the CSF samples. Full-length and soluble protein levels were normalized to the protein levels of calnexin, which was used as a loading control. Six independent experiments were included for the statistical analysis, applying a one-way ANOVA test.
CSF Collection From Adult Mice
The CSF was isolated from the cisterna magna of 4 month old animals according to the protocol from DeMattos et al. (4). Mice were anesthetized via intraperitoneal injection of a mixture containing ketamine (Bayer, 100 mg/kg body weight) and Rompun (Ratiopharm, 10 mg/kg body weight). A dorsal excision along the base of the skull to the dorsal thorax up to Th1 was made. The musculature was displaced and the meninges on top of the cisterna magna were exposed. The area was cleaned using cotton swabs. The cisterna magna was punctuated and the CSF collected using glass micropipettes (Stoelting, Wood Dale, IL, #50614). CSF samples were subjected to centrifugation on a benchtop centrifuge and visually inspected for the presence of blood in the form of pelleted residual erythrocytes. A total of 5–20 μl of blood-free CSF was collected from each animal and stored at −80 °C. The animals were sacrificed afterward. Samples affected by visible blood contamination were excluded from the analysis. Overall, five BACE1−/− and eight wild-type mice (males and females) were used for the analysis.
Human CSF Samples
Human CSF samples were provided by the Karolinska University Hospital & Institute, Dept. of Clinical Neuroscience, Neuroimmunology Unit. The ethical review board of the Karolinska Institute approved the study (Diary Number: 2009/2107–31-2) and written informed consent was obtained from all patients. Six human CSF samples of patients with minor symptoms were chosen for the analysis.
In-solution Digestion and Peptide Purification
The in-solution digestion was performed according to Olsen et al. (14). All steps were performed at room temperature. Briefly, 5 μl of CSF were solubilized in denaturation buffer (6 m urea (Sigma, U5128) in 10 mm HEPES pH 8.0, sample to buffer ratio 1:10) in low protein binding tubes (Eppendorf). 1 μl of reduction buffer (10 mm dithiothreitol in 0.05 m ammonium bicarbonate) was added for 10 μl of digestion buffer, followed by incubation for 30 min. Then, 1 μl of iodoacetamide solution for 10 μl of digestion buffer was added, followed by incubation for 20 min. 0.5 μg Lys-C solution (Promega, Life Technologies, Darmstadt, Germany) was added and incubated for 4 h. The sample was diluted 4-fold with 50 mm ammonium bicarbonate, 0.4 μg trypsin (Promega, Life Technologies, Darmstadt, Germany) was added and incubated for 16 h. The digestion reaction was stopped using 1 μl of 100% trifluoroacetic acid. Homemade C18 STAGE Tips were used for purification of the in-solution digested samples. Cleanup was done according to Rappsilber et al. (15). The eluted peptides from one digestion reaction (5 μl of CSF) were split into two aliquots and processed on individual STAGE Tips, each representing one technical replicate. Peptides were eluted in 60% acetonitrile, dried by speedvac, resuspended in 10 μl 0.1% trifluoroacetic acid in water and loaded onto the autosampler. 8 μl of the peptide solution was injected per run. Thus, each technical replicate contained the peptides of 2 μl of digested CSF.
Comparison of In-solution Digestion and Filter Aided Sample Preparation (FASP)
Pooled human CSF from six individuals was used to test the efficiency and reproducibility of in-solution digestion and FASP. Three replicates, 5 μl human CSF each, was digested with the in-solution digestion protocol described above and with FASP according to Wisniewski et al. (16). A double digestion with 0.1 μg LysC and 0.1 μg trypsin was performed.
Mass Spectrometry
Samples were analyzed by LC-MS/MS, coupling an Easy nLC1000 nanoflow HPLC system to the Q Exactive benchtop mass spectrometer (both Thermo Fisher Scientific). A two-column setup was used. The pre-column (Acclaim Pep Map 100, 75 μm × 2 cm, nano Viper C18, 3 μm, Thermo Fisher Scientific), waste line and the analytical column (Pep Map RSLC, C18, 2 μm, 75 μm × 50 cm, Thermo Fisher Scientific) were interconnected using a three way tee connector. Peptides were eluted in a trilinear gradient at 50 °C at a maximum pressure of 800 bar. The aqueous solvent (solvent A) consisted of HPLC grade 0.1% formic acid in water, whereas the organic solvent was pure HPLC grade acetonitrile (solvent B) (both Sigma). The column was equilibrated with at least 10 column volumes of solvent A, followed by loading of the sample at maximum pressure of 800 bar (in solvent A). The trilinear gradient consisted of the following linear increases in solvent B: 5–25% 175 min, 25–35% 45 min, 35–60% 20 min. The layout of the gradient was provided by Nagarjuna Nagaraj (Max Planck Institute of Biochemistry, Martinsried, Germany). At the end of the gradient, the column was washed with at least 10 column volumes of 95% solvent B in order to avoid sample carryover. The analysis of BACE1−/− and wild-type samples was alternated.
Online electrospray of the eluting peptides into the Q Exactive mass spectrometer was achieved with the Easy Spray ion source (Thermo Fisher Scientific). Full MS spectra were recorded at a resolution of 70,000 over a mass range between m/z 400–1800. The automatic gain control target was set to 3 × 106 and a maximum injection time of 50 ms was allowed. The 10 most intense peptide ions were chosen for fragmentation. The MS/MS spectra were recorded at a resolution of 17,500 with the automatic gain control target set to 100,000 and a maximum injection time of 50 ms. A mass window of 2.0 m/z was applied to precursor selection. Normalized collisional energy for the higher-energy collision-induced dissociation fragmentation was set to 25%. A dynamic exclusion with a time window of 40 s was applied. Singly charged molecules were not selected for fragmentation and the monoisotopic precursor selection was enabled. The underfill ratio (minimum percentage of the estimated target value at maximum fill time) was set to 0.1%.
Data Analysis
Overall, 26 LC-MS/MS files (five BACE1−/− and eight wild-type CSF samples with two technical replicates each) were subjected to data analysis using the MaxQuant software environment (version 1.3.0.5). The implemented Andromeda search engine was used for matching the peak lists against a concatenated forward and reverse database including the complete UniProt-SwissProt mouse database (release 2012–08, with a total of 59345 database entries) and the standard MaxQuant contaminant database. The following settings were chosen for the MaxQuant software environment: Oxidation of methionines and N-terminal acetylation were set as variable modifications. Mass deviation was set to 20 ppm for the first and 6 ppm for the main search. The maximum number of peptide modifications was set to 5, the maximum number of missed cleavages was set to 2. Peptide and site false discovery rate (FDR) were set to 0.01. The search for co-fragmented peptides in the MS/MS spectra was enabled (“second peptides” option). Quantification was achieved using the LFQ (Label-Free Quantification) and iBAQ (intensity Based Absolute Quantification) algorithms (17, 18). Razor and unique peptides were used for LFQ quantification. Reproducibility of retention times was checked manually. According to this inspection the match between runs option was enabled, allowing a time window of 2 min to search for already identified peptides in all obtained chromatograms. Human CSF samples were searched with the same settings against a human database including all known isoforms from UniProt (reference database, release 2014–04-14).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD001514 (19). Annotated MS/MS spectra are available at the UCSF MS-Viewer (20) (http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer) under following search key: “sxmp8fxdhm.”
Statistical evaluation of the data was performed with the freely available Perseus statistics software (version 1.2.0.17) and Microsoft Excel. Common contaminants and reverse decoy matches were removed from the protein identification list. At least 2 unique peptides per protein were required for a protein identification. Only proteins that were identified and quantifiable in at least one technical of at least three biological replicates in each group were used for relative quantification. The arithmetic mean was used to obtain the average LFQ intensity within each biological group. For statistical evaluation a two-sided t test was used. The p value was corrected using false discovery rate (FDR) based multiple hypothesis testing. Both t test and FDR based multiple hypothesis testing were carried out with the default settings of the Perseus statistics software.
Plasmids pcDNA3.1-CD5-SLIC-FLAG was generated by exchanging RHA-MBP (EcoRV/Not1) of pcDNA3.1-CD5-SLIC-MBP for the PCR product RHA-FLAG amplified with CMV-F and RHA FLAG Not1 rev RHA FLAG Not1 C-terminal from pcDNA3.1-CD5-SLIC-MBP. Subsequently, pcDNA3.1-CD5-HA-SLIC-FLAG was generated by exchanging CD5-LHA in between (HindIII/EcoRV) of pcDNA3.1-CD5-SLIC-FLAG for 5′UTR-CD5-HA-SLIC generated with CMV-F and CD5-HA-LHA rev from pcDNA3.1-HA-IL1R2-FLAG template (21). pcDNA3.1-CD5-HA-SLIC-FLAG-ENPP5, pcDNA3.1-CD5-HA-SLIC-FLAG-PTPRN2 and pcDNA3.1-HA-CD5-SLIC-FLAG-PLXDC2 were generated by cloning PCR products of murine ENPP5, PTPRN2, and PLXDC2 lacking the respective signal peptide and containing the left homology arm (LHA) and the right homology arm (RHA) into EcoRV linearized pcDNA3.1-CD5-HA-SLIC-FLAG vector via Gibson assembly. The resulting plasmids are coding for respectively PLXDC2, PTPRN2 and ENPP5 with an HA epitope tag (YPYDVPDYA) at the N terminus and a FLAG (DYKDDDDK) epitope tag at the C terminus. The generation of peak12-BACE1 and peak12-control vectors has been described (22).
Cell Culture, Transfections, Treatments
Human embryonic kidney 293T cells (HEK293T) were cultured in Dulbecco's modified Eagle medium (DMEM, Gibco, Life Technologies, Darmstadt, Germany) supplemented with 10% fetal calf serum (FCS/Gibco) and 1% Penicillin/Streptomycin (P/S). Transfections of HEK293T cells were done with Lipofectamine 2000. Selection was achieved with Zeocin (Invitrogen) at a concentration of 200 μg/ml. 2 days after transient BACE1 transfection the medium was replaced with either BACE1 inhibitor C3 (2 μm) containing selection medium or DMSO containing selection medium as a control. After 48 h of treatment, conditioned media and cell lysates (150 mm NaCl, 50 mm Tris pH 7.5, 1% Triton X-100, protease inhibitor mixture from Roche) were collected. Protein measurement assay was performed and volumes for immunoprecipitation and immunoblotting were calculated accordingly. To enrich the secreted ectodomains from the supernatants immunoprecipitation using HA.7 agarose (Sigma) was performed overnight at 4 °C.
BACE1 In Vitro Assay
Recombinant proteins HA-PLXDC2-FLAG, HA-PTPRN2-FLAG, and HA-ENPP5-FLAG were purified from cell lysates of stably expressing HEK293T cells using anti-HA-agarose (Sigma). Proteins were eluted by adding 0.1 μg/μl HPLC purified HA peptide (Sigma). The eluted proteins were incubated with recombinant mouse BACE1 (R&D Systems, Minneapolis, MN) in 50 mm Na+ acetate buffer pH 4.4 from 4 to 16 h as described (22). The samples were boiled in reducing Laemmli buffer and applied to Western blot analysis as described above.
RESULTS
For the quantitative proteomic analysis of murine CSF from adult mice, we first evaluated the performance of in-solution digestion versus FASP (16, 23). Given that the amount of blood-free CSF ranged between 10–15 μl per mouse, we used human CSF for the initial comparison of in-solution digestion and FASP. Human CSF has a similar protein concentration as murine CSF, but is available in milliliter quantities. Five μl each of a pooled human CSF sample were used in three replicates for in-solution digestion and FASP. Results were analyzed independently with MaxQuant. In-solution digestion gave slightly better results than FASP regarding the number of identified and quantified proteins (Fig. 1A). On average, 297 and 273 proteins were identified by at least two unique peptides with in-solution digestion and FASP, respectively. The number of quantified proteins in three out of three replicates was 270 for in-solution digestion and 255 for FASP. The Venn diagram (Fig. 1B) shows that FASP is rather complementary to in-solution digestion as the overlap of quantified proteins was only 61%. We also tested the reproducibility of protein quantification using the LFQ intensity values. Both methods performed equally well with an average correlation coefficient R better than 0.99 for log10 transformed LFQ intensities.
Fig. 1.
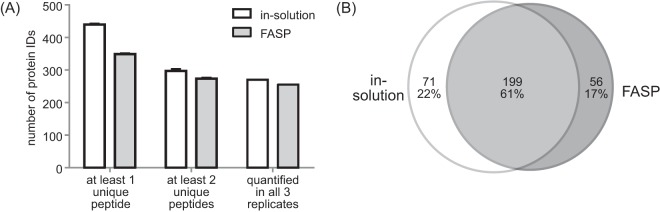
Comparison of in-solution digestion with FASP. A, Comparison of identified and quantified proteins in human CSF. Identifications with one or two unique peptides are shown as average values of three replicates with standard deviation error bars. The third comparison shows proteins that were quantified in three out of three replicates. B, Comparison of the overlap of quantified proteins in all replicates. 61% of the proteins were quantified with both methods, whereas 22 and 17% were only quantified by in-solution digestion or FASP, respectively.
For the quantitative analysis of adult mouse CSF, we chose in-solution digestion because it is less time consuming and gave slightly better results compared with FASP. Five four month old BACE1−/− mice and eight four month old wild-type mice were analyzed. Five μl CSF of each mouse was used for LC-MS/MS analysis. The resulting peptides were split into 2 aliquots, representing one technical replicate each, and were analyzed by LC-MS/MS. Subsequent protein identification and label-free quantification of the raw data was performed in the MaxQuant software environment (Fig. 2).
Fig. 2.

Workflow for the analysis of individual BACE1−/− and wild-type CSF proteomes. The CSF of BACE1−/− mice and wild-type littermates was obtained from the cisterna magna. CSF was digested with Lys-C and trypsin in-solution. Peptides were collected and purified on C18 STAGE Tips. Samples were measured on a 50 cm column using 4 h gradients in two technical replicates on the Q Exactive mass spectrometer, followed by label-free analysis using the MaxQuant algorithm.
Chromatographic conditions between the different runs were highly reproducible, resulting in a high degree of correlation of label-free quantification (LFQ) intensities between technical and biological replicates (Fig. 3A, supplemental Tables S1, S2). Overall, 817 proteins were identified at a false discovery rate of under 1% at the peptide and protein level. 715 proteins were identified with at least 2 unique peptides (Fig. 3B and supplemental Table S1). Five hundred twenty-two of those proteins were quantifiable in at least three BACE1−/− and at least three wild-type mice. Thus, our method allows the accurate identification and quantification of more than 500 proteins in the CSF of individual mice (Fig. 3 B). The majority of proteins quantified in the CSF did not show a gross change in their relative abundance between BACE1−/− and wild-type mice (Fig. 3C). The difference in protein levels was less than twofold for 95% of all quantified proteins and the majority of them clustered around zero (changes in the protein levels is expressed as the log2 fold ratio of BACE1−/− over wild-type samples, Fig. 3C). Therefore, the absence of BACE1 does not lead to major changes in the composition of murine CSF, which is an important finding for the BACE-inhibitors currently under development as a therapeutic approach for Alzheimer's disease.
Fig. 3.

Quality control of the label-free comparative CSF analysis of BACE1−/− mice. A, Upper panel: The log10 LFQ intensities of two selected technical replicates are plotted against each other. Lower panel: The log10 LFQ intensities of two selected biological replicates (two individual BACE1−/− mice) are plotted against each other. Note the high correlation for both biological and technical replicates as indicated by the coefficient of determination being close to 1. B, Number of identified proteins that were detected by at least 1 unique peptide, at least 2 unique peptides and at least 2 unique peptides that were quantifiable in 3 or more biological replicates. LFQ: label-free quantification. ID: protein identification. C, Histogram displaying the relative protein abundances in the BACE1−/− CSF. The log2 fold change of the mean BACE1−/− : wild-type ratio is shown on the x axis. The bin size for the log2 fold change is 0.2. The vast majority of all quantified proteins remained unchanged. Ko: BACE1−/−. Wt: wild type.
The usage of in-solution digestion with a long gradient for LC separation of peptides was superior compared with previous mouse CSF studies and identified more than 2.5 times as many proteins as in previous studies (6, 7) (Fig. 4A), thus providing the most comprehensive list of the murine CSF proteome to date. Our data was analyzed according to UniProt subcellular location terms. Among the 715 identified proteins, 213 (33%) are annotated as secreted and 236 (33%) as cytoplasmic proteins (Fig. 4B), whereas a smaller number are annotated as nuclear (88; 12%) or mitochondrial (59; 6%) proteins. A similar percentage of nuclear (10%) and mitochondrial (2%) proteins has been detected in human CSF and may be partially explained by exosomal secretion, apoptosis of ependymal cells or contamination during CSF extraction (3). One hundred sixty-seven proteins were annotated as membrane proteins (Fig. 4C), with 100 of them being integral membrane proteins (either transmembrane or GPI-anchored). Integral membrane proteins may not only be released as intact membrane proteins by one of the mechanisms above, but alternatively may undergo ectodomain shedding, which is a general physiological process, in which the ectodomain of membrane proteins is proteolytically released from cells for the purpose of cell-cell communication (24). To test this possibility, we analyzed the identified peptides from all 100 integral membrane proteins with our recently developed QARIP (Quantitative Analysis of Regulated Intramembrane Proteolysis) web server (25). QARIP maps all identified peptides of an intrinsic membrane protein to its intracellular-, extracellular- or transmembrane domains. For the majority of proteins annotated as transmembrane or GPI anchored by UniProt (87 out of 100) the peptides mapped exclusively to the extracellular domains (supplemental Table S3–S5). As the majority of proteins feature large extracellular domains and rather small intracellular domains, the discovery of ectodomain derived peptides is statistically more likely. However, also for proteins such as PTPRN2 or NCAM1, where the intracellular domain makes up 39 and 35% of the entire protein (supplemental Table S3), only ectodomain-derived peptides were identified, speaking against a statistical effect. In conclusion, our data suggests that the presence of integral or GPI-anchored protein fragments in the CSF is dependent on proteolytic shedding and not simply because of the release of full-length proteins.
Fig. 4.
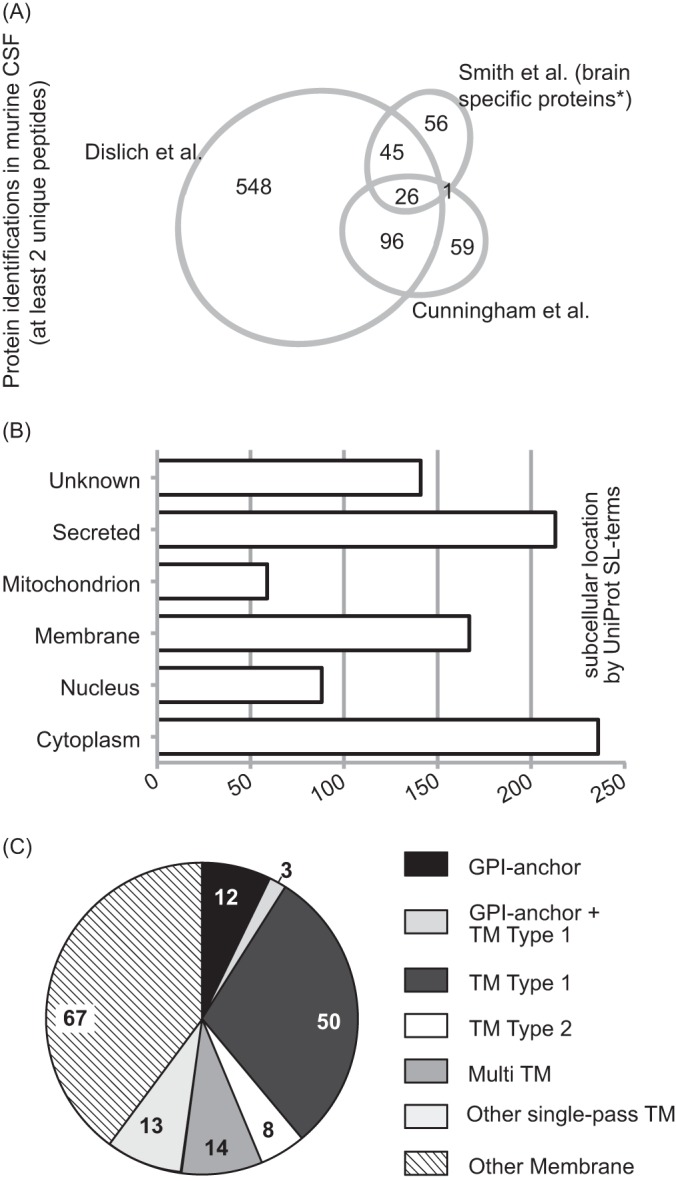
Comparison and distribution of protein identifications with at least 2 unique peptides. A, An area-proportional Venn diagram displays the number of protein identifications of our and two recent murine CSF analyses (6, 7). * Smith et al. identified a total of 261 proteins with at least two unique peptides. One hundred twenty-eight of them were previously found in mouse brain. Only those proteins were used for this Venn diagram, as the other non brain-specific proteins were not supplied in the supplementary table of their manuscript. B, UniProt Keyword subcellular location analysis for cellular components of proteins identified by at least two unique peptides. C, Subanalysis of annotated membrane proteins to GPI-anchored, or transmembrane (TM) type 1 or 2 and multipass according to UniProt subcellular location.
One of the proteases contributing to the ectodomain shedding of transmembrane proteins is BACE1. The BACE1 active-site resides on the extracellular part of the protease, which results in the proteolytic release (shedding) of the substrate ectodomain into the extracellular compartment (11, 24). We therefore assumed that membrane protein ectodomains that are released into the CSF by BACE1 should be less abundant in the CSF of BACE1−/− mice and that these differences should be quantifiable. Fig. 5 shows a volcano plot with the mean fold change of all biological replicates (expressed as the log2 fold change of the BACE1−/− : wild-type ratio) plotted against their corresponding p values. Fifty-eight proteins had a p value ≤ 0.05 and are above the horizontal line. This included 42 proteins with reduced protein levels and 16 proteins with increased levels in the BACE1−/− CSF. Proteins with increased levels include several lipoproteins, such as ApoA1 and ApoC3, which may indicate a role of BACE1 activity in lipid metabolism. However, given that they are soluble proteins and that their levels are increased in BACE1−/− CSF, they are unlikely to be direct BACE1 substrates. Only membrane proteins that show reduced levels of their ectodomains in the CSF of BACE1−/− compared with wild-type mice are considered possible BACE1 substrates. Altogether, 100 transmembrane or GPI-anchored proteins were identified in the entire data set (Fig. 4C). Eighteen of them are among the 42 significantly reduced proteins in the BACE1−/− CSF and thus may be candidate BACE1 substrates (Table I). All 168 peptides detected from these proteins strictly mapped to their extracellular domains (Table I), which indicates that their release is because of ectodomain shedding (e.g. by BACE1) and not because of the release of the full-length transmembrane proteins. 10 of the 18 membrane proteins showed a significant (p value ≤ 0.05) reduction in CSF of more than 1.5-fold (APLP1, APLP2, ENPP5, PTPRN2, CHL1, PLXDC2, APP, DAG1, CDH2, CNTN2), with five of them (APLP1, APLP2, ENPP5, PTPRN2, CHL1) being reduced by even more than twofold (Fig. 5). Ten of the 18 proteins represent previously identified BACE1 substrates or substrate candidates (APP, APLP1, APLP2, CHL1, CNTN2, NCAM1, PLXDC2, PAM, PTPRN2, Sez6L2) (26–30). CELSR2 represents a G-protein coupled receptor with seven transmembrane domains, featuring a topology that is rather unusual for a BACE1 substrate. However, a previous study identified several G-protein coupled receptors (Latrophilin 1–3) as putative BACE1 substrates in primary cortical neurons (27). The additional membrane proteins (ENPP5, DAG1, CDH2, EFNB3, ATP6AP1, CDH13, and QSOX1) may be novel BACE1 substrate candidates. Among the 18 membrane proteins, the well-known BACE1 substrates APLP1 and APLP2 (27, 30–35) showed the strongest decrease (5.4- and 2.9-fold respectively) (Fig. 5). The known BACE1 substrates CHL1, APP and CNTN2 were also significantly decreased, although to a lesser extent (2-, 1.7-, and 1.5-fold respectively). Taken together, these results demonstrate a) that BACE1-dependent release of soluble ectodomains can be monitored in the CSF and b) that BACE1 substrates can be identified from mouse CSF (Fig. 5).
Fig. 5.

Identification and validation of putative BACE1 substrates in mouse CSF. Each circle represents a protein that was quantifiable in at least three biological replicates. The log2 fold change of the mean BACE1−/− : wild-type ratio is plotted against the -log10 of the p value. The vertical bars mark a fold change of at least 1.5 and greater than the twofold standard deviation (corresponding to a reduction of 1.86-fold). The horizontal bar shows the cutoff for statistical significance (p value 0.05 or below). Proteins above this line have a p value < 0.05. Proteins shown as black and gray filled circles are regarded putative BACE1 substrates, as they are decreased in the absence of BACE1 and are annotated as membrane proteins. Gene names in “bold” mark previously known BACE1 substrates. The circle filled in black indicates that this protein remains significantly changed after a false discovery rate based multiple hypothesis testing. Gene names in regular font show proteins that are significantly decreased or up-regulated in the CSF of BACE1−/− mice by at least 1.5-fold. Gene names in “italic” show integral or GPI-anchored membrane proteins that are significantly decreased in the CSF of BACE1−/− mice, but show a fold change below 1.5. One non-significant outlier is not shown in Fig. 3 in order to save space for visualizing the data (UniProt ID P05063; -3.39, 0.71).
Table I. Summary of all integral or GPI-anchored membrane proteins that are significantly decreased in the CSF of BACE1−/− mice. Proteins are ranked by their fold change (mean of BACE1−/− to wild-type ratio), thus displaying the proteins with the strongest reduction in the BACE1−/− CSF on top. Proteins shown on dark grey background remain significantly changed after a false discovery based multiple hypothesis testing. Proteins that are shown on light and dark grey background show at least a 1.5-fold reduction in the BACE1−/− CSF. Previously identified: Theses proteins have been identified as BACE1 substrates or BACE1 substrate candidates in previous studies. a) p value: All proteins shown have a p value <0.05 according to a Student's t-test comparing at least three biological replicates of wild type and BACE1−/− mice b) Topology: Protein topology as predicted by the PHOEBIUS web server. c) Peptide distribution: Shown is the graphical output of the QARIP web server. A schematic of each protein is shown, that shows the distribution of the different topological regions and the identified peptides within the corresponding protein. Peptides that were detected during LC-MS/MS measurements are shown in black. The individual protein domains are shown in brown (signal peptide), blue (extracellular domain), yellow (transmembrane domain) and green (intracellular domain). All detected peptides originate from the extracellular domains, suggesting that they are released into the CSF via ectodomain shedding.
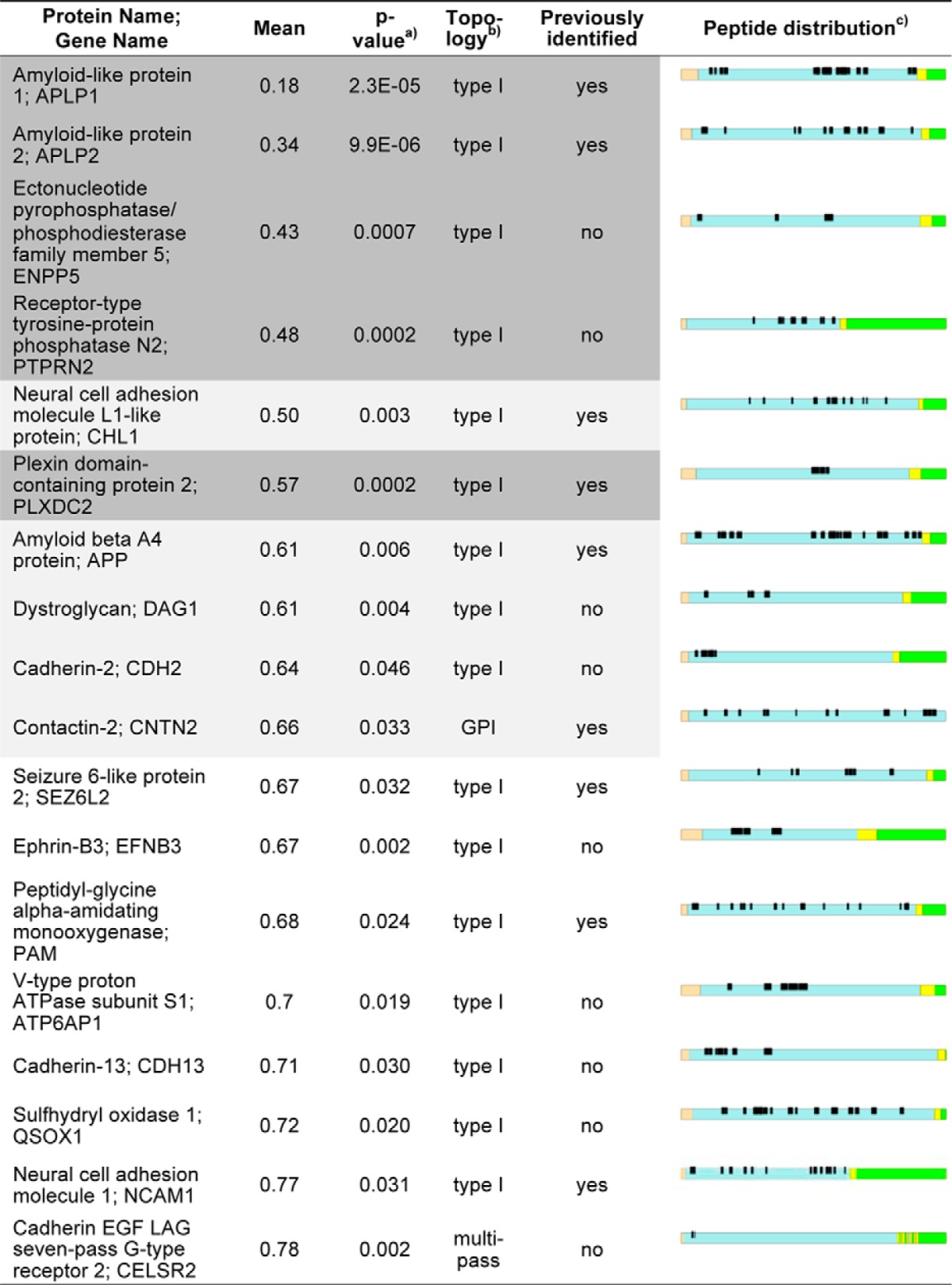
After applying a false discovery rate-based multiple hypothesis testing, five proteins (APLP1, APLP2, PTPRN2, PLXDC2, ENPP5) remained significantly decreased in BACE1−/− mice in comparison to their wild-type littermates (Fig. 5 and Table I). These five proteins, which are all type I membrane proteins, comprise the known BACE1 substrates APLP1 and APLP2 as well as the recently described BACE1 substrates PLXDC2 and PTPRN2 (26–28). In addition the type I membrane protein ENPP5 (ectonucleotide pyrophosphatase 5) was significantly reduced in BACE1−/− CSF, making ENPP5 a novel BACE1 substrate candidate (Fig. 5 and Table I).
The individual LFQ intensities measured for these top five proteins are shown in Fig. 6, as well as their relative abundance in the entire data set expressed by their iBAQ score (18). The iBAQ intensities of all quantified proteins ranged over seven orders of magnitude, with the top 5 BACE1 substrates distributed in the middle of this range. This indicates that our workflow allows accurate quantification of BACE1 substrates despite the presence of proteins which are 103–104 times more abundant. Notably, the relative protein abundances, as determined by the IBAQ scores, were similar to the protein abundances in human CSF (supplemental Fig. S1, supplemental Table S6), which was analyzed using the same workflow as the murine CSF.
Fig. 6.

Top 5 proteins that are decreased in the BACE1−/− CSF. A, The LFQ values of the top 5 significantly regulated proteins are shown for each biological replicate. LFQ values are normalized to the wild-type mean of all biological replicates (set to 1) for a given protein. B, Relative abundance of all observed proteins relative to each other as calculated by the iBAQ algorithm. Each protein identified in the CSF data set is displayed as a black circle and ranked by its relative abundance in the CSF. The iBAQ (intensity Based Absolute Quantification) score is calculated by dividing the total ion intensity of all observed peptides by the theoretical number of all observable peptides of a protein (18). The iBAQ score thus ranks the proteins in a data set by their approximate abundance, the proteins of highest abundance on top. LFQ: label-free quantification.
To further validate our findings and methodology, we obtained CSF from an independent set of three BACE1−/− and three wild-type mice. First, we validated our mass spectrometric data for the known BACE1 substrates APLP1 and APLP2. To this aim, the CSF was subjected to Western blotting. Similar to our mass spectrometry-based results, the ectodomains of both proteins were strongly reduced in BACE1−/− mouse CSF (Fig. 7A). Moreover, the densitometric quantification of the secreted ectodomains demonstrated a confirmatory correlation with our label-free quantification results, with a 7.5-fold reduction for APLP1 and a 3.5-fold reduction for APLP2 (Fig. 7B).
Fig. 7.
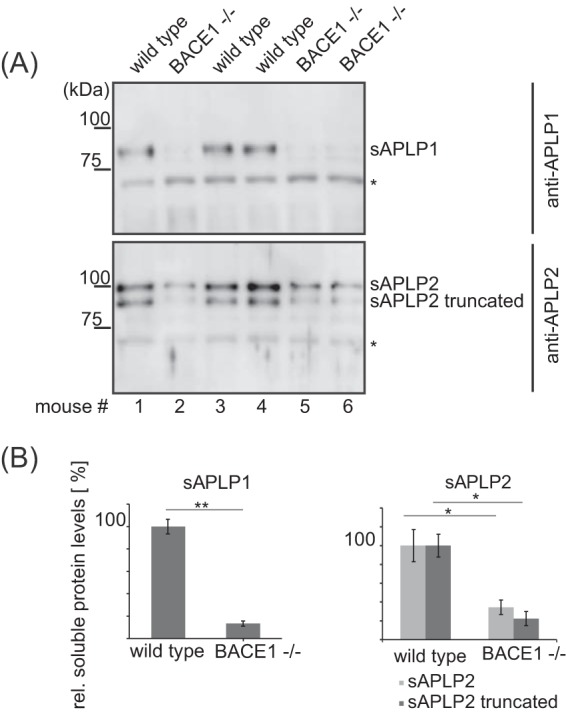
Western blot analysis of APLP1 and APLP2 in murine CSF. A, The CSF of individual mice of an independent set of animals was subjected to SDS-PAGE and analyzed by Western blotting. Antibodies against APLP1 and APLP2 were raised against the extracellular domains of these proteins and detect the secreted ectodomains (sAPLP1 and sAPLP2) in the CSF. Two bands are detected for APLP2, one of them is considered to be a truncated form of mature APLP2 (35). Unspecific bands detected by both antibodies are marked with an asterisk (*) and show that equal amounts of CSF were loaded per lane. B, Densitometric quantitation of the Western blot shown in A) (*: p < 0.05, **: p < 0.01; two-tailed Student's t-Test, n = 3).
Next we studied the three additional proteins PLXDC2, PTPRN2 and ENPP5 in order to further confirm the mass spectrometric data. In contrast to APLP1 and APLP2 no antibody was available allowing successful detection of the proteins' ectodomains in mouse CSF. Thus, an HA epitope tag was added to the N terminus of the ectodomains and additionally a FLAG epitope tag to the C terminus of the full-length proteins. The recombinant proteins were stably expressed in human embryonic kidney 293 (HEK293) cells and their processing by BACE1 was analyzed. The shed ectodomains were detected by immunoblot as faint bands in the supernatant. The full-length proteins were seen in the cell lysate and showed specific bands at their predicted apparent molecular weights (Fig. 8A–8C). As a control, these protein bands were not seen in the lysate or supernatant of cells transfected with the empty vector. Because endogenous BACE1 is highly expressed in the nervous system, but at very low levels in non-neuronal tissue (36), the HEK293 cells were additionally transiently transfected with a BACE1 or a control plasmid. Compared with the control transfected cells, transfection of BACE1 strongly increased the shedding of the ectodomains into the conditioned medium for all three proteins, PLXDC2, PTPRN2 and ENPP5 (Fig. 8). Addition of the specific BACE1 inhibitor C3 (13) largely blocked this increase in substrate ectodomain shedding, demonstrating that the proteolytic activity of BACE1 was required for the increased cleavage of the three proteins.
Fig. 8.

BACE1 increases shedding of PLXDC2, PTPRN2 and ENPP5. Shown are immunoblots of the indicated proteins. Human embryonic kidney HEK293T cells were stably transfected with HA and FLAG epitope-tagged proteins (HA-PLXDC2-FLAG (A), HA-PTPRN2-FLAG (B), HA-ENPP5-FLAG (C) or the empty pcDNA3.1 vector as control (CON). Additionally, the stable cells were transiently transfected with BACE1 or empty peak12 vector as indicated. Additionally the cells were treated with either the BACE1 inhibitor C3 or DMSO. The recombinant full-length (fl) proteins PLXDC2, PTPRN2 and ENPP5 were detected in the cell lysate (lys) using the N-terminally binding HA antibody and the C-terminally binding FLAG antibody (indicated by arrowheads). The soluble (s) ectodomains sPLXDC2, sPTPRN2, sENPP5 were immunoprecipitated from the conditioned medium with the HA.7 monoclonal antibody and detected using the HA monoclonal antibody 3F10 (indicated by arrowheads). Calnexin served as a loading control. BACE1 was detected by 3D5 antibody. Shown are representative immunoblots from three independent experiments.
To further demonstrate that the increased cleavage is because of a direct cleavage by BACE1 and not merely because of secondary effects, the epitope-tagged proteins were immunoprecipitated from the cell lysates using an antibody against the HA-tag and subject to an in vitro cleavage assay by purified, recombinant BACE1. As a result of this assay, BACE1 cleavage should increase the generation of the same shed ectodomain, which was also seen in the supernatant of the transfected HEK293 cells. Indeed, in the absence of BACE1 in the in vitro assay, only full-length PLXDC2 was detected (Fig. 9A). When BACE1 was added to the assay, the intensity of the full-length band was reduced and a cleaved PLXDC2 ectodomain was detected using the HA antibody for detection. Importantly, the cleaved PLXDC2 had the same apparent molecular weight as the shed PLXDC2 ectodomain in the supernatant of the transfected HEK293 cells (Fig. 9A compare with Fig. 8A), demonstrating that PLXDC2 cleavage in the cells occurs in the same way as in the in vitro assay. As controls, both the full-length protein and the shed ectodomain were not detected, when the assay was performed in the absence of PLXDC2 expression (Control) or when only recombinant BACE1 was loaded (BACE1).
Fig. 9.

BACE1 in vitro assay. Recombinant HA-PLXDC2-FLAG (A), HA-PTPRN2-FLAG (B), HA-ENPP5-FLAG (C) were purified from cell lysates of stably transfected HEK293T cells by immunoprecipitation (using antibody HA.7) and digested with or without BACE1 (B1) in vitro. Detection of full-length proteins and their N-terminal, soluble ectodomains (resulting from BACE1 cleavage) was done using the HA 3F10 antibody. Control: immunoprecipitation from HEK293T cells stably expressing the empty vector. The full-length (fl) proteins flPLXDC2, flPTPRN2 and flENPP5 (indicated by arrowheads) and the soluble (s) ectodomains sPLXDC2, sPTPRN2 and sENPP5 (indicated by arrowheads) were detected using the HA 3F10 monoclonal antibody. BACE1: recombinant BACE1 was loaded without any purified substrate protein. Shown is a representative blot of three independent experiments. *: PTPRN2 degradation product. ** unspecific band.
Similar results were obtained for PTPRN2 and ENPP5. In the absence of BACE1, full-length PTPRN2 (arrowhead, flPTPRN2) as well as several presumed degradation products were detected. In the presence of BACE1 the full-length protein was reduced and a 65 kDa fragment was generated as well as a less intensive 55 kDa fragment (Fig. 9B). The 65 kDa fragment corresponds exactly to the shed PTPRN2 ectodomain observed in the conditioned medium of HEK293 cells (Fig. 9B).
For ENPP5, in the absence of BACE1 the full-length protein at 80 kDa was detected as well as a smaller fragment of around 70 kDa (Fig. 9C). After addition of BACE1 the full-length protein levels were reduced, whereas the 70 kDa fragment was strongly increased, which has the same apparent molecular weight as the shed ectodomain in HEK293 cells (Fig. 8C). Given the low levels of the 70 kDa fragment in the absence of BACE1 it is likely that ENPP5 is efficiently cleaved by BACE1 in cells, such that the purification of full-length ENPP5 with the HA antibody copurified lower levels of the BACE1-generated ENPP5 ectodomain.
Taken together, the HEK293 cell experiments and the in vitro assays validate ENPP5 and confirm PTPRN2 and PLXDC2 as BACE1 substrates and demonstrate their direct cleavage by BACE1 in the in vitro assay and their cleavage by BACE1 in cells.
DISCUSSION
In this study we show for the first time that the CSF proteome of individual mice can be analyzed in a quantitative manner. Our simple workflow allows the in-depth investigation of murine CSF of individual mice with low sample volumes in ∼4.5 h per sample. It combines the simplicity of an in-solution digestion with ultra-long analytical HPLC columns in conjunction with a mass spectrometer providing fast scan speed while maintaining a high resolution. It avoids the time and sample consuming protein concentration, depletion and fractionation steps that were required for the analysis of murine CSF in the past. Additionally, we compared in-solution digestion with FASP. In-solution digestion showed to be superior to FASP with regards to the number of protein identifications as well as quantifications. However, FASP seems to be a complementary method to in-solution digestion, as the overlap of proteins identified by both methods was only 61%.
Moreover, our workflow increases proteomic depth, as the number of protein identifications were two- to threefold higher compared with previous studies (6, 7). This is a clear step forward toward the goal of determining the whole mouse CSF proteome. Additionally, 55–67% of previously identified proteins in murine CSF were confirmed in our study (Fig. 4A). Some of the previously identified proteins might only be detectable in murine CSF on previous immunodepletion or extensive fractionation, because of their low expression levels or the presence of highly abundant proteins such as albumin. This could explain why the overlap between the different studies is not complete and why some well-known BACE1 substrates such as seizure protein 6 were not quantified in our analysis (27, 28). Our simplified workflow minimizes the variance during sample processing, which is helpful for the accuracy of label-free quantification. The quantitative results of the top two BACE1 substrates in this data set were validated by Western blot analysis and demonstrate high correlation of protein levels between our mass spectrometry based methodology and immunoblotting. In our hands label-free quantification is currently the best strategy to investigate murine CSF, as SILAC labeling of entire mice is extremely expensive and time consuming, whereas chemical labeling of the small sample amounts may increase variation between samples. As our method only requires 5 μl of CSF from one mouse (for two technical replicates), the longitudinal CSF sampling of mice is possible, as up to 8 μl of CSF can be harvested from an adult mouse every 2–3 months (37). Mice are the most common animal model in clinical research, including a large variety of neurological diseases (38). Therefore, our method will be of great value for investigating the CSF of those mice. Additionally, preclinical findings in the CSF can be rapidly transferred to the clinic. For example, the identified BACE1 substrates may be used for monitoring the response to BACE inhibitors during clinical inhibition studies as we found the great majority of them to be also detectable in human CSF.
Analysis with the QARIP webserver revealed that a significant number of proteins detected in murine CSF are derived from ectodomain shedding, which has also been suggested by a previous study analyzing the human CSF peptidome and proteome (39). Thus, mouse CSF, for example from wild-type and protease-deficient mice, is generally well suited for the identification of protease substrates in vivo.
This is the first proteomic analysis of the CSF of the BACE1−/− mouse model and demonstrates that the activity of the BACE1 protease can be monitored by quantifying the amount of released BACE1 substrate ectodomains into this body fluid. Our data suggests that BACE1 is not an exclusive sheddase for many of the proteins released into the CSF, as their shedding is not completely abolished in the absence of BACE1. This was also confirmed on analysis by Western blotting. Although APLP1 was almost absent in CSF, APLP2 was reduced less drastically, which is in agreement with our previous proteomics study of primary cortical neurons treated with a BACE1 inhibitor (27). This is most likely because of the compensatory or parallel proteolysis by alternative sheddases.
BACE1 is the rate-limiting enzyme of Aβ production and is a major drug target for the therapy of Alzheimer's disease (40). The interest in BACE1 is because of two reasons: On the one hand, orally available BACE1 inhibitors have recently shown their Aβ-lowering potential in humans and non-human primates (41, 42). On the other hand, an increasing number of BACE1 substrates is described, together with the elucidation of a complex phenotype in BACE1−/− mice, raising concerns about mechanism-based side effects (11). Our study validates previously identified BACE1 substrates in the CSF. In addition, we identify ENPP5 as a novel BACE1 substrate. Moreover, we confirm the recently described substrates PLXDC2 and PTPRN2 (26–28) in vitro and in cell culture. ENPP5 belongs to the family of nucleotide pyrophosphatases and is highly expressed in rat brain. However, in contrast to other nucleotide pyrophosphatase members, its catalytic activity could not be demonstrated so far and no other physiological function has been described (43, 44).
PLXDC2 has been identified as a BACE1 substrate by two independent proteomic studies, where the pharmacologic inhibition of BACE1 led to a decrease of detectable PLXDC2 in the conditioned media of HEK293 cell and cultivated primary neurons (26, 29). It has been shown to regulate neuronal cell fate and proliferation. Interestingly, the ectodomain itself is sufficient for the observed effects on neurogenesis (45). BACE1 could thus control the function of this protein by regulating its cell surface levels as well as the amount and distribution of soluble, functional ectodomains.
PTPRN2 has been first described as a BACE1 substrate in the murine endocrine pancreas. This was demonstrated by a reduction of PTPRN2 protein levels in the conditioned media and the according increase of protein levels in the cell lysate of BACE1 deficient islets using SRM/MRM-based mass spectrometry (28). In addition, PTPRN2 has been shown to regulate insulin secretion (46). It is expressed throughout the CNS, however its role in the nervous system has not been investigated so far (47). The functional analysis of these proteins in future studies may broaden our understanding of the complex phenotype of BACE1−/− mice as well as mechanism-based side effects on therapeutic inhibition of BACE1. Furthermore, the expanding panel of physiologic BACE1 substrates in the CSF may aid in the development of prognostic and diagnostic biomarkers and drug development. Our methodology is well-suited to also analyze CSF samples derived from non-human primates or patients treated with BACE1 inhibitors in drug development and clinical studies, respectively. This may allow the determination of which membrane proteins are predominant BACE1 substrates in humans. Such substrates may also lead to the development of APP-specific BACE inhibitors that spare other substrates, as it has been described for Notch-sparing γ-secretase inhibitors (48). Furthermore, a panel of physiological substrates that can routinely be monitored in the CSF will aid in evaluating the drug response in individual patients undergoing therapy and improving personalized medicine approaches for the treatment of Alzheimer's disease.
Supplementary Material
Acknowledgments
We thank Katrin Moschke for excellent technical support. We also thank Prof. Tomas Olsson (Karolinska University Hospital & Institute, Dept. of Clinical Neuroscience, Neuroimmunology Unit) and Franziska Hoffmann (Institute for Clinical Neuroimmunology, University Hospital LMU Munich, Max-Planck-Institute) for providing us the human CSF samples.
Footnotes
Author Contributions: The manuscript was written by Bastian Dislich and Stefan F. Lichtenthaler. The study was guided and planned by Bastian Dislich, Sebastian Hogl, Melanie Meyer-Luehmann and Stefan F. Lichtenthaler. Bastian Dislich established the mass spectrometry of murine CSF and performed the proteomic analysis of the BACE1−/− CSF. Felix Wohlrab performed all other experiments, except for the CSF extraction, which was performed by Teresa Bachhuber, and the analysis of human CSF, which was carried out by Stephan Müller. Peer-Hendrik Kuhn developed the expression vectors/plasmids that were used in this study. All authors have given approval to the final version of the manuscript.
* This work was supported by the DFG (SyNergy), the BMBF (RiMOD-FTD), the Verum Foundation, IWT, the Breuer Foundation Research Prize, and the TUM School of Medicine.
 This article contains supplemental Fig. S1 and Tables S1 to S6.
This article contains supplemental Fig. S1 and Tables S1 to S6.
1 The abbreviations used are:
- CSF
- cerebrospinal fluid
- APP
- amyloid precursor protein
- BACE1
- β-site APP cleaving enzyme 1
- FASP
- filter aided sample preparation
- FDR
- false discovery rate
- iBAQ
- intensity based absolute quantification
- LFQ
- label-free quantification.
REFERENCES
- 1. Romeo M. J., Espina V., Lowenthal M., Espina B. H., Petricoin E. F. 3rd, Liotta L. A. (2005) CSF proteome: a protein repository for potential biomarker identification. Expert Rev. Proteomics 2, 57–70 [DOI] [PubMed] [Google Scholar]
- 2. Craft G. E., Chen A., Nairn A. C. (2013) Recent advances in quantitative neuroproteomics. Methods 61, 186–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schutzer S. E., Liu T., Natelson B. H., Angel T. E., Schepmoes A. A., Purvine S. O., Hixson K. K., Lipton M. S., Camp D. G., Coyle P. K., Smith R. D., Bergquist J. (2010) Establishing the proteome of normal human cerebrospinal fluid. PLoS ONE 5, e10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeMattos R. B., Bales K. R., Parsadanian M., O'Dell M. A., Foss E. M., Paul S. M., Holtzman D. M. (2002) Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J. Neurochem. 81, 229–236 [DOI] [PubMed] [Google Scholar]
- 5. Rudick R. A., Zirretta D. K., Herndon R. M. (1982) Clearance of albumin from mouse subarachnoid space: a measure of CSF bulk flow. J. Neurosci. Meth. 6, 253–259 [DOI] [PubMed] [Google Scholar]
- 6. Cunningham R., Jany P., Messing A., Li L. (2013) Protein changes in immunodepleted cerebrospinal fluid from a transgenic mouse model of Alexander disease detected using mass spectrometry. J. Proteome Res. 12, 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith J. S., Angel T. E., Chavkin C., Orton D. J., Moore R. J., Smith R. D. (2014) Characterization of individual mouse cerebrospinal fluid proteomes. Proteomics 14, 1102–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dislich B., Lichtenthaler S. F. (2012) The Membrane-Bound Aspartyl Protease BACE1: Molecular and Functional Properties in Alzheimer's Disease and Beyond. Front. Physiol. 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 10. Ghosh A. K., Osswald H. L. (2014) BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer's disease. Chem. Soc. Rev. 43, 6765–6813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vassar R., Kuhn P. H., Haass C., Kennedy M. E., Rajendran L., Wong P. C., Lichtenthaler S. F. (2014) Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem. 130, 4–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao J., Fu Y., Yasvoina M., Shao P., Hitt B., O'Connor T., Logan S., Maus E., Citron M., Berry R., Binder L., Vassar R. (2007) Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J. Neurosci. 27, 3639–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stachel S. J., Coburn C. A., Steele T. G., Jones K. G., Loutzenhiser E. F., Gregro A. R., Rajapakse H. A., Lai M. T., Crouthamel M. C., Xu M., Tugusheva K., Lineberger J. E., Pietrak B. L., Espeseth A. S., Shi X. P., Chen-Dodson E., Holloway M. K., Munshi S., Simon A. J., Kuo L., Vacca J. P. (2004) Structure-based design of potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1). J. Med. Chem. 47, 6447–6450 [DOI] [PubMed] [Google Scholar]
- 14. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 15. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 16. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 17. Luber C. A., Cox J., Lauterbach H., Fancke B., Selbach M., Tschopp J., Akira S., Wiegand M., Hochrein H., O'Keeffe M., Mann M. (2010) Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32, 279–289 [DOI] [PubMed] [Google Scholar]
- 18. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 19. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baker P. R., Chalkley R. J. (2014) MS-Viewer: A Web-based Spectral Viewer for Proteomics Results. Mol. Cell. Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuhn P. H., Marjaux E., Imhof A., De Strooper B., Haass C., Lichtenthaler S. F. (2007) Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J. Biol. Chem. 282, 11982–11995 [DOI] [PubMed] [Google Scholar]
- 22. Lichtenthaler S. F., Dominguez D. I., Westmeyer G. G., Reiss K., Haass C., Saftig P., De Strooper B., Seed B. (2003) The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J. Biol. Chem. 278, 48713–48719 [DOI] [PubMed] [Google Scholar]
- 23. Wiśniewski J. R., Ostasiewicz P., Mann M. (2011) High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 10, 3040–3049 [DOI] [PubMed] [Google Scholar]
- 24. Lichtenthaler S. F., Haass C., Steiner H. (2011) Regulated intramembrane proteolysis–lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796 [DOI] [PubMed] [Google Scholar]
- 25. Ivankov D. N., Bogatyreva N. S., Hönigschmid P., Dislich B., Hogl S., Kuhn P. H., Frishman D., Lichtenthaler S. F. (2013) QARIP: a web server for quantitative proteomic analysis of regulated intramembrane proteolysis. Nucleic Acids Res. 41, W459–W464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hemming M. L., Elias J. E., Gygi S. P., Selkoe D. J. (2009) Identification of β-Secretase (BACE1) Substrates Using Quantitative Proteomics. PLoS ONE 4, e8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuhn P. H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., Volbracht C., Schepers U., Imhof A., Hoffmeister A., Haass C., Roßner S., Bräse S., Lichtenthaler S. F. (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 31, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stützer I., Selevsek N., Esterházy D., Schmidt A., Aebersold R., Stoffel M. (2013) Systematic proteomic analysis identifies beta-site amyloid precursor protein cleaving enzyme 2 and 1 (BACE2 and BACE1) substrates in pancreatic beta-cells. J. Biol. Chem. 288, 10536–10547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou L., Barão S., Laga M., Bockstael K., Borgers M., Gijsen H., Annaert W., Moechars D., Mercken M., Gevaert K., De Strooper B. (2012) The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J. Biol. Chem. 287, 25927–25940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hogl S., van Bebber F., Dislich B., Kuhn P. H., Haass C., Schmid B., Lichtenthaler S. F. (2013) Label-free quantitative analysis of the membrane proteome of Bace1 protease knock-out zebrafish brains. Proteomics 13, 1519–1527 [DOI] [PubMed] [Google Scholar]
- 31. Eggert S., Paliga K., Soba P., Evin G., Masters C. L., Weidemann A., Beyreuther K. (2004) The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J. Biol. Chem. 279, 18146–18156 [DOI] [PubMed] [Google Scholar]
- 32. Li Q., Südhof T. C. (2004) Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J. Biol. Chem. 279, 10542–10550 [DOI] [PubMed] [Google Scholar]
- 33. Pastorino L., Ikin A. F., Lamprianou S., Vacaresse N., Revelli J. P., Platt K., Paganetti P., Mathews P. M., Harroch S., Buxbaum J. D. (2004) BACE (beta-secretase) modulates the processing of APLP2 in vivo. Mol Cell Neurosci 25, 642–649 [DOI] [PubMed] [Google Scholar]
- 34. Sala Frigerio C., Fadeeva J. V., Minogue A. M., Citron M., Van Leuven F., Staufenbiel M., Paganetti P., Selkoe D. J., Walsh D. M. (2010) beta-Secretase cleavage is not required for generation of the intracellular C-terminal domain of the amyloid precursor family of proteins. FEBS J. 277, 1503–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hogl S., Kuhn P. H., Colombo A., Lichtenthaler S. F. (2011) Determination of the proteolytic cleavage sites of the amyloid precursor-like protein 2 by the proteases ADAM10, BACE1 and gamma-secretase. PLoS ONE 6, e21337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 37. Liu L., Duff K. (2008) A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J. Vis. Exp. 21, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chesselet M. F., Carmichael S. T. (2012) Animal models of neurological disorders. Neurotherapeutics 9, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zougman A., Pilch B., Podtelejnikov A., Kiehntopf M., Schnabel C., Kumar C., Mann M. (2008) Integrated analysis of the cerebrospinal fluid peptidome and proteome. J. Proteome Res. 7, 386–399 [DOI] [PubMed] [Google Scholar]
- 40. Yan R., Vassar R. (2014) Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 13, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. May P. C., Dean R. A., Lowe S. L., Martenyi F., Sheehan S. M., Boggs L. N., Monk S. A., Mathes B. M., Mergott D. J., Watson B. M., Stout S. L., Timm D. E., Smith Labell E., Gonzales C. R., Nakano M., Jhee S. S., Yen M., Ereshefsky L., Lindstrom T. D., Calligaro D. O., Cocke P. J., Greg Hall D., Friedrich S., Citron M., Audia J. E. (2011) Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J. Neurosci. 31, 16507–16516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sankaranarayanan S., Holahan M. A., Colussi D., Crouthamel M. C., Devanarayan V., Ellis J., Espeseth A., Gates A. T., Graham S. L., Gregro A. R., Hazuda D., Hochman J. H., Holloway K., Jin L., Kahana J., Lai M. T., Lineberger J., McGaughey G., Moore K. P., Nantermet P., Pietrak B., Price E. A., Rajapakse H., Stauffer S., Steinbeiser M. A., Seabrook G., Selnick H. G., Shi X. P., Stanton M. G., Swestock J., Tugusheva K., Tyler K. X., Vacca J. P., Wong J., Wu G., Xu M., Cook J. J., Simon A. J. (2009) First demonstration of cerebrospinal fluid and plasma A beta lowering with oral administration of a beta-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates. J. Pharmacol. Exp. Ther. 328, 131–140 [DOI] [PubMed] [Google Scholar]
- 43. Massé K., Bhamra S., Allsop G., Dale N., Jones E. A. (2010) Ectophosphodiesterase/nucleotide phosphohydrolase (Enpp) nucleotidases: cloning, conservation and developmental restriction. Int. J. Dev. Biol. 54, 181–193 [DOI] [PubMed] [Google Scholar]
- 44. Ohe Y., Ohnishi H., Okazawa H., Tomizawa K., Kobayashi H., Okawa K., Matozaki T. (2003) Characterization of nucleotide pyrophosphatase-5 as an oligomannosidic glycoprotein in rat brain. Biochem. Biophys. Res. Commun. 308, 719–725 [DOI] [PubMed] [Google Scholar]
- 45. Miller S. F., Summerhurst K., Rünker A. E., Kerjan G., Friedel R. H., Chédotal A., Murphy P., Mitchell K. J. (2007) Expression of Plxdc2/TEM7R in the developing nervous system of the mouse. Gene Expression Patterns 7, 635–644 [DOI] [PubMed] [Google Scholar]
- 46. Caromile L. A., Oganesian A., Coats S. A., Seifert R. A., Bowen-Pope D. F. (2010) The neurosecretory vesicle protein phogrin functions as a phosphatidylinositol phosphatase to regulate insulin secretion. J. Biol. Chem. 285, 10487–10496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wasmeier C., Hutton J. C. (1996) Molecular cloning of phogrin, a protein-tyrosine phosphatase homologue localized to insulin secretory granule membranes. J. Biol. Chem. 271, 18161–18170 [DOI] [PubMed] [Google Scholar]
- 48. Augelli-Szafran C. E., Wei H. X., Lu D., Zhang J., Gu Y., Yang T., Osenkowski P., Ye W., Wolfe M. S. (2010) Discovery of notch-sparing gamma-secretase inhibitors. Curr. Alzheimer Res. 7, 207–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.